
Adaptor Protein Complexes in HIV-1 Pathogenesis: Mechanisms and Therapeutic Potential

 , , , , and
, , , , and
Abstract
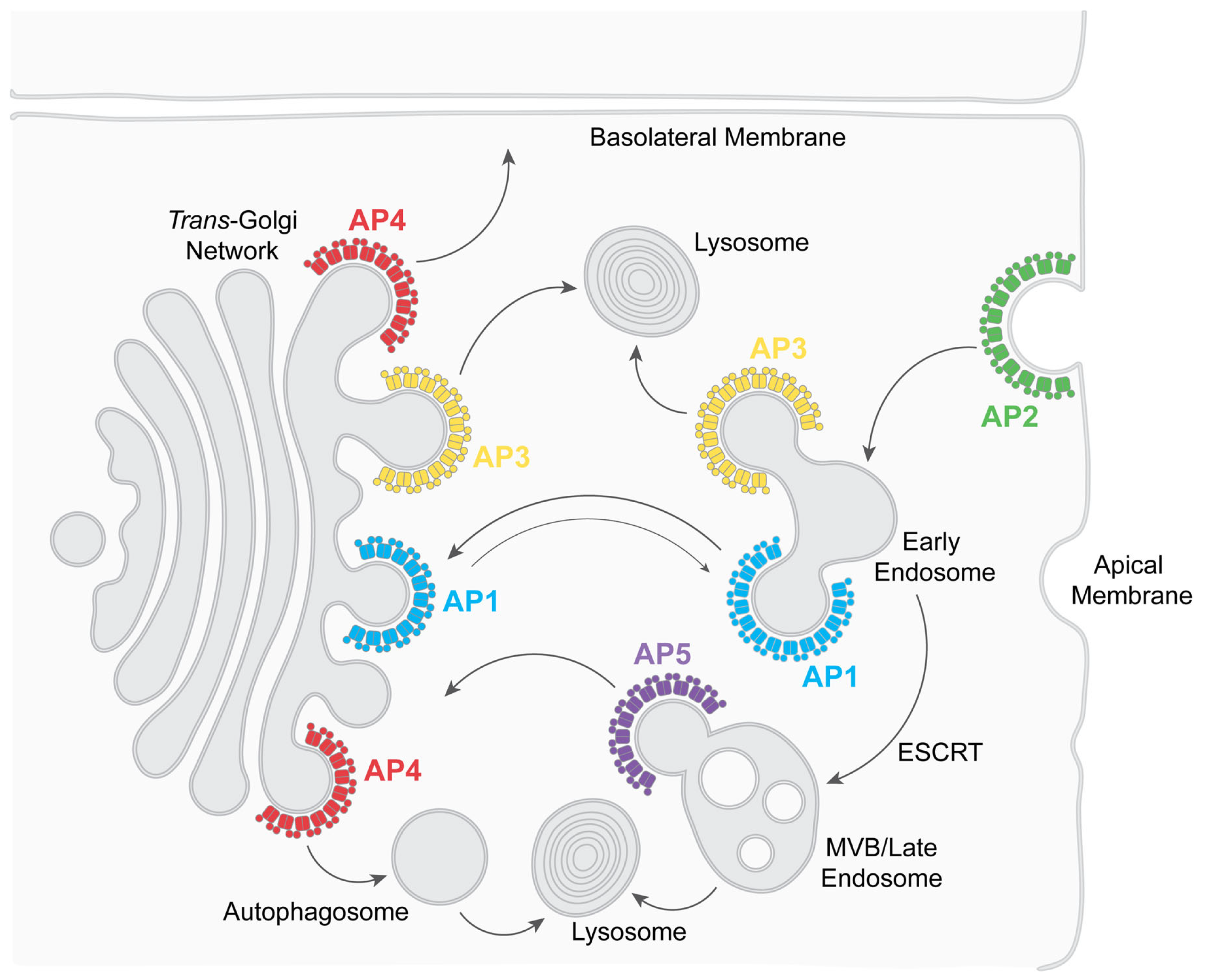
1. Introduction to Adaptor Protein Complexes
2. HIV-1 and AP Complexes
2.1. Modulating Innate and Adaptive Immunity
2.1.1. Lentiviral Nef Interacts with AP1 to Modulate MHC-I, Rendering Infected Cells Less Susceptible to Virus-Specific Cytotoxic T Lymphocytes (CTL)
2.1.2. Nef Interacts with AP2 to Modulate CD4, Rendering Infected Cells Less Susceptible to Antibody-Dependent Cellular Cytotoxicity (ADCC)
2.1.3. Nef Interacts with AP1γ2 to Send MHC-I and CD4 to Lysosomes for Degradation
2.1.4. Vpu Interacts with AP1 and AP2 to Counteract the Interferon-Induced Protein BST2
2.2. Assembly and Release of Infectious Virions
2.2.1. Nef-Mediated Modulation of CD4 and SERINC Proteins via AP2 Increases Virion Infectivity
2.2.2. While HIV-1 Vpu Uses AP1 and AP2 to Counteract Virion Entrapment by BST2, SIV Accomplishes This Using Nef and AP2
2.2.3. Env Interacts with AP Complexes: Immune Evasion and Virion-Incorporation
2.2.4. Gag Interaction with AP3: Support of Virion Assembly at MVBs
2.3. Structural Basis of HIV-1 AP Interactions
2.3.1. How Nef and Vpu Involve Sequences Both in Their Cellular Targets and in Themselves to Interact with AP Complexes
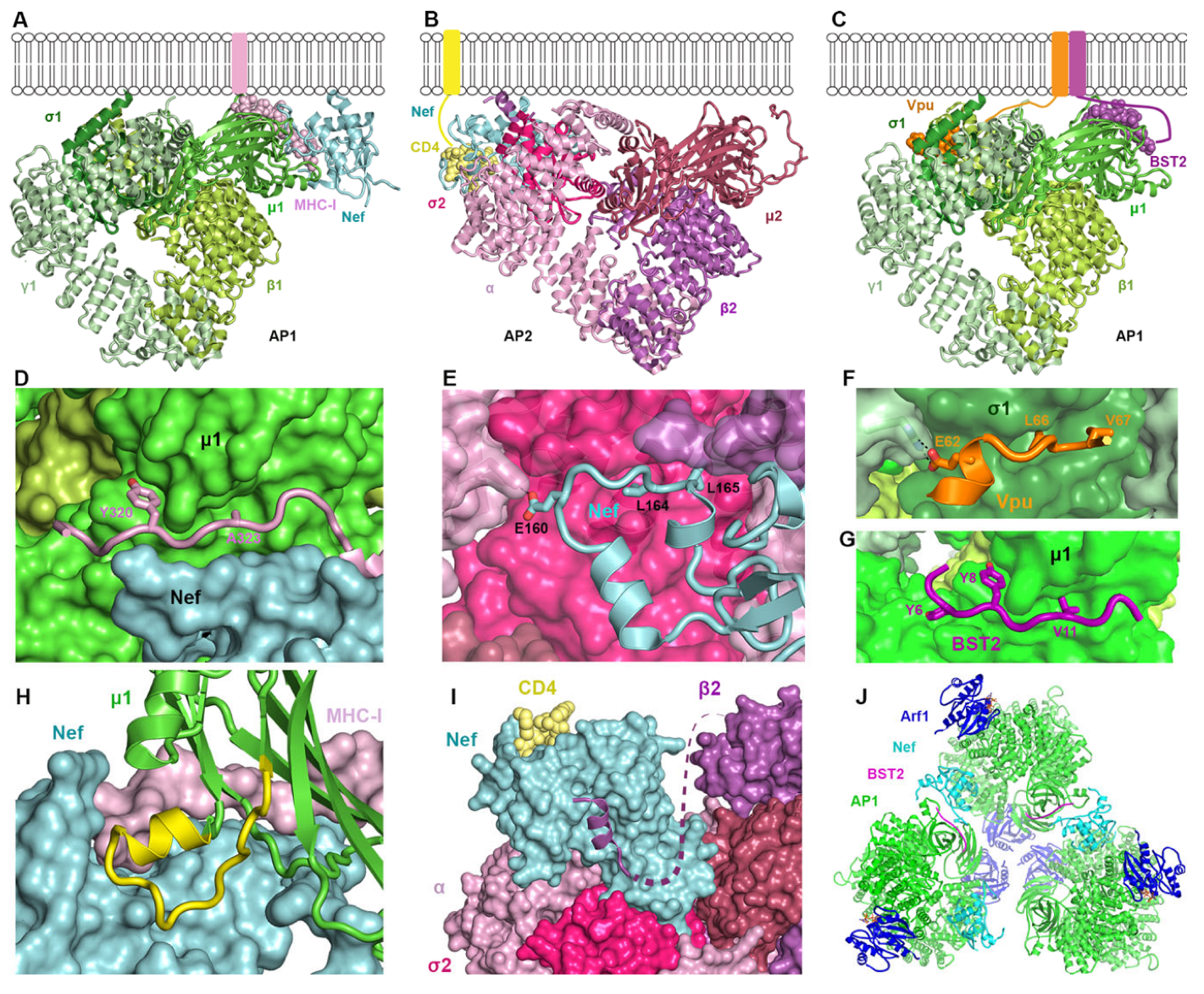
2.3.2. How Nef, Vpu, and Their Targets Change the Conformation of AP Complexes
2.3.3. How Nef and Vpu Informed on the Binding Partner of Acidic Cluster Sorting Motifs
3. Controversies, Open Questions, and Future Directions
4. Opportunity for Therapeutic Intervention
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Sanger, A.; Hirst, J.; Davies, A.K.; Robinson, M.S. Adaptor Protein Complexes and Disease at a Glance. J. Cell Sci. 2019, 132, jcs222992. [Google Scholar] [CrossRef] [PubMed]
- Boehm, M.; Bonifacino, J.S. Adaptins. Mol. Biol. Cell 2001, 12, 2907–2920. [Google Scholar] [CrossRef] [PubMed]
- Bonifacino, J.S.; Traub, L.M. Signals for Sorting of Transmembrane Proteins to Endosomes and Lysosomes. Annu. Rev. Biochem. 2003, 72, 395–447. [Google Scholar] [CrossRef] [PubMed]
- Dell’Angelica, E.C.; Ohno, H.; Ooi, C.E.; Rabinovich, E.; Roche, K.W.; Bonifacino, J.S. AP-3: An Adaptor-like Protein Complex with Ubiquitous Expression. EMBO J. 1997, 16, 917–928. [Google Scholar] [CrossRef]
- Mettlen, M.; Chen, P.-H.; Srinivasan, S.; Danuser, G.; Schmid, S.L. Regulation of Clathrin-Mediated Endocytosis. Annu. Rev. Biochem. 2018, 87, 871–896. [Google Scholar] [CrossRef]
- Maldonado-Báez, L.; Wendland, B. Endocytic Adaptors: Recruiters, Coordinators and Regulators. Trends Cell Biol. 2006, 16, 505–513. [Google Scholar] [CrossRef]
- Collins, B.M.; McCoy, A.J.; Kent, H.M.; Evans, P.R.; Owen, D.J. Molecular Architecture and Functional Model of the Endocytic AP2 Complex. Cell 2002, 109, 523–535. [Google Scholar] [CrossRef]
- Ricotta, D.; Conner, S.D.; Schmid, S.L.; von Figura, K.; Honing, S. Phosphorylation of the AP2 Mu Subunit by AAK1 Mediates High Affinity Binding to Membrane Protein Sorting Signals. J. Cell Biol. 2002, 156, 791–795. [Google Scholar] [CrossRef]
- Motley, A.M.; Berg, N.; Taylor, M.J.; Sahlender, D.A.; Hirst, J.; Owen, D.J.; Robinson, M.S. Functional Analysis of AP-2 Alpha and Mu2 Subunits. Mol. Biol. Cell 2006, 17, 5298–5308. [Google Scholar] [CrossRef]
- Höning, S.; Ricotta, D.; Krauss, M.; Späte, K.; Spolaore, B.; Motley, A.; Robinson, M.; Robinson, C.; Haucke, V.; Owen, D.J. Phosphatidylinositol-(4,5)-Bisphosphate Regulates Sorting Signal Recognition by the Clathrin-Associated Adaptor Complex AP2. Mol. Cell 2005, 18, 519–531. [Google Scholar] [CrossRef]
- Jackson, L.P.; Kelly, B.T.; McCoy, A.J.; Gaffry, T.; James, L.C.; Collins, B.M.; Höning, S.; Evans, P.R.; Owen, D.J. A Large-Scale Conformational Change Couples Membrane Recruitment to Cargo Binding in the AP2 Clathrin Adaptor Complex. Cell 2010, 141, 1220–1229. [Google Scholar] [CrossRef] [PubMed]
