
Genetic Diversity in the Capsid Protein-Coding Region of HIV-1 Circulating in Benguela, Angola: Implications for Primary Resistance to the Novel Capsid Inhibitor Lenacapavir


Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. FTA® Card Disc Preparation
2.3. Nested PCR and Sequencing of the Capsid-Encoding Region
2.4. Sequence Editing and Analysis
3. Results
3.1. Sociodemographic and Epidemiological Analysis of the Sample Group
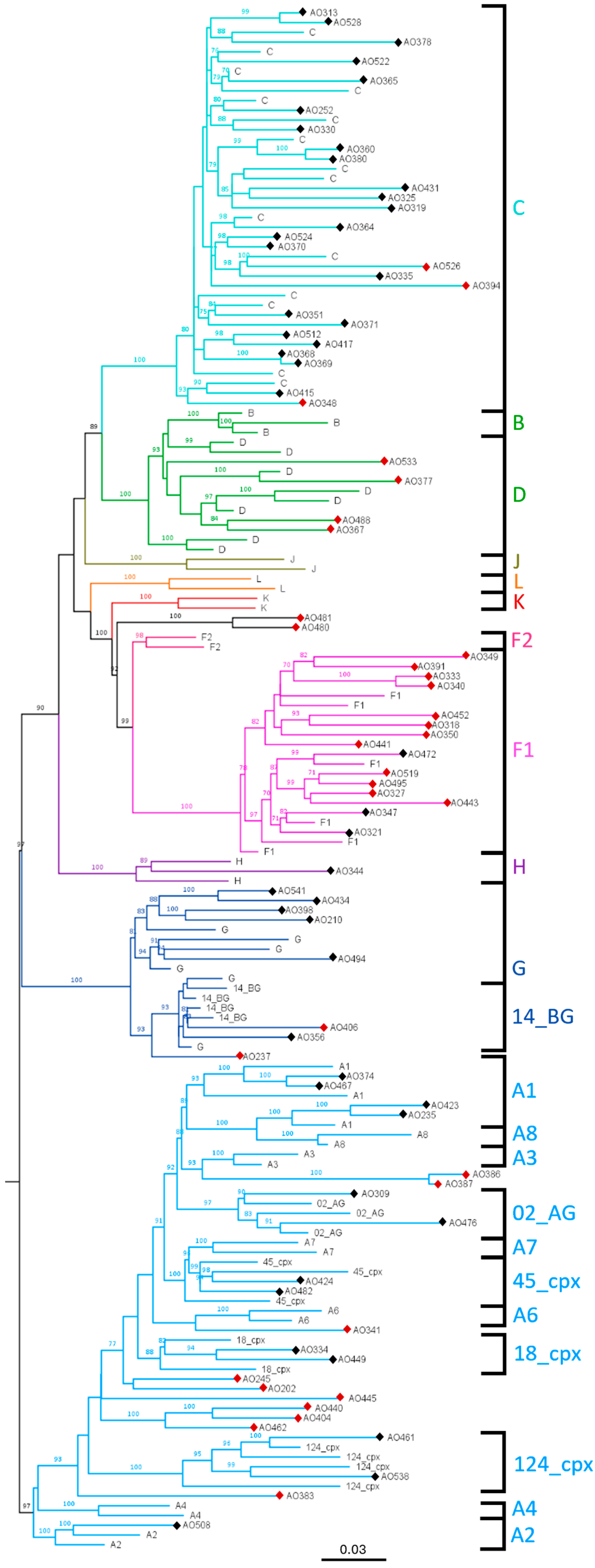
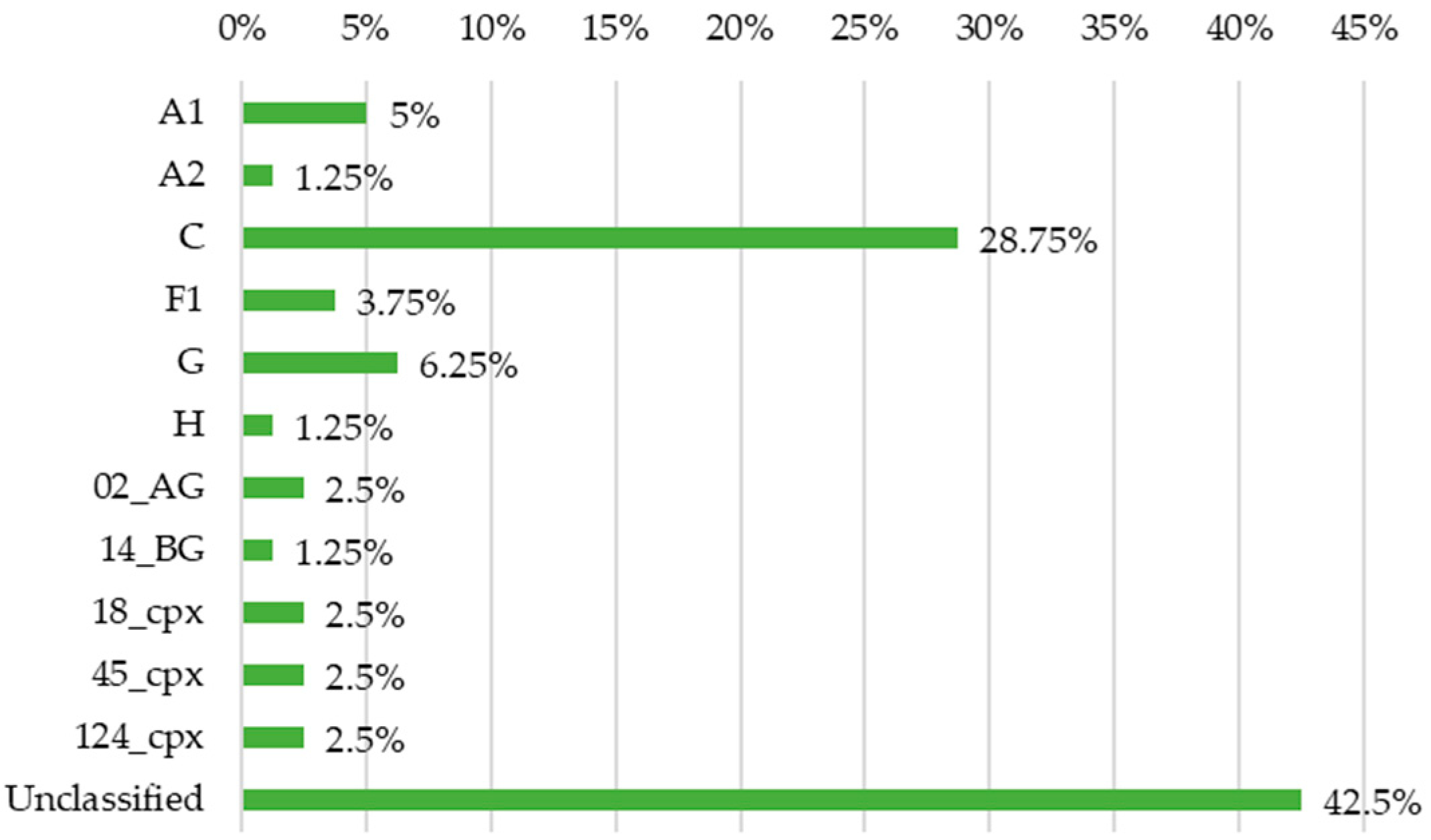
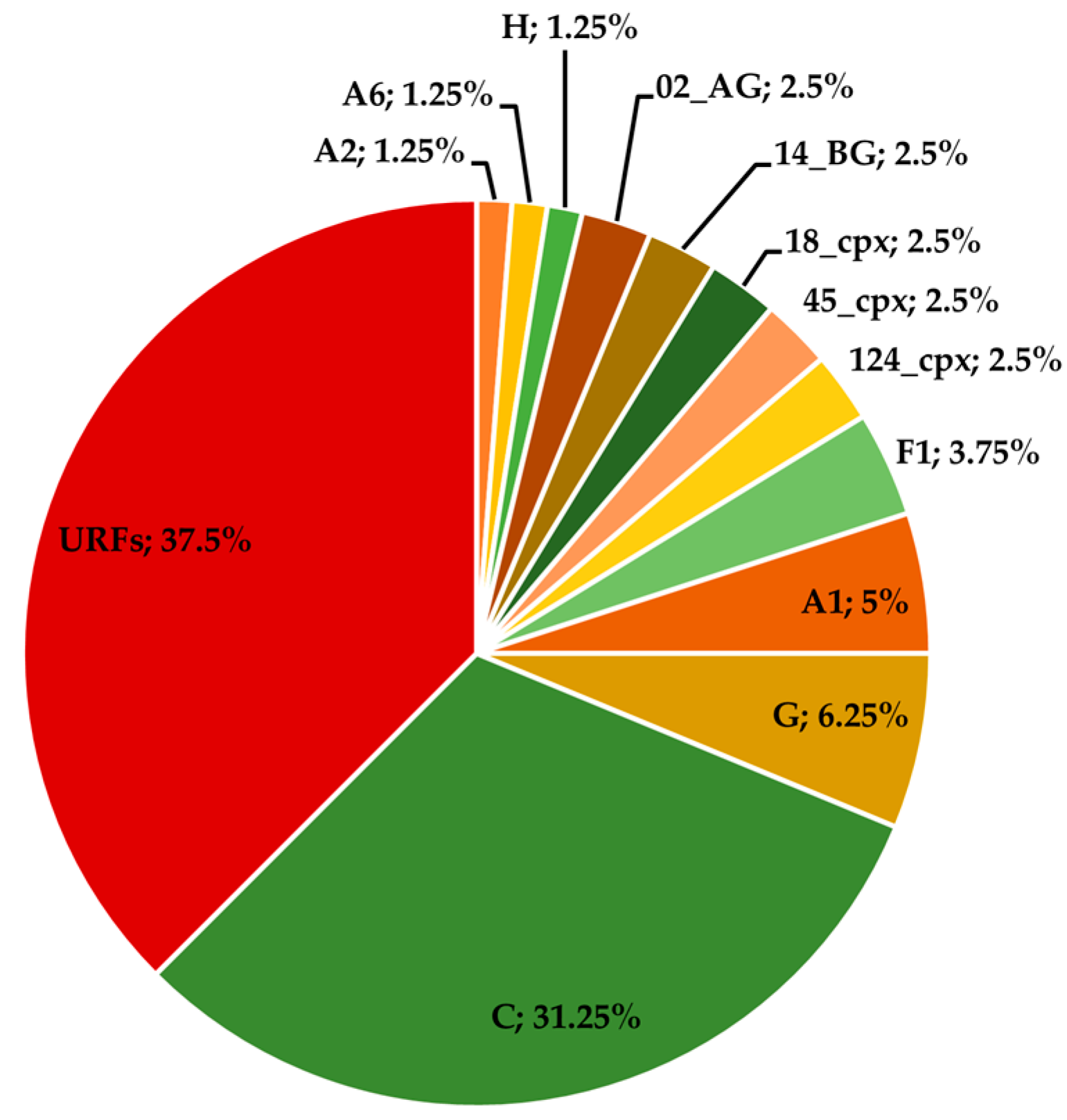
3.2. Phylogenetic Characterisation of the Sequences
3.3. Resistance Analysis to LEN
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Joint United Nations Programme on HIV/AIDS. The Urgency of Now: AIDS at a Crossroads; Joint United Nations Programme on HIV/AIDS: Geneva, Switzerland, 2024. [Google Scholar]
- Hemelaar, J. The Origin and Diversity of the HIV-1 Pandemic. Trends Mol. Med. 2012, 18, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Afonso, J.M.; Bello, G.; Guimarães, M.L.; Sojka, M.; Morgado, M.G. HIV-1 Genetic Diversity and Transmitted Drug Resistance Mutations Among Patients from the North, Central and South Regions of Angola. PLoS ONE 2012, 7, e42996. [Google Scholar] [CrossRef] [PubMed]
- Quitéria, A. Diversidade Genética e Resistência aos Anti-Retrovirais no Vírus da Imunodeficiência Humana Tipo 1 em Circulação em Benguela, Angola. Master’s Dissertation, Instituto de Higiene e Medicina Tropical, Universidade Nova de Lisboa, Lisboa, Portugal, 2019. [Google Scholar]
- Country Factsheets—Angola. Available online: https://www.unaids.org/en/regionscountries/countries/angola (accessed on 20 August 2024).
