
TAR RNA Mimicry of INI1 and Its Influence on Non-Integration Function of HIV-1 Integrase

Abstract

1. Introduction
2. Relevant Sections
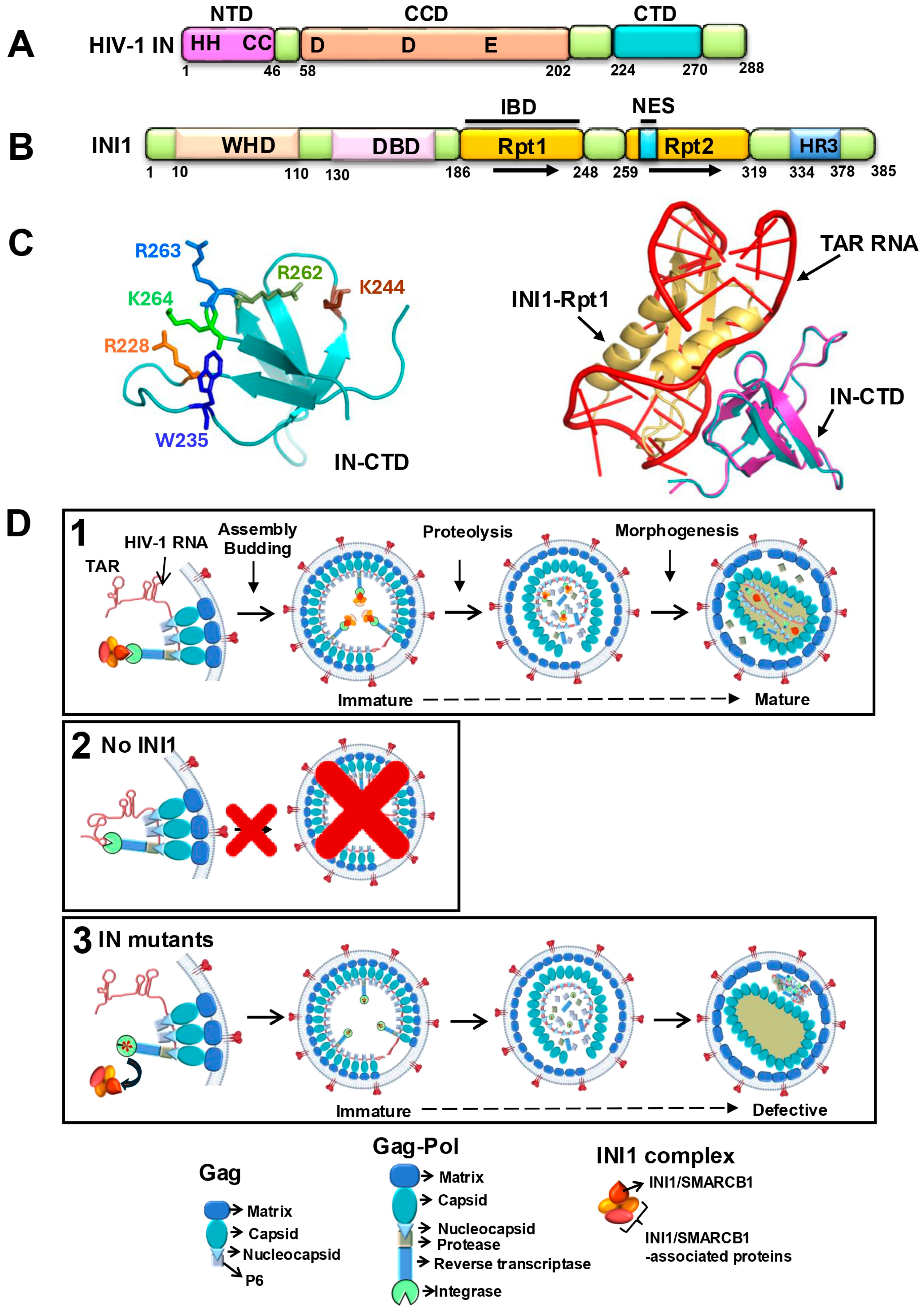
2.1. Integrase as a Target for Inhibiting HIV-1 Late Events
2.2. INI1 Is an IN-Binding Host Factor Essential for Viral Late Events
2.3. Structure of the Rpt1 Domain of INI1 and Structural Modeling of IN-CTD/INI1-Rpt1 Interactions
2.4. Structural Mimicry Between INI1-Rpt1 and TAR RNA
- (i)
- TAR RNA and INI1183–304 bind to the same residues of IN: A panel of IN-CTD substitution mutations that span the interface residues of the IN-CTD/INI1-Rpt1 complex were tested for their ability to interact with TAR RNA using a protein–RNA interaction Alpha assay. The interaction profiles of TAR RNA and INI1183–304 with IN-CTD mutants were identical, indicating that these molecules recognize the same residues of IN [32] (see Table 1).