- Beacham, G.M.; Partlow, E.A.; Hollopeter, G. Conformational Regulation of AP1 and AP2 Clathrin Adaptor Complexes. Traffic 2019, 20, 741–751. [Google Scholar] [CrossRef] [PubMed]
- Collins, D.R.; Collins, K.L. HIV-1 Accessory Proteins Adapt Cellular Adaptors to Facilitate Immune Evasion. PLoS Pathog. 2014, 10, e1003851. [Google Scholar] [CrossRef] [PubMed]
- Strazic Geljic, I.; Kucan Brlic, P.; Musak, L.; Karner, D.; Ambriović-Ristov, A.; Jonjic, S.; Schu, P.; Rovis, T.L. Viral Interactions with Adaptor-Protein Complexes: A Ubiquitous Trait among Viral Species. Int. J. Mol. Sci. 2021, 22, 5274. [Google Scholar] [CrossRef]
- Robinson, M.S. 100-kD Coated Vesicle Proteins: Molecular Heterogeneity and Intracellular Distribution Studied with Monoclonal Antibodies. J. Cell Biol. 1987, 104, 887–895. [Google Scholar] [CrossRef]
- Simpson, F.; Peden, A.A.; Christopoulou, L.; Robinson, M.S. Characterization of the Adaptor-Related Protein Complex, AP-3. J. Cell Biol. 1997, 137, 835–845. [Google Scholar] [CrossRef]
- Hirst, J.; Bright, N.A.; Rous, B.; Robinson, M.S. Characterization of a Fourth Adaptor-Related Protein Complex. Mol. Biol. Cell 1999, 10, 2787–2802. [Google Scholar] [CrossRef]
- Hirst, J.; Itzhak, D.N.; Antrobus, R.; Borner, G.H.H.; Robinson, M.S. Role of the AP-5 Adaptor Protein Complex in Late Endosome-to-Golgi Retrieval. PLoS Biol. 2018, 16, e2004411. [Google Scholar] [CrossRef]
- Simmen, T.; Höning, S.; Icking, A.; Tikkanen, R.; Hunziker, W. AP-4 Binds Basolateral Signals and Participates in Basolateral Sorting in Epithelial MDCK Cells. Nat. Cell Biol. 2002, 4, 154–159. [Google Scholar] [CrossRef]
- Ramirez, P.W.; Sharma, S.; Singh, R.; Stoneham, C.A.; Vollbrecht, T.; Guatelli, J. Plasma Membrane-Associated Restriction Factors and Their Counteraction by HIV-1 Accessory Proteins. Cells 2019, 8, 1020. [Google Scholar] [CrossRef]
- Collins, K.L.; Chen, B.K.; Kalams, S.A.; Walker, B.D.; Baltimore, D. HIV-1 Nef Protein Protects Infected Primary Cells against Killing by Cytotoxic T Lymphocytes. Nature 1998, 391, 397–401. [Google Scholar] [CrossRef]
- Le Gall, S.; Buseyne, F.; Trocha, A.; Walker, B.D.; Heard, J.M.; Schwartz, O. Distinct Trafficking Pathways Mediate Nef-Induced and Clathrin-Dependent Major Histocompatibility Complex Class I down-Regulation. J. Virol. 2000, 74, 9256–9266. [Google Scholar] [CrossRef] [PubMed]
- Roeth, J.F.; Williams, M.; Kasper, M.R.; Filzen, T.M.; Collins, K.L. HIV-1 Nef Disrupts MHC-I Trafficking by Recruiting AP-1 to the MHC-I Cytoplasmic Tail. J. Cell Biol. 2004, 167, 903–913. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, M.R.; Wonderlich, E.R.; Roeth, J.F.; Leonard, J.A.; Collins, K.L. HIV-1 Nef Targets MHC-I and CD4 for Degradation via a Final Common Beta-COP-Dependent Pathway in T Cells. PLoS Pathog. 2008, 4, e1000131. [Google Scholar] [CrossRef] [PubMed]
- Hurley, J.H.; Bonifacino, J.S. Nef-Arious Goings-on at the Golgi. Nat. Struct. Mol. Biol. 2012, 19, 661–662. [Google Scholar] [CrossRef]
- Robinson, M.S.; Antrobus, R.; Sanger, A.; Davies, A.K.; Gershlick, D.C. The Role of the AP-1 Adaptor Complex in Outgoing and Incoming Membrane Traffic. J. Cell Biol. 2024, 223, e202310071. [Google Scholar] [CrossRef]
- Tavares, L.A.; de Carvalho, J.V.; Costa, C.S.; Silveira, R.M.; de Carvalho, A.N.; Donadi, E.A.; daSilva, L.L.P. Two Functional Variants of AP-1 Complexes Composed of Either Γ2 or Γ1 Subunits Are Independently Required for Major Histocompatibility Complex Class I Downregulation by HIV-1 Nef. J. Virol. 2020, 94, e02039-19. [Google Scholar] [CrossRef]
- Jia, X.; Singh, R.; Homann, S.; Yang, H.; Guatelli, J.; Xiong, Y. Structural Basis of Evasion of Cellular Adaptive Immunity by HIV-1 Nef. Nat. Struct. Mol. Biol. 2012, 19, 701–706. [Google Scholar] [CrossRef]
- Craig, H.M.; Pandori, M.W.; Guatelli, J.C. Interaction of HIV-1 Nef with the Cellular Dileucine-Based Sorting Pathway Is Required for CD4 down-Regulation and Optimal Viral Infectivity. Proc. Natl. Acad. Sci. USA 1998, 95, 11229–11234. [Google Scholar] [CrossRef]
