
Phylogenetic Analysis and Spread of HPAI H5N1 in Middle Eastern Countries Based on Hemagglutinin and Neuraminidase Gene Sequences


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Methodology
2.1. Study Design and Inclusion/Exclusion Criteria
2.1.1. Nucleotide Sequences
2.1.2. Amino Acid Sequences
2.2. Molecular Evolutionary Genetics Analysis (MEGA) Version 11
2.3. Phylogenetic Analyses
2.4. Amino Acid Analysis
3. Results
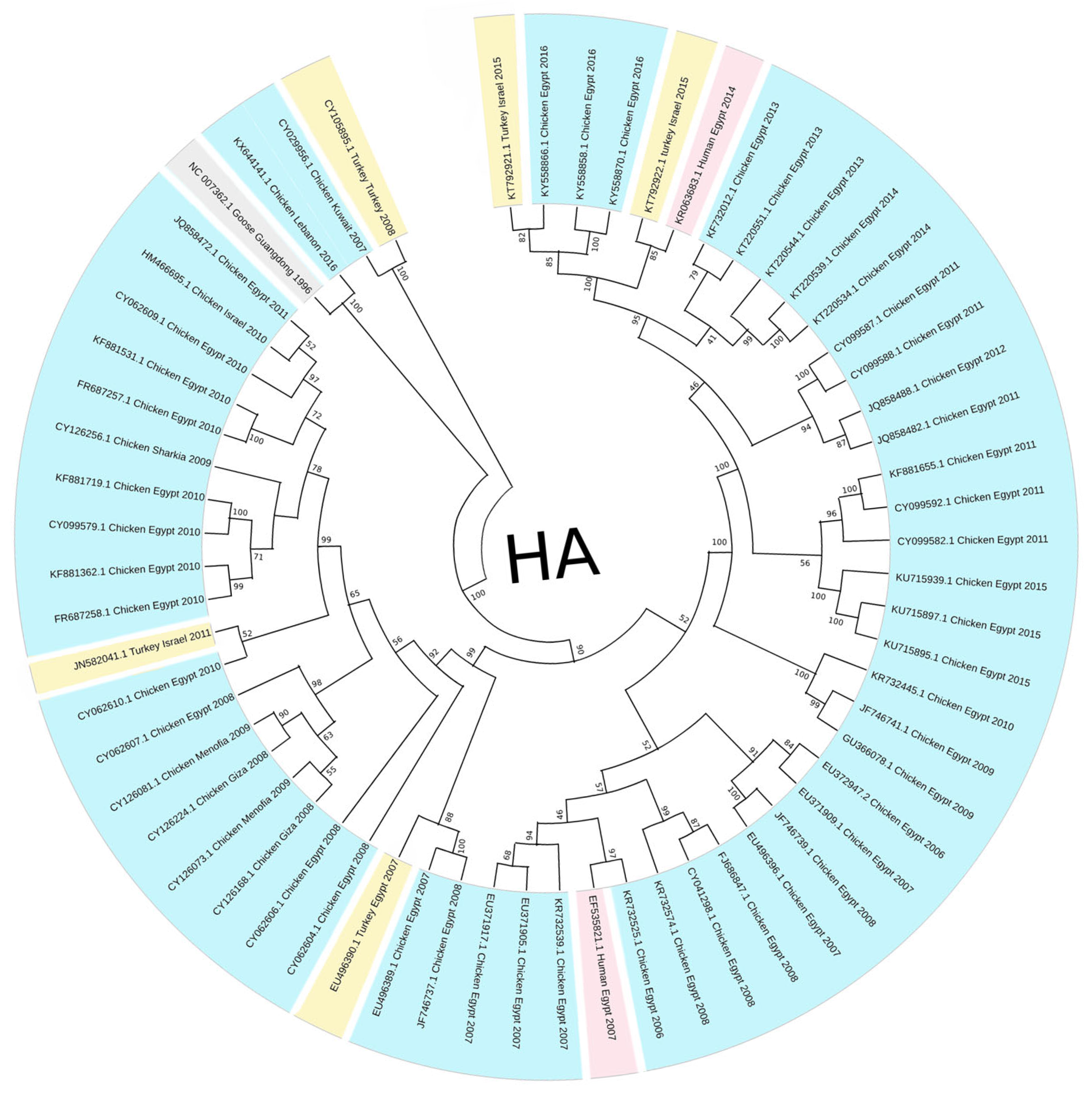
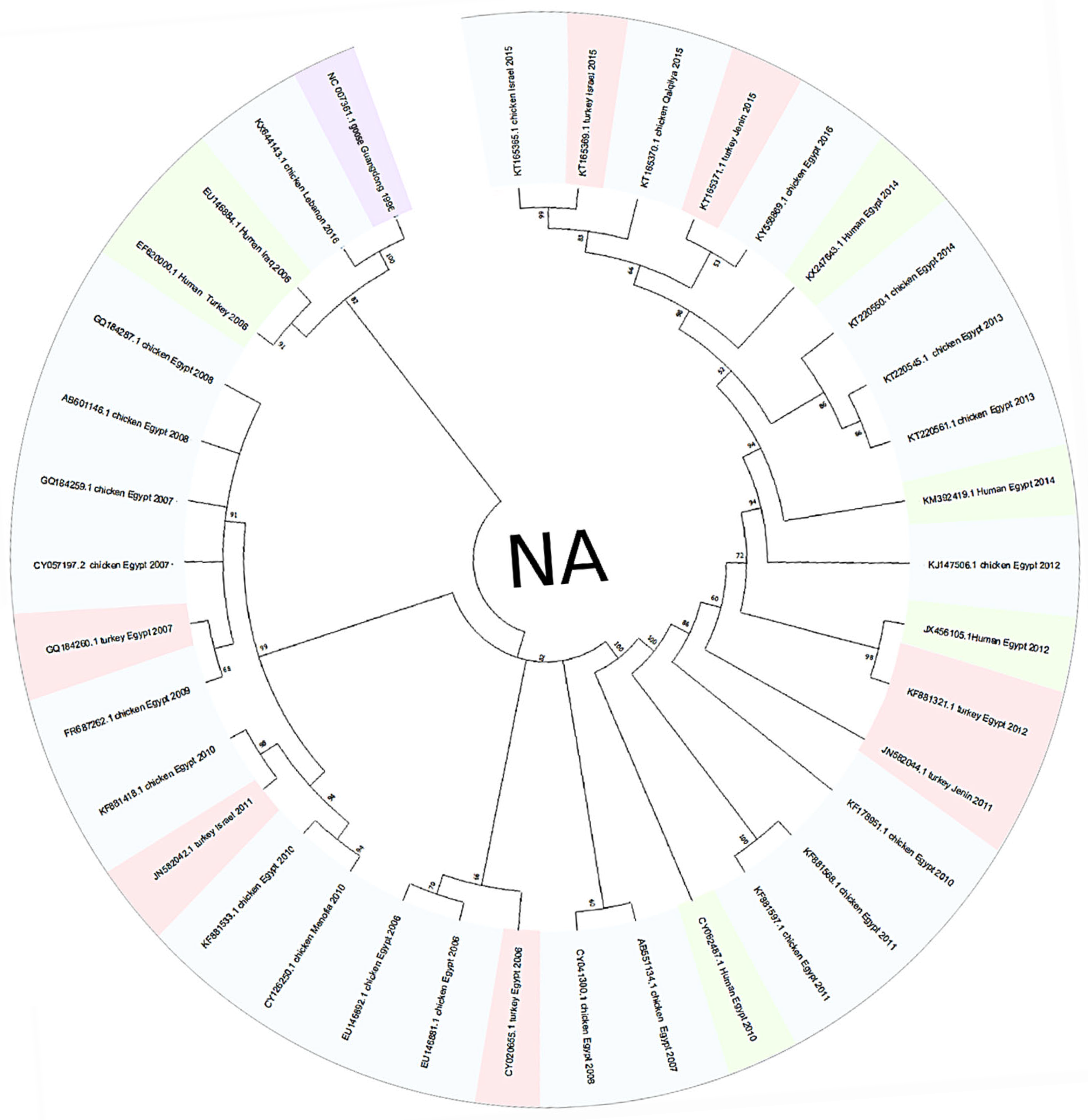
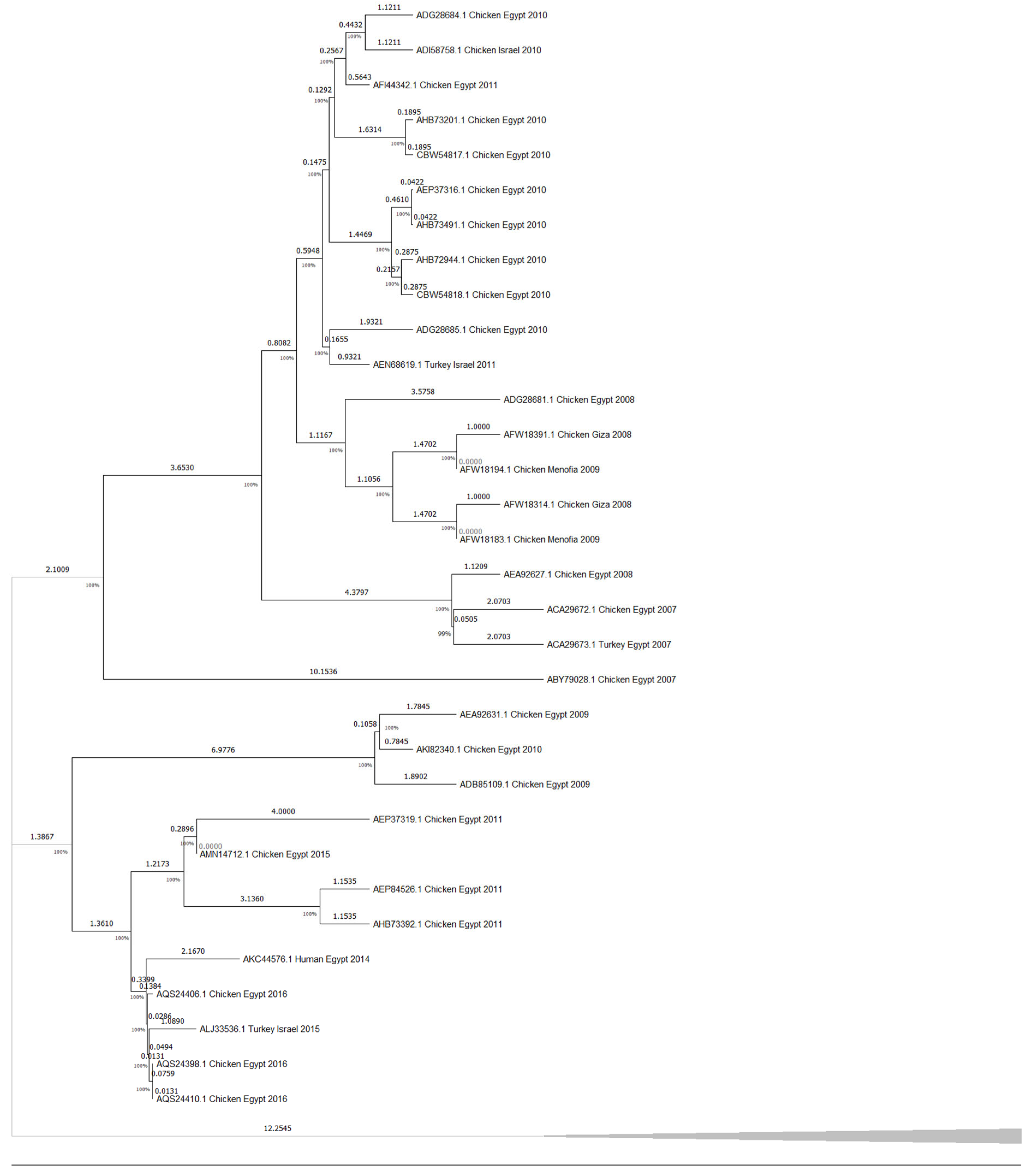
3.1. Phylogenetic Tree Analysis
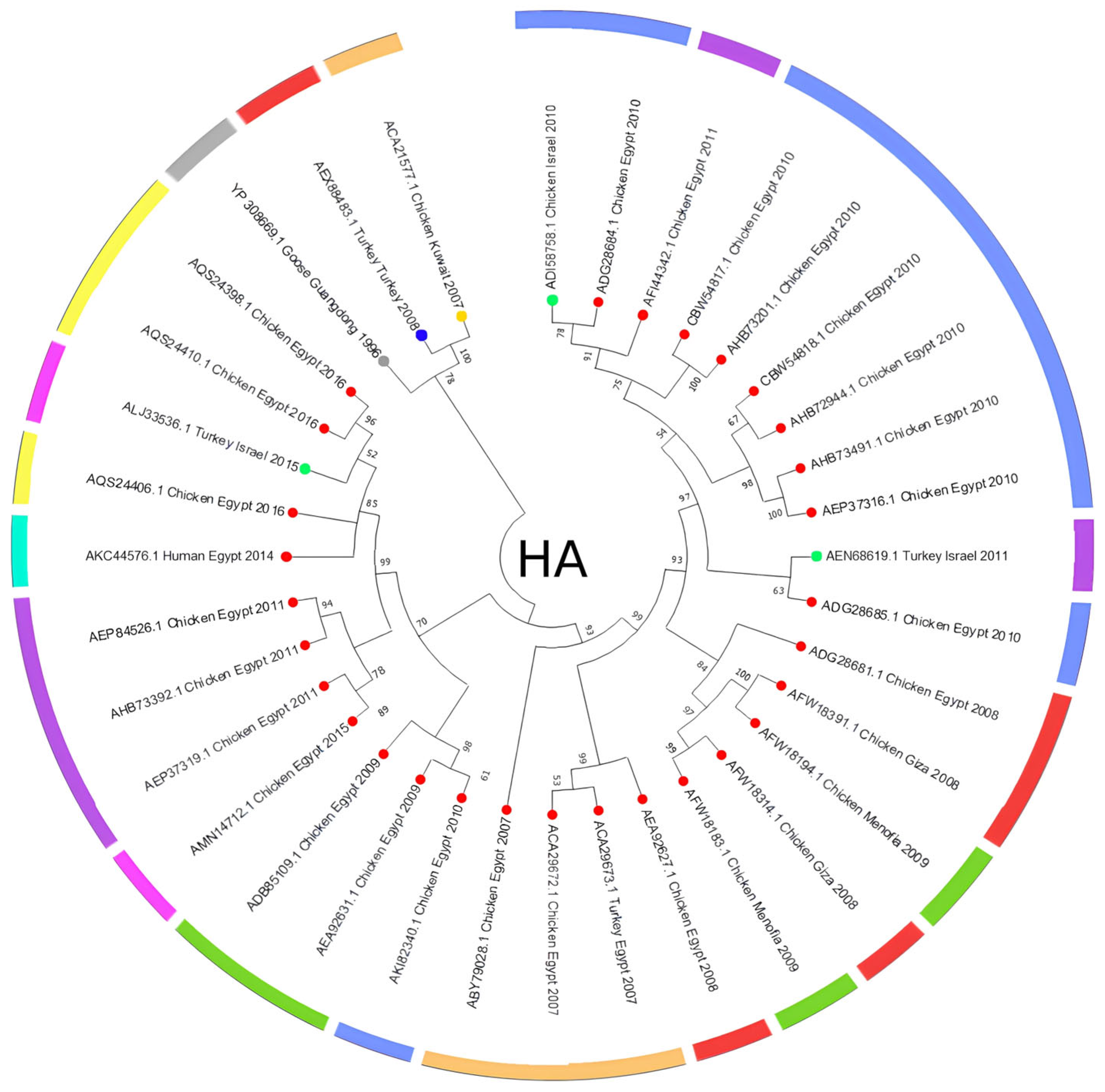
3.2. Amino Acid Analysis
4. Discussion
5. Surveillance, Policy, and Public Health Response to H5N1
6. Study Limitations
7. Future Directions and Recommended Strategies
8. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AIV | Avian Influenza Virus |
| BLAST | Basic Local Alignment Search Tool |
| FAO | Food and Agriculture Organization |
| HA | Hemagglutinin |
| HPAI | Highly Pathogenic Avian Influenza |
| IAV | Influenza A Virus |
| LPAI | Low-Pathogenicity Avian Influenza |