- Sebastião, C.S.; Morais, J.; Brito, M. Clinical and Public Health Implications of HIV- Genetic Diversity and Drug Resistance Mutations in Angola: A Systematic Review. AIDS Rev. 2020, 23, 48–56. [Google Scholar] [CrossRef]
- Bester, S.M.; Wei, G.; Zhao, H.; Adu-Ampratwum, D.; Iqbal, N.; Courouble, V.V.; Francis, A.C.; Annamalai, A.S.; Singh, P.K.; Shkriabai, N.; et al. Structural and Mechanistic Bases for a Potent HIV-1 Capsid Inhibitor. Science 2020, 370, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, S.; Torbett, B.E. Interactions of HIV-1 Capsid with Host Factors and Their Implications for Developing Novel Therapeutics. Viruses 2021, 13, 417. [Google Scholar] [CrossRef]
- National Institutes of Health. Gilead Sciences Study to Evaluate Safety, Pharmacokinetics, and Antiviral Activity of Lenacapavir Administered Subcutaneously in Human Immunodeficiency Virus (HIV) -1 Infected Adults; National Institutes of Health: Bethesda, MD, USA, 2021.
- National Institutes of Health. Gilead Sciences Study to Evaluate the Safety and Efficacy of Lenacapavir (GS-6207) in Combination with an Optimized Background Regimen (OBR) in Heavily Treatment Experienced Participants Living with HIV-1 Infection with Multidrug Resistance (CAPELLA); National Institutes of Health: Bethesda, MD, USA, 2024.
- Margot, N. Resistance Analysis of Long-Acting Lenacapavir in Heavily Treatment-Experienced People with HIV After 104 Weeks of Treatment. In Proceedings of the 19th European AIDS Conference, Warsaw, Poland, 18–21 October 2023. [Google Scholar]
- Margot, N.; Vanderveen, L.; Naik, V.; Ram, R.; Parvangada, P.C.; Martin, R.; Rhee, M.; Callebaut, C. Phenotypic Resistance to Lenacapavir and Monotherapy Efficacy in a Proof-of-Concept Clinical Study. J. Antimicrob. Chemother. 2022, 77, 989–995. [Google Scholar] [CrossRef]
- Nka, A.D.; Bouba, Y.; Teto, G.; Semengue, E.N.J.; Takou, D.K.; Ngueko, A.M.K.; Fabeni, L.; Carioti, L.; Armenia, D.; Pabo, W.; et al. Evaluation of HIV-1 Capsid Genetic Variability and Lenacapavir (GS-6207) Drug Resistance-Associated Mutations According to Viral Clades Among Drug-Naive Individuals. J. Antimicrob. Chemother. 2023, 78, 272–275. [Google Scholar] [CrossRef]
- Troyano-Hernáez, P.; Reinosa, R.; Holguín, Á. HIV Capsid Protein Genetic Diversity Across HIV-1 Variants and Impact on New Capsid-Inhibitor Lenacapavir. Front. Microbiol. 2022, 13, 854974. [Google Scholar] [CrossRef]
- Margot, N.; Naik, V.; Nekkalapudi, A.; Boopathy, A.; Falkard, B.; Callebaut, C. Rapid HIV-1 Genotyping Assay for the Detection of Capsid Mutations. J. Med. Virol. 2023, 95, e29292. [Google Scholar] [CrossRef]
- Hall, T.A. BioEdit: A User-Friendly Biological Sequence Alignment Editor and Analysis Program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT Online Service: Multiple Sequence Alignment, Interactive Sequence Choice and Visualization. Brief. Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2016, 44, D67–D72. [Google Scholar] [CrossRef]
- Castresana, J. Selection of Conserved Blocks from Multiple Alignments for Their Use in Phylogenetic Analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef]
- Trifinopoulos, J.; Nguyen, L.-T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A Fast Online Phylogenetic Tool for Maximum Likelihood Analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef]