- Greenberg, M.E.; Bronson, S.; Lock, M.; Neumann, M.; Pavlakis, G.N.; Skowronski, J. Co-Localization of HIV-1 Nef with the AP-2 Adaptor Protein Complex Correlates with Nef-Induced CD4 down-Regulation. EMBO J. 1997, 16, 6964–6976. [Google Scholar] [CrossRef]
- Chaudhuri, R.; Lindwasser, O.W.; Smith, W.J.; Hurley, J.H.; Bonifacino, J.S. Downregulation of CD4 by Human Immunodeficiency Virus Type 1 Nef Is Dependent on Clathrin and Involves Direct Interaction of Nef with the AP2 Clathrin Adaptor. J. Virol. 2007, 81, 3877–3890. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.-J.; Cai, C.Y.; Zhang, X.; Zhang, H.-T.; Hirst, J.A.; Burakoff, S.J. HIV Nef-Mediated CD4 down-Regulation Is Adaptor Protein Complex 2 Dependent. J. Immunol. 2005, 175, 3157–3164. [Google Scholar] [CrossRef] [PubMed]
- Wildum, S.; Schindler, M.; Münch, J.; Kirchhoff, F. Contribution of Vpu, Env, and Nef to CD4 down-Modulation and Resistance of Human Immunodeficiency Virus Type 1-Infected T Cells to Superinfection. J. Virol. 2006, 80, 8047–8059. [Google Scholar] [CrossRef]
- Lama, J.; Mangasarian, A.; Trono, D. Cell-Surface Expression of CD4 Reduces HIV-1 Infectivity by Blocking Env Incorporation in a Nef- and Vpu-Inhibitable Manner. Curr. Biol. 1999, 9, 622–631. [Google Scholar] [CrossRef] [PubMed]
- Veillette, M.; Désormeaux, A.; Medjahed, H.; Gharsallah, N.-E.; Coutu, M.; Baalwa, J.; Guan, Y.; Lewis, G.; Ferrari, G.; Hahn, B.H.; et al. Interaction with Cellular CD4 Exposes HIV-1 Envelope Epitopes Targeted by Antibody-Dependent Cell-Mediated Cytotoxicity. J. Virol. 2014, 88, 2633–2644. [Google Scholar] [CrossRef] [PubMed]
- Pham, T.N.Q.; Lukhele, S.; Hajjar, F.; Routy, J.-P.; Cohen, É.A. HIV Nef and Vpu Protect HIV-Infected CD4+ T Cells from Antibody-Mediated Cell Lysis through down-Modulation of CD4 and BST2. Retrovirology 2014, 11, 15. [Google Scholar] [CrossRef]
- Veillette, M.; Coutu, M.; Richard, J.; Batraville, L.-A.; Dagher, O.; Bernard, N.; Tremblay, C.; Kaufmann, D.E.; Roger, M.; Finzi, A. The HIV-1 Gp120 CD4-Bound Conformation Is Preferentially Targeted by Antibody-Dependent Cellular Cytotoxicity-Mediating Antibodies in Sera from HIV-1-Infected Individuals. J. Virol. 2015, 89, 545–551. [Google Scholar] [CrossRef]
- Amorim, N.A.; da Silva, E.M.L.; de Castro, R.O.; da Silva-Januário, M.E.; Mendonça, L.M.; Bonifacino, J.S.; da Costa, L.J.; daSilva, L.L.P. Interaction of HIV-1 Nef Protein with the Host Protein Alix Promotes Lysosomal Targeting of CD4 Receptor. J. Biol. Chem. 2014, 289, 27744–27756. [Google Scholar] [CrossRef]
- Tavares, L.A.; da Silva, E.M.L.; da Silva-Januário, M.E.; Januário, Y.C.; de Cavalho, J.V.; Czernisz, É.S.; Mardones, G.A.; daSilva, L.L.P. CD4 Downregulation by the HIV-1 Protein Nef Reveals Distinct Roles for the Γ1 and Γ2 Subunits of the AP-1 Complex in Protein Trafficking. J. Cell Sci. 2017, 130, 429–443. [Google Scholar] [CrossRef]
- Bissig, C.; Gruenberg, J. ALIX and the Multivesicular Endosome: ALIX in Wonderland. Trends Cell Biol. 2014, 24, 19–25. [Google Scholar] [CrossRef]
- Kwon, Y.; Kaake, R.M.; Echeverria, I.; Suarez, M.; Karimian Shamsabadi, M.; Stoneham, C.; Ramirez, P.W.; Kress, J.; Singh, R.; Sali, A.; et al. Structural Basis of CD4 Downregulation by HIV-1 Nef. Nat. Struct. Mol. Biol. 2020, 27, 822–828. [Google Scholar] [CrossRef] [PubMed]
- Marassi, F.M.; Ma, C.; Gratkowski, H.; Straus, S.K.; Strebel, K.; Oblatt-Montal, M.; Montal, M.; Opella, S.J. Correlation of the Structural and Functional Domains in the Membrane Protein Vpu from HIV-1. Proc. Natl. Acad. Sci. USA 1999, 96, 14336–14341. [Google Scholar] [CrossRef] [PubMed]
- Skasko, M.; Wang, Y.; Tian, Y.; Tokarev, A.; Munguia, J.; Ruiz, A.; Stephens, E.B.; Opella, S.J.; Guatelli, J. HIV-1 Vpu Protein Antagonizes Innate Restriction Factor BST-2 via Lipid-Embedded Helix-Helix Interactions. J. Biol. Chem. 2012, 287, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Van Damme, N.; Goff, D.; Katsura, C.; Jorgenson, R.L.; Mitchell, R.; Johnson, M.C.; Stephens, E.B.; Guatelli, J. The Interferon-Induced Protein BST-2 Restricts HIV-1 Release and Is Downregulated from the Cell Surface by the Viral Vpu Protein. Cell Host Microbe 2008, 3, 245–252. [Google Scholar] [CrossRef]