| MEGA | Molecular Evolutionary Genetics Analysis |
| MENA | Middle East and North Africa |
| NA | Neuraminidase |
| NCBI | National Center for Biotechnology Information |
| NJ | Neighbor-Joining (phylogenetic method) |
| NNI | Nearest-Neighbor-Interchange |
| OIE | World Organization for Animal Health |
| Pi | Parsimony-Informative Sites |
| WHO | World Health Organization |
References
- Kim, J.H.; Cho, C.H.; Shin, J.H.; Yang, J.C.; Park, T.J.; Park, J.; Park, J.P. Highly sensitive and label-free detection of influenza H5N1 viral proteins using affinity peptide and porous BSA/MXene nanocomposite electrode. Anal. Chim. Acta. 2023, 1251, 341018. [Google Scholar] [CrossRef] [PubMed]
- Sendor, A.B.; Weerasuriya, D.; Sapra, A. Avian Influenza. In StatPearls; StatPearls: Treasure Island, FL, USA, 2024. [Google Scholar]
- Rehman, S.; Effendi, M.H.; Witaningruma, A.M.; Nnabuikeb, U.E.; Bilal, M.; Abbas, A.; Abbas, R.Z.; Hussain, K. Avian influenza (H5N1) virus, epidemiology and its effects on backyard poultry in Indonesia: A review. F1000Research 2022, 11, 1321. [Google Scholar] [CrossRef] [PubMed]