- (ii)
- TAR RNA and INI1183–304 compete for binding to IN-CTD: TAR RNA and INI1183–304 competed for binding to IN-CTD with similar IC50 values (IC50 ≈ 5 nM) in an Alpha assay [32]. Furthermore, the inhibition of the IN-CTD/INI1-Rpt1 interaction by TAR was specific, as a scrambled RNA or a different fragment of HIV-1 genomic RNA (nts 237–279) did not inhibit CTD/INI1183–304 binding [32]. Together, these results indicated that INI1 Rpt1 and TAR require the same surface of IN-CTD for binding.
- (iii)
- Structural similarity between INI1 Rpt1 and HIV-1 TAR RNA: To understand this further, the complex between IN-CTD and TAR RNA was computationally modeled using MdockPP [32,60,61]. It was found that the same set of hydrophobic and positively charged IN-CTD residues is involved in interaction with both INI1-Rpt1 and TAR RNA, confirming the biochemical studies (Figure 1C, left panel). When the complexes of IN-CTD/INI1-Rpt1 were superimposed onto the complex of IN-CTD/TAR, INI1-Rpt1 and TAR overlapped with each other in three-dimensional space (Figure 1C right panel) [32]. A close examination of the Rpt1 NMR structures indicated that it has a string of surface-exposed, negatively charged residues that are positioned in a specific manner. An examination of the position of phosphate groups on TAR, which overlap with INI1-Rpt1 in the superimposed structure, indicated that these phosphate groups are positioned in a manner resembling the arrangement of the negatively charged residues on the INI1-Rpt1 surface in three-dimensional space [32]. These analyses indicated that TAR RNA and INI-Rpt1 have overall similar shape and electrostatic charge distribution on the surface, explaining how these two molecules could contact the same residues on the surface of IN-CTD. This is consistent with the similarity in binding of these two molecules to IN [32].
2.5. A Model to Explain the Role of INI1 in HIV-1 Late Events Based on Its RNA Mimicry
2.6. Role of RNA and/or INI1 in Particle Morphogenesis
3. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 2D | Two-dimensional |
| 3D | Three-dimensional |
| aa | Amino acid |
| AIDS | Acquired immunodeficiency syndrome |
| ALLINI | Allosteric inhibitors of integrase |
| ART | Anti-retroviral therapy |
| ATP | Adenosine triphosphate |
| BAF47 | Bramha-related gene (BRG)1-associated factor 47 |
| CA | Capsid |
| cDNA | Complementary deoxyribonucleic acid |
| CTD | C-terminal domain |
| DBD | DNA-binding domain |
| DNA | Deoxyribonucleic acid |
| GST | Glutathione S-transferase |
| HADDOCK | High ambiguity driven protein–protein docking |
| HDAC1 | Histone deacetylase 1 |
| HIV | Human immunodeficiency virus |
| HR3 | Homology region III |
| hSNF5 | Human sucrose non-fermenting |
| IBD | Integrase-binding domain |
| IC50 | Half-maximal inhibitory concentration |
| IID | INI1-interaction-defective |
| IN | Integrase |
| INI1 | Integrase interactor 1 |
| LEDGF | Lens epithelium–derived growth factor |
| LTR | Long terminal repeat |
| MA | Matrix |
| NC | Nucleocapsid |
| ND | Not determined |
| NES | Nuclear export signal |
| NMR | Nuclear magnetic resonance |
| nts | Nucleotides |
| PDB | Protein Data Bank |
| PPI | Protein–protein interaction |
| PR | Protease |
| RNA | Ribonucleic acid |
| RNP | Ribonucleoprotein |
| Rpt1 | Repeat 1 |
| Rpt2 | Repeat 2 |
| RT | Reverse transcriptase |
| SAP18 | Sin3A associated protein 18 |
| shRNA | Short hairpin ribonucleic acid |
| SIV | Simian immunodeficiency virus |
| SMARCB1 | SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily B member 1 |
| SWI/SNF | Switch/sucrose non-fermenting |
| TAR | Trans-activation response |
| Tat | Trans-activator of transcription |
| Vpr | Viral protein R |
| WHD | Winged-Helix DNA-binding domain |
| WT | Wild-type |
References