- Neil, S.J.D.; Zang, T.; Bieniasz, P.D. Tetherin Inhibits Retrovirus Release and Is Antagonized by HIV-1 Vpu. Nature 2008, 451, 425–430. [Google Scholar] [CrossRef]
- Blasius, A.L.; Giurisato, E.; Cella, M.; Schreiber, R.D.; Shaw, A.S.; Colonna, M. Bone Marrow Stromal Cell Antigen 2 Is a Specific Marker of Type I IFN-Producing Cells in the Naive Mouse, but a Promiscuous Cell Surface Antigen Following IFN Stimulation. J. Immunol. 2006, 177, 3260–3265. [Google Scholar] [CrossRef]
- Kupzig, S.; Korolchuk, V.; Rollason, R.; Sugden, A.; Wilde, A.; Banting, G. Bst-2/HM1.24 Is a Raft-Associated Apical Membrane Protein with an Unusual Topology. Traffic 2003, 4, 694–709. [Google Scholar] [CrossRef]
- Margottin, F.; Bour, S.P.; Durand, H.; Selig, L.; Benichou, S.; Richard, V.; Thomas, D.; Strebel, K.; Benarous, R. A Novel Human WD Protein, h-Beta TrCp, That Interacts with HIV-1 Vpu Connects CD4 to the ER Degradation Pathway through an F-Box Motif. Mol. Cell 1998, 1, 565–574. [Google Scholar] [CrossRef]
- Kueck, T.; Foster, T.L.; Weinelt, J.; Sumner, J.C.; Pickering, S.; Neil, S.J.D. Serine Phosphorylation of HIV-1 Vpu and Its Binding to Tetherin Regulates Interaction with Clathrin Adaptors. PLoS Pathog. 2015, 11, e1005141. [Google Scholar] [CrossRef]
- Lewinski, M.K.; Jafari, M.; Zhang, H.; Opella, S.J.; Guatelli, J. Membrane Anchoring by a C-Terminal Tryptophan Enables HIV-1 Vpu to Displace Bone Marrow Stromal Antigen 2 (BST2) from Sites of Viral Assembly. J. Biol. Chem. 2015, 290, 10919–10933. [Google Scholar] [CrossRef]
- Stoneham, C.A.; Singh, R.; Jia, X.; Xiong, Y.; Guatelli, J. Endocytic Activity of HIV-1 Vpu: Phosphoserine-Dependent Interactions with Clathrin Adaptors. Traffic 2017, 18, 545–561. [Google Scholar] [CrossRef]
- Mitchell, R.S.; Katsura, C.; Skasko, M.A.; Fitzpatrick, K.; Lau, D.; Ruiz, A.; Stephens, E.B.; Margottin-Goguet, F.; Benarous, R.; Guatelli, J.C. Vpu Antagonizes BST-2-Mediated Restriction of HIV-1 Release via Beta-TrCP and Endo-Lysosomal Trafficking. PLoS Pathog. 2009, 5, e1000450. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Weber, E.; Tokarev, A.; Lewinski, M.; Rizk, M.; Suarez, M.; Guatelli, J.; Xiong, Y. Structural Basis of HIV-1 Vpu-Mediated BST2 Antagonism via Hijacking of the Clathrin Adaptor Protein Complex 1. eLife 2014, 3, e02362. [Google Scholar] [CrossRef] [PubMed]
- Pujol, F.M.; Laketa, V.; Schmidt, F.; Mukenhirn, M.; Müller, B.; Boulant, S.; Grimm, D.; Keppler, O.T.; Fackler, O.T. HIV-1 Vpu Antagonizes CD317/Tetherin by Adaptor Protein-1-Mediated Exclusion from Virus Assembly Sites. J. Virol. 2016, 90, 6709–6723. [Google Scholar] [CrossRef] [PubMed]
- McNatt, M.W.; Zang, T.; Bieniasz, P.D. Vpu Binds Directly to Tetherin and Displaces It from Nascent Virions. PLoS Pathog. 2013, 9, e1003299. [Google Scholar] [CrossRef]
- Sharma, S.; Jafari, M.; Bangar, A.; William, K.; Guatelli, J.; Lewinski, M.K. The C-Terminal End of HIV-1 Vpu Has a Clade-Specific Determinant That Antagonizes BST-2 and Facilitates Virion Release. J. Virol. 2019, 93, e02315-18. [Google Scholar] [CrossRef]
- Douglas, J.L.; Viswanathan, K.; McCarroll, M.N.; Gustin, J.K.; Früh, K.; Moses, A.V. Vpu Directs the Degradation of the Human Immunodeficiency Virus Restriction Factor BST-2/Tetherin via a {beta}TrCP-Dependent Mechanism. J. Virol. 2009, 83, 7931–7947. [Google Scholar] [CrossRef]
- Tokarev, A.A.; Munguia, J.; Guatelli, J.C. Serine-Threonine Ubiquitination Mediates Downregulation of BST-2/Tetherin and Relief of Restricted Virion Release by HIV-1 Vpu. J. Virol. 2011, 85, 51–63. [Google Scholar] [CrossRef]
- Magadán, J.G.; Pérez-Victoria, F.J.; Sougrat, R.; Ye, Y.; Strebel, K.; Bonifacino, J.S. Multilayered Mechanism of CD4 Downregulation by HIV-1 Vpu Involving Distinct ER Retention and ERAD Targeting Steps. PLoS Pathog. 2010, 6, e1000869. [Google Scholar] [CrossRef]