- International Committee on Taxonomy of Viruses (ICTV). Orthomyxoviridae—Virus Taxonomy: 2023 Release. Available online: https://ictv.global/taxonomy/taxondetails?taxnode_id=202403953&taxon_name=Orthomyxoviridae (accessed on 1 March 2025).
- Centers for Disease Control and Prevention (CDC). How Flu Viruses Can Change: “Drift” and “Shift”. Available online: https://www.cdc.gov/flu/php/viruses/change.html?CDC_AAref_Val=https://www.cdc.gov/flu/about/viruses/change.htm (accessed on 3 March 2025).
- World Health Organization (WHO). Global Influenza Surveillance and Response System (GISRS). Available online: https://www.who.int/initiatives/global-influenza-surveillance-and-response-system (accessed on 8 March 2025).
- Krammer, F.; Smith, G.J.D.; Fouchier, R.A.M.; Peiris, M.; Kedzierska, K.; Doherty, P.C.; Palese, P.; Shaw, M.L.; Treanor, J.; Webster, R.G.; et al. Influenza. Nat. Rev. Dis. Primers 2018, 4, 3. [Google Scholar] [CrossRef]
- Hurt, A.C. The epidemiology and spread of drug resistant human influenza viruses. Curr. Opin. Virol. 2014, 8, 22–29. [Google Scholar] [CrossRef] [PubMed]