- World Health Organization. HIV and AIDS; World Health Organization: Geneva, Switzerland, 2023. [Google Scholar]
- Lichterfeld, M.; Gao, C.; Yu, X.G. An ordeal that does not heal: Understanding barriers to a cure for HIV-1 infection. Trends Immunol. 2022, 43, 608–616. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Narayanan, E.; Liu, Q.; Tsybovsky, Y.; Boswell, K.; Ding, S.; Hu, Z.; Follmann, D.; Lin, Y.; Miao, H.; et al. A multiclade env-gag VLP mRNA vaccine elicits tier-2 HIV-1-neutralizing antibodies and reduces the risk of heterologous SHIV infection in macaques. Nat. Med. 2021, 27, 2234–2245. [Google Scholar] [CrossRef] [PubMed]
- Back, D.; Marzolini, C. The challenge of HIV treatment in an era of polypharmacy. J. Int. AIDS Soc. 2020, 23, e25449. [Google Scholar] [CrossRef] [PubMed]
- Clavel, F.; Hance, A.J. HIV drug resistance. New Engl. J. Med. 2004, 350, 1023–1035. [Google Scholar] [CrossRef]
- Roux, H.; Chomont, N. Measuring Human Immunodeficiency Virus Reservoirs: Do We Need to Choose Between Quantity and Quality? J. Infect. Dis. 2024, 229, 635–643. [Google Scholar] [CrossRef]
- Henderson, L.J.; Reoma, L.B.; Kovacs, J.A.; Nath, A. Advances toward Curing HIV-1 Infection in Tissue Reservoirs. J. Virol. 2020, 94, e00375-19. [Google Scholar] [CrossRef]
- Sun, Y.; Wang, L. Development of Anti-HIV Therapeutics: From Conventional Drug Discovery to Cutting-Edge Technology. Pharmaceuticals 2024, 17, 887. [Google Scholar] [CrossRef]
- Woollard, S.M.; Kanmogne, G.D. Maraviroc: A review of its use in HIV infection and beyond. Drug Des. Devel Ther. 2015, 9, 5447–5468. [Google Scholar] [CrossRef]
- Engelman, A.; Englund, G.; Orenstein, J.M.; Martin, M.A.; Craigie, R. Multiple effects of mutations in human immunodeficiency virus type 1 integrase on viral replication. J. Virol. 1995, 69, 2729–2736. [Google Scholar] [CrossRef]
- Elliott, J.L.; Eschbach, J.E.; Koneru, P.C.; Li, W.; Puray-Chavez, M.; Townsend, D.; Lawson, D.Q.; Engelman, A.N.; Kvaratskhelia, M.; Kutluay, S.B. Integrase-RNA interactions underscore the critical role of integrase in HIV-1 virion morphogenesis. Elife 2020, 9, e54311. [Google Scholar] [CrossRef]
- Elliott, J.L.; Kutluay, S.B. Going beyond Integration: The Emerging Role of HIV-1 Integrase in Virion Morphogenesis. Viruses 2020, 12, 1005. [Google Scholar] [CrossRef] [PubMed]
- Engelman, A.N.; Kvaratskhelia, M. Multimodal Functionalities of HIV-1 Integrase. Viruses 2022, 14, 926. [Google Scholar] [CrossRef] [PubMed]
- Kleinpeter, A.; Freed, E.O. How to package the RNA of HIV-1. Elife 2020, 9, e63585. [Google Scholar] [CrossRef] [PubMed]
- Yung, E.; Sorin, M.; Pal, A.; Craig, E.; Morozov, A.; Delattre, O.; Kappes, J.; Ott, D.; Kalpana, G.V. Inhibition of HIV-1 virion production by a transdominant mutant of integrase interactor 1. Nat. Med. 2001, 7, 920–926. [Google Scholar] [CrossRef]
- Feng, L.; Larue, R.C.; Slaughter, A.; Kessl, J.J.; Kvaratskhelia, M. HIV-1 integrase multimerization as a therapeutic target. Curr. Top. Microbiol. Immunol. 2015, 389, 93–119. [Google Scholar] [CrossRef]
- Kessl, J.J.; Kutluay, S.B.; Townsend, D.; Rebensburg, S.; Slaughter, A.; Larue, R.C.; Shkriabai, N.; Bakouche, N.; Fuchs, J.R.; Bieniasz, P.D.; et al. HIV-1 Integrase Binds the Viral RNA Genome and Is Essential during Virion Morphogenesis. Cell 2016, 166, 1257–1268.e12. [Google Scholar] [CrossRef]
- Cherepanov, P.; Maertens, G.; Proost, P.; Devreese, B.; Van Beeumen, J.; Engelborghs, Y.; De Clercq, E.; Debyser, Z. HIV-1 integrase forms stable tetramers and associates with LEDGF/p75 protein in human cells. J. Biol. Chem. 2003, 278, 372–381. [Google Scholar] [CrossRef]
- Engelman, A.; Cherepanov, P. The lentiviral integrase binding protein LEDGF/p75 and HIV-1 replication. PLoS Pathog. 2008, 4, e1000046. [Google Scholar] [CrossRef]