- Lole, K.S.; Bollinger, R.C.; Paranjape, R.S.; Gadkari, D.; Kulkarni, S.S.; Novak, N.G.; Ingersoll, R.; Sheppard, H.W.; Ray, S.C. Full-Length Human Immunodeficiency Virus Type 1 Genomes from Subtype C-Infected Seroconverters in India, with Evidence of Intersubtype Recombination. J. Virol. 1999, 73, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.F.; Shafer, R.W. Web Resources for HIV Type 1 Genotypic-Resistance Test Interpretation. Clin. Infect. Dis. 2006, 42, 1608–1618. [Google Scholar] [CrossRef]
- Zhou, J.; Price, A.J.; Halambage, U.D.; James, L.C.; Aiken, C. HIV-1 Resistance to the Capsid-Targeting Inhibitor PF74 Results in Altered Dependence on Host Factors Required for Virus Nuclear Entry. J. Virol. 2015, 89, 9068–9079. [Google Scholar] [CrossRef]
- Shi, J.; Zhou, J.; Shah, V.B.; Aiken, C.; Whitby, K. Small-Molecule Inhibition of Human Immunodeficiency Virus Type 1 Infection by Virus Capsid Destabilization. J. Virol. 2011, 85, 542–549. [Google Scholar] [CrossRef]
- Sowd, G.A.; Shi, J.; Fulmer, A.; Aiken, C. HIV-1 Capsid Stability Enables Inositol Phosphate-Independent Infection of Target Cells and Promotes Integration into Genes. PLoS Pathog. 2023, 19, e1011423. [Google Scholar] [CrossRef]
- Pineda-Peña, A.-C.; Faria, N.R.; Imbrechts, S.; Libin, P.; Abecasis, A.B.; Deforche, K.; Gómez-López, A.; Camacho, R.J.; de Oliveira, T.; Vandamme, A.-M. Automated Subtyping of HIV-1 Genetic Sequences for Clinical and Surveillance Purposes: Performance Evaluation of the New REGA Version 3 and Seven Other Tools. Infect. Genet. Evol. 2013, 19, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, N.; von Siebenthal, C.; Vongrad, V.; Turk, T.; Neumann, K.; Beerenwinkel, N.; Bogojeska, J.; Fellay, J.; Roth, V.; Kok, Y.L.; et al. Determinants of HIV-1 reservoir size and long-term dynamics during suppressive ART. Nat. Commun. 2019, 10, 3193. [Google Scholar] [CrossRef] [PubMed]
- Abidi, S.H.; Aibekova, L.; Davlidova, S.; Amangeldiyeva, A.; Foley, B.; Ali, S. Origin and Evolution of HIV-1 Subtype A6. PLoS ONE 2021, 16, e0260604. [Google Scholar] [CrossRef] [PubMed]
- de Pina-Araujo, I.I.M.; Delatorre, E.; Guimarães, M.L.; Morgado, M.G.; Bello, G. Origin and Population Dynamics of a Novel HIV-1 Subtype G Clade Circulating in Cape Verde and Portugal. PLoS ONE 2015, 10, e0127384. [Google Scholar] [CrossRef]
- Zhang, C.; Ding, N.; Wei, J.-F. Different Sliding Window Sizes and Inappropriate Subtype References Result in Discordant Mosaic Maps and Breakpoint Locations of HIV-1 CRFs. Infect. Genet. Evol. 2008, 8, 693–697. [Google Scholar] [CrossRef]
- Smyth, R.P.; Schlub, T.E.; Grimm, A.J.; Waugh, C.; Ellenberg, P.; Chopra, A.; Mallal, S.; Cromer, D.; Mak, J.; Davenport, M.P. Identifying Recombination Hot Spots in the HIV-1 Genome. J. Virol. 2014, 88, 2891–2902. [Google Scholar] [CrossRef]
- Da Silva, R.K.M.; Morais, J.; Foley, B.T.; Bello, G.; Morgado, M.G.; Guimarães, M.L. Identification of a New Circulating Recombinant Form of Human Immunodeficiency Virus Type 1, CRF124_cpx Involving Subtypes A, G, H, and CRF27_cpx in Angola. Front. Microbiol. 2022, 13, 992640. [Google Scholar] [CrossRef]
- Margot, N.; Pennetzdorfer, N.; Naik, V.; Rhee, M.; Callebaut, C. Cross-resistance to entry inhibitors and lenacapavir resistance through Week 52 in study CAPELLA. Antivir. Ther. 2023, 28, 13596535231220754. [Google Scholar] [CrossRef]