- WHO/OIE/FAO H5N1 Evolution Working Group. Continued evolution of highly pathogenic avian influenza A (H5N1): Updated nomenclature. Influenza Other Respir. Viruses 2012, 6, 1–5. [Google Scholar] [CrossRef]
- Ort, J.T.; Zolnoski, S.A.; Lam, T.T.; Neher, R.; Moncla, L.H. Development of avian influenza A(H5) virus datasets for Nextclade enables rapid and accurate clade assignment. bioRxiv 2025. [Google Scholar] [CrossRef]
- Cattoli, G.; Fusaro, A.; Monne, I.; Coven, F.; Joannis, T.; El-Hamid, H.S.; Hussein, A.A.; Cornelius, C.; Amarin, N.M.; Mancin, M.; et al. Evidence for differing evolutionary dynamics of A/H5N1 viruses among countries applying or not applying avian influenza vaccination in poultry. Vaccine 2011, 29, 9368–9375. [Google Scholar] [CrossRef]
- Alexander, D.J. An overview of the epidemiology of avian influenza. Vaccine 2007, 25, 5637–5644. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention (CDC). Highly Pathogenic Asian Avian Influenza A(H5N1) Virus. Centers for Disease Control and Prevention. Available online: https://archive.cdc.gov/#/details?url=https://www.cdc.gov/flu/avianflu/h5n1-virus.htm (accessed on 9 March 2025).