- Arias, J.F.; Heyer, L.N.; von Bredow, B.; Weisgrau, K.L.; Moldt, B.; Burton, D.R.; Rakasz, E.G.; Evans, D.T. Tetherin Antagonism by Vpu Protects HIV-Infected Cells from Antibody-Dependent Cell-Mediated Cytotoxicity. Proc. Natl. Acad. Sci. USA 2014, 111, 6425–6430. [Google Scholar] [CrossRef]
- Galão, R.P.; Le Tortorec, A.; Pickering, S.; Kueck, T.; Neil, S.J.D. Innate Sensing of HIV-1 Assembly by Tetherin Induces NFκB-Dependent Proinflammatory Responses. Cell Host Microbe 2012, 12, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Cocka, L.J.; Bates, P. Identification of Alternatively Translated Tetherin Isoforms with Differing Antiviral and Signaling Activities. PLoS Pathog. 2012, 8, e1002931. [Google Scholar] [CrossRef] [PubMed]
- Ross, T.M.; Oran, A.E.; Cullen, B.R. Inhibition of HIV-1 Progeny Virion Release by Cell-Surface CD4 Is Relieved by Expression of the Viral Nef Protein. Curr. Biol. 1999, 9, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Usami, Y.; Wu, Y.; Göttlinger, H.G. SERINC3 and SERINC5 Restrict HIV-1 Infectivity and Are Counteracted by Nef. Nature 2015, 526, 218–223. [Google Scholar] [CrossRef]
- Rosa, A.; Chande, A.; Ziglio, S.; De Sanctis, V.; Bertorelli, R.; Goh, S.L.; McCauley, S.M.; Nowosielska, A.; Antonarakis, S.E.; Luban, J.; et al. HIV-1 Nef Promotes Infection by Excluding SERINC5 from Virion Incorporation. Nature 2015, 526, 212–217. [Google Scholar] [CrossRef]
- Leonhardt, S.A.; Purdy, M.D.; Grover, J.R.; Yang, Z.; Poulos, S.; McIntire, W.E.; Tatham, E.A.; Erramilli, S.K.; Nosol, K.; Lai, K.K.; et al. Antiviral HIV-1 SERINC Restriction Factors Disrupt Virus Membrane Asymmetry. Nat. Commun. 2023, 14, 4368. [Google Scholar] [CrossRef]
- Raghunath, G.; Abbott, E.H.; Marin, M.; Wu, H.; Reyes Ballista, J.M.; Brindley, M.A.; Melikyan, G.B. Disruption of Transmembrane Phosphatidylserine Asymmetry by HIV-1 Incorporated SERINC5 Is Not Responsible for Virus Restriction. Biomolecules 2024, 14, 570. [Google Scholar] [CrossRef]
- Shi, J.; Xiong, R.; Zhou, T.; Su, P.; Zhang, X.; Qiu, X.; Li, H.; Li, S.; Yu, C.; Wang, B.; et al. HIV-1 Nef Antagonizes SERINC5 Restriction by Downregulation of SERINC5 via the Endosome/Lysosome System. J. Virol. 2018, 92, e00196-18. [Google Scholar] [CrossRef]
- Singh, R.; Stoneham, C.; Lim, C.; Jia, X.; Guenaga, J.; Wyatt, R.; Wertheim, J.O.; Xiong, Y.; Guatelli, J. Phosphoserine Acidic Cluster Motifs Bind Distinct Basic Regions on the μ Subunits of Clathrin Adaptor Protein Complexes. J. Biol. Chem. 2018, 293, 15678–15690. [Google Scholar] [CrossRef]
- Stoneham, C.A.; Ramirez, P.W.; Singh, R.; Suarez, M.; Debray, A.; Lim, C.; Jia, X.; Xiong, Y.; Guatelli, J. A Conserved Acidic-Cluster Motif in SERINC5 Confers Partial Resistance to Antagonism by HIV-1 Nef. J. Virol. 2020, 94, e01554-19. [Google Scholar] [CrossRef]
- Zhang, F.; Landford, W.N.; Ng, M.; McNatt, M.W.; Bieniasz, P.D.; Hatziioannou, T. SIV Nef Proteins Recruit the AP-2 Complex to Antagonize Tetherin and Facilitate Virion Release. PLoS Pathog. 2011, 7, e1002039. [Google Scholar] [CrossRef] [PubMed]
- Buffalo, C.Z.; Stürzel, C.M.; Heusinger, E.; Kmiec, D.; Kirchhoff, F.; Hurley, J.H.; Ren, X. Structural Basis for Tetherin Antagonism as a Barrier to Zoonotic Lentiviral Transmission. Cell Host Microbe 2019, 26, 359–368.e8. [Google Scholar] [CrossRef] [PubMed]
- Egan, M.A.; Carruth, L.M.; Rowell, J.F.; Yu, X.; Siliciano, R.F. Human Immunodeficiency Virus Type 1 Envelope Protein Endocytosis Mediated by a Highly Conserved Intrinsic Internalization Signal in the Cytoplasmic Domain of Gp41 Is Suppressed in the Presence of the Pr55gag Precursor Protein. J. Virol. 1996, 70, 6547–6556. [Google Scholar] [CrossRef]
- Ohno, H.; Aguilar, R.C.; Fournier, M.C.; Hennecke, S.; Cosson, P.; Bonifacino, J.S. Interaction of Endocytic Signals from the HIV-1 Envelope Glycoprotein Complex with Members of the Adaptor Medium Chain Family. Virology 1997, 238, 305–315. [Google Scholar] [CrossRef] [PubMed]