- Yant, S.R. In Vitro Resistance Profile of GS-6207, a First-in-Class Picomolar HIV Capsid Inhibitor in Clinical Development as a Novel Long-Acting Antiretroviral Agent. In Proceedings of the 10th IAS Conference on HIV Science (IAS 2019), Mexico City, Mexico, 21–24 July 2019. [Google Scholar]
- Bbosa, N.; Kaleebu, P.; Ssemwanga, D. HIV Subtype Diversity Worldwide. Curr. Opin. HIV AIDS 2019, 14, 153. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Sequence 5′–3′ | Location (in HXB2) | CA-Encoding Region (in HXB2) | |
|---|---|---|---|
| Outer primers | CA_S1 GAS ATA AAA GAC ACC AAG GAA GC | 1066–1088 | 1186–1879 |
| CA_AS1 CAT TTC CAA CAG CCC TTT TTC C | 2015–2036 | ||
| Inner primers | CA_S2 GCA GCA GCT GMC ACA GG | 1141–1157 | |
| CA_AS2 CCT AAA ATT GCY TCT CTG CAT C | 1920–1941 |
| Sequence 5′–3′ | Location (in HXB2) | CA-Encoding Region (in HXB2) |
|---|---|---|
| 3MA-F2 CAG TAG CAA YCC TCT ATT GTG TRC | 1031–1054 | 1186–1879 |
| 5NC-R CCT AGG GGC CCT GCA ATT T | 1998–2016 |
| LEN Resistance Mutations (Validated) | Polymorphisms of Unknown Effect (Stanford HIVdb) | Literature-Reported Mutations | ||
|---|---|---|---|---|
| Stanford HIVdb | IAS-USA | ANRS-MIE | ||
| L56I | L56I | L56I | T107A/C/S | S41A [24] |
| N57S | M66I | M66I | E45A [7] | |
| M66I | Q67H | Q67H/K/N | Q67A [25] | |
| Q67H/K/N/Y | K70H/N/R/S | K70H/N/R/S | L172I [24] | |
| K70H/N/R/S | N74D/S | N74D/H/K/S | E180A [7] | |
| N74D/K/S | A105T | A105S/T | T200I [26] | |
| A105E/S/T | T107N | T107C/N | ||
| T107C/N | ||||
| Characteristics | |
|---|---|
| Age (years) | |
| Mean (Min–Max) | 39 (0–80) |
| Sex (%) | |
| Female | 54 |
| Male | 39 |
| Unspecified | 7 |
| Marital status (%) | |
| Single | 36 |
| Married | 35 |
| Divorced | 12 |
| Widowed | 10 |
| Unspecified | 7 |
| Education level (%) | |
| No education | 28 |
| Primary education | 31 |
| Secondary education | 28 |
| Higher education | 6 |
| Unspecified | 7 |
| Previous STIs (%) | |
| Yes | 12 |
| No | 42 |
| Does not know/Did not answer | 46 |
| Present STI co-infection (%) | |
| Syphilis | 9 |
| HBV | 7 |
| Syphilis + HBV | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Queirós, G.; Yefimenko, L.; Pereira, F.M.; Piedade, J. Genetic Diversity in the Capsid Protein-Coding Region of HIV-1 Circulating in Benguela, Angola: Implications for Primary Resistance to the Novel Capsid Inhibitor Lenacapavir. Viruses 2025, 17, 711. https://doi.org/10.3390/v17050711
Queirós G, Yefimenko L, Pereira FM, Piedade J. Genetic Diversity in the Capsid Protein-Coding Region of HIV-1 Circulating in Benguela, Angola: Implications for Primary Resistance to the Novel Capsid Inhibitor Lenacapavir. Viruses. 2025; 17(5):711. https://doi.org/10.3390/v17050711
Chicago/Turabian StyleQueirós, Gonçalo, Lesya Yefimenko, Filomena M. Pereira, and João Piedade. 2025. "Genetic Diversity in the Capsid Protein-Coding Region of HIV-1 Circulating in Benguela, Angola: Implications for Primary Resistance to the Novel Capsid Inhibitor Lenacapavir" Viruses 17, no. 5: 711. https://doi.org/10.3390/v17050711
APA StyleQueirós, G., Yefimenko, L., Pereira, F. M., & Piedade, J. (2025). Genetic Diversity in the Capsid Protein-Coding Region of HIV-1 Circulating in Benguela, Angola: Implications for Primary Resistance to the Novel Capsid Inhibitor Lenacapavir. Viruses, 17(5), 711. https://doi.org/10.3390/v17050711