- Peiris, J.S.; de Jong, M.D.; Guan, Y. Avian influenza virus (H5N1): A threat to human health. Clin. Microbiol. Rev. 2007, 20, 243–267. [Google Scholar] [CrossRef]
- Fauci, A.S. Pandemic influenza threat and preparedness. Emerg. Infect. Dis. 2006, 12, 73–77. [Google Scholar] [CrossRef]
- Ceyhan, M.; Yildirim, I.; Ferraris, O.; Bouscambert-Duchamp, M.; Frobert, E.; Uyar, N.; Tezer, H.; Oner, A.F.; Buzgan, T.; Torunoglu, M.A.; et al. Serosurveillance study on transmission of H5N1 virus during a 2006 avian influenza epidemic. Epidemiol. Infect. 2010, 138, 1274–1280. [Google Scholar] [CrossRef] [PubMed]
- Tourky, W.; Ibrahim, M.; Said, A.; El Naggar, R.; AboElkhair, M. Avian Influenza Virus Characteristics, Epidemiology, and Pathogenesis in Poultry in Egypt. Damanhour J. Vet. Sci. 2024, 11, 23–41. [Google Scholar] [CrossRef]
- World Health Organization. Avian influenza—Situation in Iraq—Update 4. 30 January 2006. Available online: https://www.who.int/emergencies/disease-outbreak-news/item/2006_03_01a-en (accessed on 2 March 2025).
- Oner, A.F.; Bay, A.; Arslan, S.; Akdeniz, H.; Sahin, H.A.; Cesur, Y.; Epcacan, S.; Yilmaz, N.; Deger, I.; Kizilyildiz, B.; et al. Avian Influenza A (H5N1) infection in eastern Turkey in 2006. N. Engl. J. Med. 2006, 355, 2179–2185. [Google Scholar] [CrossRef]
- Kayali, G.; Webby, R.J.; Ducatez, M.F.; El Shesheny, R.A.; Kandeil, A.M.; Govorkova, E.A.; Mostafa, A.; Ali, M.A. The epidemiological and molecular aspects of influenza H5N1 viruses at the human-animal interface in Egypt. PLoS ONE 2011, 6, e17730. [Google Scholar] [CrossRef] [PubMed]
- Abdelwhab, E.M.; Hafez, H.M. An overview of the epidemic of highly pathogenic H5N1 avian influenza virus in Egypt: Epidemiology and control challenges. Epidemiol. Infect. 2011, 139, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H. Challenge for One Health: Co-Circulation of Zoonotic H5N1 and H9N2 Avian Influenza Viruses in Egypt. Viruses 2018, 10, 121. [Google Scholar] [CrossRef]
- To, K.K.; Ng, K.H.; Que, T.L.; Chan, J.M.; Tsang, K.Y.; Tsang, A.K.; Chen, H.; Yuen, K.Y. Avian influenza A H5N1 virus: A continuous threat to humans. Emerg. Microbes. Infect. 2012, 1, e25. [Google Scholar] [CrossRef]
- Abubakar, A.; Malik, M.; Pebody, R.G.; Elkholy, A.A.; Khan, W.; Bellos, A.; Mala, P. Burden of acute respiratory disease of epidemic and pandemic potential in the WHO Eastern Mediterranean Region: A literature review. East Mediterr. Health J. 2016, 22, 513–526. [Google Scholar] [CrossRef]
- Sooryanarain, H.; Elankumaran, S. Environmental role in influenza virus outbreaks. Annu. Rev. Anim. Biosci. 2015, 3, 347–373. [Google Scholar] [CrossRef]
- Horimoto, T.; Kawaoka, Y. Pandemic threat posed by avian influenza A viruses. Clin. Microbiol. Rev. 2001, 14, 129–149. [Google Scholar] [CrossRef]
- Jiao, C.; Wang, B.; Chen, P.; Jiang, Y.; Liu, J. Analysis of the conserved protective epitopes of hemagglutinin on influenza A viruses. Front. Immunol. 2023, 14, 1086297. [Google Scholar] [CrossRef]
- Air, G.M. Influenza neuraminidase. Influenza Other Respir. Viruses 2012, 6, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef]
- Nei, M.; Kumar, S. Molecular Evolution and Phylogenetics; Oxford University Press: New York, NY, USA, 2000; Available online: https://global.oup.com/academic/product/molecular-evolution-and-phylogenetics-9780195135855 (accessed on 19 January 2025).