- McCune, J.M.; Rabin, L.B.; Feinberg, M.B.; Lieberman, M.; Kosek, J.C.; Reyes, G.R.; Weissman, I.L. Endoproteolytic Cleavage of Gp160 Is Required for the Activation of Human Immunodeficiency Virus. Cell 1988, 53, 55–67. [Google Scholar] [CrossRef]
- Rowell, J.F.; Stanhope, P.E.; Siliciano, R.F. Endocytosis of Endogenously Synthesized HIV-1 Envelope Protein. Mechanism and Role in Processing for Association with Class II MHC. J. Immunol. 1995, 155, 473–488. [Google Scholar] [CrossRef]
- Boge, M.; Wyss, S.; Bonifacino, J.S.; Thali, M. A Membrane-Proximal Tyrosine-Based Signal Mediates Internalization of the HIV-1 Envelope Glycoprotein via Interaction with the AP-2 Clathrin Adaptor. J. Biol. Chem. 1998, 273, 15773–15778. [Google Scholar] [CrossRef]
- Berlioz-Torrent, C.; Shacklett, B.L.; Erdtmann, L.; Delamarre, L.; Bouchaert, I.; Sonigo, P.; Dokhelar, M.C.; Benarous, R. Interactions of the Cytoplasmic Domains of Human and Simian Retroviral Transmembrane Proteins with Components of the Clathrin Adaptor Complexes Modulate Intracellular and Cell Surface Expression of Envelope Glycoproteins. J. Virol. 1999, 73, 1350–1361. [Google Scholar] [CrossRef]
- Day, J.R.; Münk, C.; Guatelli, J.C. The Membrane-Proximal Tyrosine-Based Sorting Signal of Human Immunodeficiency Virus Type 1 Gp41 Is Required for Optimal Viral Infectivity. J. Virol. 2004, 78, 1069–1079. [Google Scholar] [CrossRef]
- Bhakta, S.J.; Shang, L.; Prince, J.L.; Claiborne, D.T.; Hunter, E. Mutagenesis of Tyrosine and Di-Leucine Motifs in the HIV-1 Envelope Cytoplasmic Domain Results in a Loss of Env-Mediated Fusion and Infectivity. Retrovirology 2011, 8, 37. [Google Scholar] [CrossRef]
- Wyss, S.; Berlioz-Torrent, C.; Boge, M.; Blot, G.; Höning, S.; Benarous, R.; Thali, M. The Highly Conserved C-Terminal Dileucine Motif in the Cytosolic Domain of the Human Immunodeficiency Virus Type 1 Envelope Glycoprotein Is Critical for Its Association with the AP-1 Clathrin Adaptor [Correction of Adapter]. J. Virol. 2001, 75, 2982–2992. [Google Scholar] [CrossRef] [PubMed]
- Anokhin, B.; Spearman, P. Viral and Host Factors Regulating HIV-1 Envelope Protein Trafficking and Particle Incorporation. Viruses 2022, 14, 1729. [Google Scholar] [CrossRef] [PubMed]
- Breed, M.W.; Elser, S.E.; Torben, W.; Jordan, A.P.O.; Aye, P.P.; Midkiff, C.; Schiro, F.; Sugimoto, C.; Alvarez-Hernandez, X.; Blair, R.V.; et al. Elite Control, Gut CD4 T Cell Sparing, and Enhanced Mucosal T Cell Responses in Macaca Nemestrina Infected by a Simian Immunodeficiency Virus Lacking a Gp41 Trafficking Motif. J. Virol. 2015, 89, 10156–10175. [Google Scholar] [CrossRef]
- Noble, B.; Abada, P.; Nunez-Iglesias, J.; Cannon, P.M. Recruitment of the Adaptor Protein 2 Complex by the Human Immunodeficiency Virus Type 2 Envelope Protein Is Necessary for High Levels of Virus Release. J. Virol. 2006, 80, 2924–2932. [Google Scholar] [CrossRef] [PubMed]
- Hauser, H.; Lopez, L.A.; Yang, S.J.; Oldenburg, J.E.; Exline, C.M.; Guatelli, J.C.; Cannon, P.M. HIV-1 Vpu and HIV-2 Env Counteract BST-2/Tetherin by Sequestration in a Perinuclear Compartment. Retrovirology 2010, 7, 51. [Google Scholar] [CrossRef]
- Dong, X.; Li, H.; Derdowski, A.; Ding, L.; Burnett, A.; Chen, X.; Peters, T.R.; Dermody, T.S.; Woodruff, E.; Wang, J.-J.; et al. AP-3 Directs the Intracellular Trafficking of HIV-1 Gag and Plays a Key Role in Particle Assembly. Cell 2005, 120, 663–674. [Google Scholar] [CrossRef]
- Kyere, S.K.; Mercredi, P.Y.; Dong, X.; Spearman, P.; Summers, M.F. The HIV-1 Matrix Protein Does Not Interact Directly with the Protein Interactive Domain of AP-3δ. Virus Res. 2012, 169, 411–414. [Google Scholar] [CrossRef]
- Liu, L.; Sutton, J.; Woodruff, E.; Villalta, F.; Spearman, P.; Dong, X. Defective HIV-1 Particle Assembly in AP-3-Deficient Cells Derived from Patients with Hermansky-Pudlak Syndrome Type 2. J. Virol. 2012, 86, 11242–11253. [Google Scholar] [CrossRef]