- Hordijk, W.; Gascuel, O. Improving the efficiency of SPR moves in phylogenetic tree search methods based on maximum likelihood. Bioinformatics 2005, 21, 4338–4347. [Google Scholar] [CrossRef]
- Yang, Z. Maximum likelihood phylogenetic estimation from DNA sequences with variable rates over sites: Approximate methods. J. Mol. Evol. 1994, 39, 306–314. [Google Scholar] [CrossRef]
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef]
- Hall, B.G. Building Phylogenetic Trees from Molecular Data with MEGA. Mol. Biol. Evol. 2013, 30, 1229–1235. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- University of California Museum of Paleontology. Understanding Evolution: Phylogenetics—Branches. Available online: https://evolution.berkeley.edu/evolibrary/article/phylogenetics_01 (accessed on 19 January 2025).
- Hovmoller, R.; Alexandrov, B.; Hardman, J.; Janies, D. Tracking the geographical spread of avian influenza (H5N1) with multiple phylogenetic trees. Cladistics 2010, 26, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Food and Agriculture Organization of the United Nations. Approaches to Controlling, Preventing and Eliminating H5N1 Highly Pathogenic Avian Influenza in Endemic Countries; Animal Production and Health Paper No. 171; FAO: Rome, Italy, 2011. [Google Scholar]
- Kayali, G.; Kandeil, A.; El-Shesheny, R.; Kayed, A.S.; Maatouq, A.M.; Cai, Z.; McKenzie, P.P.; Webby, R.J.; Refaey, S.E.; Kandeel, A.; et al. Avian Influenza A(H5N1) Virus in Egypt. Emerg. Infect. Dis. 2016, 22, 379. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.A.; Peterson, A.T. Ecology and geography of avian influenza (HPAI H5N1) transmission in the Middle East and northeastern Africa. Int. J. Health Geogr. 2009, 8, 47. [Google Scholar] [CrossRef]
- El-Shesheny, R.; Mostafa, A.; Kandeil, A.; Mahmoud, S.H.; Bagato, O.; Naguib, A.; Refaey, S.E.; Webby, R.J.; Ali, M.A.; Kayali, G. Biological characterization of highly pathogenic avian influenza H5N1 viruses that infected humans in Egypt in 2014–2015. Arch. Virol. 2017, 162, 687–700. [Google Scholar] [CrossRef]
- WHO/OIE/FAO H5N1 Evolution Working Group. Continuing progress towards a unified nomenclature for the highly pathogenic H5N1 avian influenza viruses: Divergence of clade 2.2 viruses. Influenza Other Respir. Viruses 2009, 3, 59–62. [Google Scholar] [CrossRef]
- Arafa, A.; El-Masry, I.; Kholosy, S.; Hassan, M.K.; Dauphin, G.; Lubroth, J.; Makonnen, Y.J. Phylodynamics of avian influenza clade 2.2.1 H5N1 viruses in Egypt. Virol. J. 2016, 13, 49. [Google Scholar] [CrossRef]
- Akpinar, E.; Saatci, E. Avian influenza in Turkey—Will it influence health in all Europe? Croat. Med. J. 2006, 47, 7–15. [Google Scholar]
- Salaheldin, A.H.; Veits, J.; Abd El-Hamid, H.S.; Harder, T.C.; Devrishov, D.; Mettenleiter, T.C.; Hafez, H.M.; Abdelwhab, E.M. Isolation and genetic characterization of a novel 2.2.1.2a H5N1 virus from a vaccinated meat-turkeys flock in Egypt. Virol. J. 2017, 14, 48. [Google Scholar] [CrossRef]
- Skehel, J.J.; Wiley, D.C. Receptor binding and membrane fusion in virus entry: The influenza hemagglutinin. Annu. Rev. Biochem. 2000, 69, 531–569. [Google Scholar] [CrossRef]
- Wiley, D.C.; Skehel, J.J. The structure and function of the hemagglutinin membrane glycoprotein of influenza virus. Annu. Rev. Biochem. 1987, 56, 365–394. [Google Scholar] [CrossRef]
- Lin, T.H.; Zhu, X.; Wang, S.; Zhang, D.; McBride, R.; Yu, W.; Babarinde, S.; Paulson, J.C.; Wilson, I.A. A single mutation in bovine influenza H5N1 hemagglutinin switches specificity to human receptors. Science 2024, 386, 1128–1134. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Guo, Y.-H.; Jiang, T.; Wang, Y.-D.; Chan, K.-H.; Li, X.-F.; Yu, W.; McBride, R.; Paulson, J.C.; Yuen, K.-Y.; et al. A Unique and Conserved Neutralization Epitope in H5N1 Influenza Viruses Identified by an Antibody against the A/Goose/Guangdong/1/96 Hemagglutinin. J. Virol. 2013, 87, 12619–12635. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, K.R.; Lee, J.; Watanabe, A.; Kuraoka, M.; Robinson-Mccarthy, L.R.; Georgiou, G.; Kelsoe, G.; Harrison, S.C. A Prevalent Focused Human Antibody Response to the Influenza Virus Hemagglutinin Head Interface. mBio 2021, 12, 10–1128. [Google Scholar] [CrossRef]
- Gubareva, L.V. Molecular mechanisms of influenza virus resistance to neuraminidase inhibitors. Virus Res. 2004, 103, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Li, K.S.; Guan, Y.; Wang, J.; Smith, G.J.; Xu, K.M.; Duan, L.; Rahardjo, A.P.; Puthavathana, P.; Buranathai, C.; Nguyen, T.D.; et al. Genesis of a highly pathogenic and potentially pandemic H5N1 influenza virus in eastern Asia. Nature 2004, 430, 209–213. [Google Scholar] [CrossRef]
- Bloom, J.D.; Gong, L.I.; Baltimore, D. Permissive secondary mutations enable the evolution of influenza oseltamivir resistance. Science 2010, 328, 1272–1275. [Google Scholar] [CrossRef]
- Dudley, J.P. Age-specific infection and death rates for human A(H5N1) avian influenza in Egypt. Eurosurveillance 2009, 14, 19198. [Google Scholar] [CrossRef]
- El-Shesheny, R.; Kandeil, A.; Bagato, O.; Maatouq, A.M.; Moatasim, Y.; Rubrum, A.; Song, M.S.; Webby, R.J.; Ali, M.A.; Kayali, G. Molecular characterization of avian influenza H5N1 virus in Egypt and the emergence of a novel endemic subclade. J. Gen. Virol. 2014, 95, 1444–1463. [Google Scholar] [CrossRef]
- Kayali, G.; El-Shesheny, R.; Kutkat, M.A.; Kandeil, A.M.; Mostafa, A.; Ducatez, M.F.; McKenzie, P.P.; Govorkova, E.A.; Nasraa, M.H.; Webster, R.G.; et al. Continuing threat of influenza (H5N1) virus circulation in Egypt. Emerg. Infect. Dis. 2011, 17, 2306–2308. [Google Scholar] [CrossRef]
- Shirah, B.H.; Zafar, S.H.; Alferaidi, O.A.; Sabir, A.M.M. Mass gathering medicine (Hajj Pilgrimage in Saudi Arabia): The clinical pattern of pneumonia among pilgrims during Hajj. J. Infect. Public Health 2017, 10, 277–286. [Google Scholar] [CrossRef]
- Alp, E.; Leblebicioglu, H.; Doganay, M.; Voss, A. Infection control practice in countries with limited resources. Ann. Clin. Microbiol. Antimicrob. 2011, 10, 36. [Google Scholar] [CrossRef]
- Lukaszuk, E.; Dziewulska, D.; Stenzel, T. Occurrence and Phylogenetic Analysis of Avian Coronaviruses in Domestic Pigeons (Columba livia domestica) in Poland between 2016 and 2020. Pathogens 2022, 11, 646. [Google Scholar] [CrossRef] [PubMed]
- Hill, E.M.; House, T.; Dhingra, M.S.; Kalpravidh, W.; Morzaria, S.; Osmani, M.G.; Brum, E.; Yamage, M.; Kalam, M.A.; Prosser, D.J.; et al. The impact of surveillance and control on highly pathogenic avian influenza outbreaks in poultry in Dhaka division, Bangladesh. PLoS Comput. Biol. 2018, 14, e1006439. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, J.S.; Jeggo, M. The One Health Approach—Why Is It So Important? Trop. Med. Infect. Dis. 2019, 4, 88. [Google Scholar] [CrossRef] [PubMed]
- FAO/WHO/WOAH. Updated Joint FAO/WHO/WOAH Assessment of Recent Influenza A(H5N1) Virus Events in Animals and People. 14 August 2024. Available online: https://www.who.int/publications/m/item/updated-joint-fao-who-woah-assessment-of-recent-influenza-a(h5n1)-virus-events-in-animals-and-people (accessed on 19 January 2025).
- Lurie, N.; Keusch, G.T.; Dzau, V.J. Urgent lessons from COVID-19: Why the world needs a standing, coordinated system and sustainable financing for global research and development. Lancet 2021, 397, 1229–1236. [Google Scholar] [CrossRef]
- Lele, D.V. Risk assessment: A neglected tool for health, safety, and environment management. Indian J. Occup. Environ. Med. 2012, 16, 57–58. [Google Scholar] [CrossRef]
- Kung, N.Y.; Morris, R.S.; Perkins, N.R.; Sims, L.D.; Ellis, T.M.; Bissett, L.; Chow, M.; Shortridge, K.F.; Guan, Y.; Peiris, M.J. Risk for infection with highly pathogenic influenza A virus (H5N1) in chickens, Hong Kong, 2002. Emerg. Infect. Dis. 2007, 13, 412–418. [Google Scholar] [CrossRef]
- Tilli, G.; Laconi, A.; Galuppo, F.; Mughini-Gras, L.; Piccirillo, A. Assessing Biosecurity Compliance in Poultry Farms: A Survey in a Densely Populated Poultry Area in North East Italy. Animals 2022, 12, 1409. [Google Scholar] [CrossRef]
- Rimi, N.A.; Sultana, R.; Muhsina, M.; Uddin, B.; Haider, N.; Nahar, N.; Zeidner, N.; Sturm-Ramirez, K.; Luby, S.P. Biosecurity Conditions in Small Commercial Chicken Farms, Bangladesh 2011–2012. Ecohealth 2017, 14, 244–258. [Google Scholar] [CrossRef]
- Jang, Y.; Lee, J.; So, B.; Lee, K.; Yun, S.; Lee, M.; Choe, N. Evaluation of changes induced by temperature, contact time, and surface in the efficacies of disinfectants against avian influenza virus. Poult. Sci. 2014, 93, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhao, D.; Zhang, J.; Huang, X.; Han, K.; Liu, Q.; Yang, J.; Zhang, L.; Li, Y. Development of an Inactivated Avian Influenza Virus Vaccine against Circulating H9N2 in Chickens and Ducks. Vaccines 2023, 11, 596. [Google Scholar] [CrossRef]
- Kandeil, A.; Mostafa, A.; El-Shesheny, R.; El-Taweel, A.N.; Gomaa, M.; Galal, H.; Kayali, G.; Ali, M.A. Avian influenza H5N1 vaccination efficacy in Egyptian backyard poultry. Vaccine 2017, 35, 6195–6201. [Google Scholar] [CrossRef] [PubMed]
- Byrne, J. To What Extent is Vaccination Against Avian Influenza Increasing Globally? Available online: https://www.feednavigator.com/article/2024/02/14/boehringer-talks-vaccination-campaigns-against-bird-flu (accessed on 19 January 2025).