- Alford, J.E.; Marongiu, M.; Watkins, G.L.; Anderson, E.C. Human Immunodeficiency Virus Type 2 (HIV-2) Gag Is Trafficked in an AP-3 and AP-5 Dependent Manner. PLoS ONE 2016, 11, e0158941. [Google Scholar] [CrossRef]
- Morris, K.L.; Buffalo, C.Z.; Stürzel, C.M.; Heusinger, E.; Kirchhoff, F.; Ren, X.; Hurley, J.H. HIV-1 Nefs Are Cargo-Sensitive AP-1 Trimerization Switches in Tetherin Downregulation. Cell 2018, 174, 659–671.e14. [Google Scholar] [CrossRef]
- Canagarajah, B.J.; Ren, X.; Bonifacino, J.S.; Hurley, J.H. The Clathrin Adaptor Complexes as a Paradigm for Membrane-Associated Allostery. Protein Sci. 2013, 22, 517–529. [Google Scholar] [CrossRef] [PubMed]
- Heldwein, E.E.; Macia, E.; Wang, J.; Yin, H.L.; Kirchhausen, T.; Harrison, S.C. Crystal Structure of the Clathrin Adaptor Protein 1 Core. Proc. Natl. Acad. Sci. USA 2004, 101, 14108–14113. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.-T.; Ren, X.; Zhang, R.; Lee, I.-H.; Hurley, J.H. HIV-1 Nef Hijacks Clathrin Coats by Stabilizing AP-1:Arf1 Polygons. Science 2015, 350, aac5137. [Google Scholar] [CrossRef] [PubMed]
- Hooy, R.M.; Iwamoto, Y.; Tudorica, D.A.; Ren, X.; Hurley, J.H. Self-Assembly and Structure of a Clathrin-Independent AP-1:Arf1 Tubular Membrane Coat. Sci. Adv. 2022, 8, eadd3914. [Google Scholar] [CrossRef]
- Foti, M.; Mangasarian, A.; Piguet, V.; Lew, D.P.; Krause, K.H.; Trono, D.; Carpentier, J.L. Nef-Mediated Clathrin-Coated Pit Formation. J. Cell Biol. 1997, 139, 37–47. [Google Scholar] [CrossRef]
- Janvier, K.; Craig, H.; Hitchin, D.; Madrid, R.; Sol-Foulon, N.; Renault, L.; Cherfils, J.; Cassel, D.; Benichou, S.; Guatelli, J. HIV-1 Nef Stabilizes the Association of Adaptor Protein Complexes with Membranes. J. Biol. Chem. 2003, 278, 8725–8732. [Google Scholar] [CrossRef]
- Voorhees, P.; Deignan, E.; van Donselaar, E.; Humphrey, J.; Marks, M.S.; Peters, P.J.; Bonifacino, J.S. An Acidic Sequence within the Cytoplasmic Domain of Furin Functions as a Determinant of Trans-Golgi Network Localization and Internalization from the Cell Surface. EMBO J. 1995, 14, 4961–4975. [Google Scholar] [CrossRef]
- Crump, C.M.; Xiang, Y.; Thomas, L.; Gu, F.; Austin, C.; Tooze, S.A.; Thomas, G. PACS-1 Binding to Adaptors Is Required for Acidic Cluster Motif-Mediated Protein Traffic. EMBO J. 2001, 20, 2191–2201. [Google Scholar] [CrossRef]
- Navarro Negredo, P.; Edgar, J.R.; Wrobel, A.G.; Zaccai, N.R.; Antrobus, R.; Owen, D.J.; Robinson, M.S. Contribution of the Clathrin Adaptor AP-1 Subunit Μ1 to Acidic Cluster Protein Sorting. J. Cell Biol. 2017, 216, 2927–2943. [Google Scholar] [CrossRef]
- Olety, B.; Usami, Y.; Wu, Y.; Peters, P.; Göttlinger, H. AP-2 Adaptor Complex-Dependent Enhancement of HIV-1 Replication by Nef in the Absence of the Nef/AP-2 Targets SERINC5 and CD4. mBio 2023, 14, e0338222. [Google Scholar] [CrossRef]
- Jouvenet, N.; Neil, S.J.D.; Bess, C.; Johnson, M.C.; Virgen, C.A.; Simon, S.M.; Bieniasz, P.D. Plasma Membrane Is the Site of Productive HIV-1 Particle Assembly. PLoS Biol. 2006, 4, e435. [Google Scholar] [CrossRef] [PubMed]
- Mlcochova, P.; Pelchen-Matthews, A.; Marsh, M. Organization and Regulation of Intracellular Plasma Membrane-Connected HIV-1 Assembly Compartments in Macrophages. BMC Biol. 2013, 11, 89. [Google Scholar] [CrossRef] [PubMed]
- Batonick, M.; Favre, M.; Boge, M.; Spearman, P.; Höning, S.; Thali, M. Interaction of HIV-1 Gag with the Clathrin-Associated Adaptor AP-2. Virology 2005, 342, 190–200. [Google Scholar] [CrossRef]
- Camus, G.; Segura-Morales, C.; Molle, D.; Lopez-Vergès, S.; Begon-Pescia, C.; Cazevieille, C.; Schu, P.; Bertrand, E.; Berlioz-Torrent, C.; Basyuk, E. The Clathrin Adaptor Complex AP-1 Binds HIV-1 and MLV Gag and Facilitates Their Budding. Mol. Biol. Cell 2007, 18, 3193–3203. [Google Scholar] [CrossRef] [PubMed]
- Painter, M.M.; Zimmerman, G.E.; Merlino, M.S.; Robertson, A.W.; Terry, V.H.; Ren, X.; McLeod, M.R.; Gomez-Rodriguez, L.; Garcia, K.A.; Leonard, J.A.; et al. Concanamycin A Counteracts HIV-1 Nef to Enhance Immune Clearance of Infected Primary Cells by Cytotoxic T Lymphocytes. Proc. Natl. Acad. Sci. USA 2020, 117, 23835–23846. [Google Scholar] [CrossRef]