- Khalil, A.T.; Ali, M.; Tanveer, F.; Ovais, M.; Idrees, M.; Shinwari, Z.K.; Hollenbeck, J.E. Emerging Viral Infections in Pakistan: Issues, Concerns, and Future Prospects. Health Secur. 2017, 15, 268–281. [Google Scholar] [CrossRef] [PubMed]
- Modirihamedan, A.; Aghajantabar, S.; King, J.; Graaf, A.; Pohlmann, A.; Aghaiyan, L.; Ziafati Kafi, Z.; Mahfoozi, Y.; Hosseini, H.; Beer, M.; et al. Wild bird trade at live poultry markets potentiates risks of avian influenza virus introductions in Iran. Infect Ecol. Epidemiol. 2021, 11, 1992083. [Google Scholar] [CrossRef]
- Parvin, R.; Nooruzzaman, M.; Kabiraj, C.K.; Begum, J.A.; Chowdhury, E.H.; Islam, M.R.; Harder, T. Controlling Avian Influenza Virus in Bangladesh: Challenges and Recommendations. Viruses 2020, 12, 751. [Google Scholar] [CrossRef]
- Fereidouni, S.R.; Werner, O.; Starick, E.; Beer, M.; Harder, T.C.; Aghakhan, M.; Modirrousta, H.; Amini, H.; Moghaddam, M.K.; Bozorghmehrifard, M.H.; et al. Avian influenza virus monitoring in wintering waterbirds in Iran, 2003–2007. Virol. J. 2010, 7, 43. [Google Scholar] [CrossRef]
- Babakir-Mina, M.; Balestra, E.; Perno, C.F.; Aquaro, S. Influenza virus A (H5N1): A pandemic risk? New Microbiol. 2007, 30, 65–78. [Google Scholar]
- Dey, P.; Ahuja, A.; Panwar, J.; Choudhary, P.; Rani, S.; Kaur, M.; Sharma, A.; Kaur, J.; Yadav, A.K.; Sood, V.; et al. Immune Control of Avian Influenza Virus Infection and Its Vaccine Development. Vaccines 2023, 11, 593. [Google Scholar] [CrossRef]
- Charisis, N. Avian influenza biosecurity: A key for animal and human protection. Vet. Ital. 2008, 44, 657–669. [Google Scholar]
- Lee, Y.J.; Chen, X.; Marwaha, S. The Need for Biosecurity Education in Biotechnology Curricula. Biodes. Res. 2023, 5, 0008. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.R. Oseltamivir in human avian influenza infection. J. Antimicrob. Chemother. 2010, 65, ii25–ii33. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
AL-Eitan, L.N.; Almahdawi, D.L.; Khair, I.Y. Phylogenetic Analysis and Spread of HPAI H5N1 in Middle Eastern Countries Based on Hemagglutinin and Neuraminidase Gene Sequences. Viruses 2025, 17, 734. https://doi.org/10.3390/v17050734
AL-Eitan LN, Almahdawi DL, Khair IY. Phylogenetic Analysis and Spread of HPAI H5N1 in Middle Eastern Countries Based on Hemagglutinin and Neuraminidase Gene Sequences. Viruses. 2025; 17(5):734. https://doi.org/10.3390/v17050734
Chicago/Turabian StyleAL-Eitan, Laith N., Diana L. Almahdawi, and Iliya Y. Khair. 2025. "Phylogenetic Analysis and Spread of HPAI H5N1 in Middle Eastern Countries Based on Hemagglutinin and Neuraminidase Gene Sequences" Viruses 17, no. 5: 734. https://doi.org/10.3390/v17050734
APA StyleAL-Eitan, L. N., Almahdawi, D. L., & Khair, I. Y. (2025). Phylogenetic Analysis and Spread of HPAI H5N1 in Middle Eastern Countries Based on Hemagglutinin and Neuraminidase Gene Sequences. Viruses, 17(5), 734. https://doi.org/10.3390/v17050734