- Ooi, C.E.; Dell’Angelica, E.C.; Bonifacino, J.S. ADP-Ribosylation Factor 1 (ARF1) Regulates Recruitment of the AP-3 Adaptor Complex to Membranes. J. Cell Biol. 1998, 142, 391–402. [Google Scholar] [CrossRef]
- Ren, X.; Farías, G.G.; Canagarajah, B.J.; Bonifacino, J.S.; Hurley, J.H. Structural Basis for Recruitment and Activation of the AP-1 Clathrin Adaptor Complex by Arf1. Cell 2013, 152, 755–767. [Google Scholar] [CrossRef]
- He, K.; Song, E.; Upadhyayula, S.; Dang, S.; Gaudin, R.; Skillern, W.; Bu, K.; Capraro, B.R.; Rapoport, I.; Kusters, I.; et al. Dynamics of Auxilin 1 and GAK in Clathrin-Mediated Traffic. J. Cell Biol. 2020, 219, e201908142. [Google Scholar] [CrossRef]
- Ungewickell, E.J.; Hinrichsen, L. Endocytosis: Clathrin-Mediated Membrane Budding. Curr. Opin. Cell Biol. 2007, 19, 417–425. [Google Scholar] [CrossRef]
- Conner, S.D.; Schmid, S.L. Identification of an Adaptor-Associated Kinase, AAK1, as a Regulator of Clathrin-Mediated Endocytosis. J. Cell Biol. 2002, 156, 921–929. [Google Scholar] [CrossRef]
- Yuan, Y.-H.; Mao, N.-D.; Duan, J.-L.; Zhang, H.; Garrido, C.; Lirussi, F.; Gao, Y.; Xie, T.; Ye, X.-Y. Recent Progress in Discovery of Novel AAK1 Inhibitors: From Pain Therapy to Potential Anti-Viral Agents. J. Enzyme Inhib. Med. Chem. 2023, 38, 2279906. [Google Scholar] [CrossRef] [PubMed]
- Upshaw, W.C.; Richey, J.M.; Ravi, G.; Chen, A.; Ahmadzadeh, S.; Shekoohi, S.; Viswanath, O.; Kaye, A.D. An Overview of the Safety and Efficacy of LX-9211 in Treating Neuropathic Pain Conditions. Expert. Opin. Investig. Drugs 2024, 33, 829–837. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Adaptor Protein Complex | Cellular Functions | Clathrin-Association | Interactions with HIV-1 Proteins | HIV Functions Supported or Affected |
|---|---|---|---|---|
| AP1 (Adaptor protein complex 1) | Transport between endosomes and the trans-Golgi network (TGN), mostly retrograde | Yes | Nef, Vpu, Env, Gag | Modulation of class I MHC Modulation of BST2 (Tetherin) Env trafficking, assembly |
| AP2 (Adaptor protein complex 2) | Endocytosis | Yes | Nef, Vpu, Env, Gag | Downregulation of CD4 Downregulation of BST2 Endocytosis of Env, assembly |
| AP3 (Adaptor protein complex 3) | Transport to lysosomes and lysosome-related organelles | Controversial | Nef, Gag | Virion assembly in MVBs/intracellular virus containing compartments |
| AP4 (Adaptor protein complex 4) | Transport to the basolateral surface of polarized cells. Transport to pre-autophagosomes | No | Unknown | Unknown |
| AP5 (Adaptor protein complex 5) | Retrieval to the TGN; transport of proteins involved in autophagic flux | No | Unknown | Virion release (HIV-2) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barone, M.E.; Lim, A.; Woody, M.; Taklifi, P.; Yeasmin, F.; Wang, K.; Lewinski, M.K.; Singh, R.; Stoneham, C.A.; Jia, X.; et al. Adaptor Protein Complexes in HIV-1 Pathogenesis: Mechanisms and Therapeutic Potential. Viruses 2025, 17, 715. https://doi.org/10.3390/v17050715
Barone ME, Lim A, Woody M, Taklifi P, Yeasmin F, Wang K, Lewinski MK, Singh R, Stoneham CA, Jia X, et al. Adaptor Protein Complexes in HIV-1 Pathogenesis: Mechanisms and Therapeutic Potential. Viruses. 2025; 17(5):715. https://doi.org/10.3390/v17050715
Chicago/Turabian StyleBarone, Maria Elena, Alexis Lim, Madison Woody, Parisa Taklifi, Fatema Yeasmin, Kequan Wang, Mary K. Lewinski, Rajendra Singh, Charlotte A. Stoneham, Xiaofei Jia, and et al. 2025. "Adaptor Protein Complexes in HIV-1 Pathogenesis: Mechanisms and Therapeutic Potential" Viruses 17, no. 5: 715. https://doi.org/10.3390/v17050715
APA StyleBarone, M. E., Lim, A., Woody, M., Taklifi, P., Yeasmin, F., Wang, K., Lewinski, M. K., Singh, R., Stoneham, C. A., Jia, X., & Guatelli, J. (2025). Adaptor Protein Complexes in HIV-1 Pathogenesis: Mechanisms and Therapeutic Potential. Viruses, 17(5), 715. https://doi.org/10.3390/v17050715