- Turlure, F.; Devroe, E.; Silver, P.A.; Engelman, A. Human cell proteins and human immunodeficiency virus DNA integration. Front. Biosci. 2004, 9, 3187–3208. [Google Scholar] [CrossRef]
- Kalpana, G.V.; Marmon, S.; Wang, W.; Crabtree, G.R.; Goff, S.P. Binding and stimulation of HIV-1 integrase by a human homolog of yeast transcription factor SNF5. Science 1994, 266, 2002–2006. [Google Scholar] [CrossRef]
- Batisse, C.; Lapaillerie, D.; Humbert, N.; Real, E.; Zhu, R.; Mely, Y.; Parissi, V.; Ruff, M.; Batisse, J. Integrase-LEDGF/p75 complex triggers the formation of biomolecular condensates that modulate HIV-1 integration efficiency in vitro. J. Biol. Chem. 2024, 300, 107374. [Google Scholar] [CrossRef] [PubMed]
- Bedwell, G.J.; Jang, S.; Li, W.; Singh, P.K.; Engelman, A.N. rigrag: High-resolution mapping of genic targeting preferences during HIV-1 integration in vitro and in vivo. Nucleic Acids Res. 2021, 49, 7330–7346. [Google Scholar] [CrossRef] [PubMed]
- Lapaillerie, D.; Lelandais, B.; Mauro, E.; Lagadec, F.; Tumiotto, C.; Miskey, C.; Ferran, G.; Kuschner, N.; Calmels, C.; Metifiot, M.; et al. Modulation of the intrinsic chromatin binding property of HIV-1 integrase by LEDGF/p75. Nucleic Acids Res. 2021, 49, 11241–11256. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.K.; Bedwell, G.J.; Engelman, A.N. Spatial and Genomic Correlates of HIV-1 Integration Site Targeting. Cells 2022, 11, 655. [Google Scholar] [CrossRef]
- Cano, J.; Kalpana, G.V. Inhibition of early stages of HIV-1 assembly by INI1/hSNF5 transdominant negative mutant S6. J. Virol. 2011, 85, 2254–2265. [Google Scholar] [CrossRef]
- La Porte, A.; Cano, J.; Wu, X.; Mitra, D.; Kalpana, G.V. An Essential Role of INI1/hSNF5 Chromatin Remodeling Protein in HIV-1 Posttranscriptional Events and Gag/Gag-Pol Stability. J. Virol. 2016, 90, 9889–9904. [Google Scholar] [CrossRef]
- Sorin, M.; Cano, J.; Das, S.; Mathew, S.; Wu, X.; Davies, K.P.; Shi, X.; Cheng, S.W.; Ott, D.; Kalpana, G.V. Recruitment of a SAP18-HDAC1 complex into HIV-1 virions and its requirement for viral replication. PLoS Pathog. 2009, 5, e1000463. [Google Scholar] [CrossRef]
- Sorin, M.; Yung, E.; Wu, X.; Kalpana, G.V. HIV-1 replication in cell lines harboring INI1/hSNF5 mutations. Retrovirology 2006, 3, 56. [Google Scholar] [CrossRef]
- Yung, E.; Sorin, M.; Wang, E.J.; Perumal, S.; Ott, D.; Kalpana, G.V. Specificity of interaction of INI1/hSNF5 with retroviral integrases and its functional significance. J. Virol. 2004, 78, 2222–2231. [Google Scholar] [CrossRef]
- Mathew, S.; Nguyen, M.; Wu, X.; Pal, A.; Shah, V.B.; Prasad, V.R.; Aiken, C.; Kalpana, G.V. INI1/hSNF5-interaction defective HIV-1 IN mutants exhibit impaired particle morphology, reverse transcription and integration in vivo. Retrovirology 2013, 10, 66. [Google Scholar] [CrossRef]
- Dixit, U.; Bhutoria, S.; Wu, X.; Qiu, L.; Spira, M.; Mathew, S.; Harris, R.; Adams, L.J.; Cahill, S.; Pathak, R.; et al. INI1/SMARCB1 Rpt1 domain mimics TAR RNA in binding to integrase to facilitate HIV-1 replication. Nat. Commun. 2021, 12, 2743. [Google Scholar] [CrossRef] [PubMed]
- Craigie, R. The molecular biology of HIV integrase. Future Virol. 2012, 7, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Engelman, A. In vivo analysis of retroviral integrase structure and function. Adv. Virus Res. 1999, 52, 411–426. [Google Scholar] [PubMed]
- Leavitt, A.D.; Robles, G.; Alesandro, N.; Varmus, H.E. Human immunodeficiency virus type 1 integrase mutants retain in vitro integrase activity yet fail to integrate viral DNA efficiently during infection. J. Virol. 1996, 70, 721–728. [Google Scholar] [CrossRef]
- Rocchi, C.; Gouet, P.; Parissi, V.; Fiorini, F. The C-Terminal Domain of HIV-1 Integrase: A Swiss Army Knife for the Virus? Viruses 2022, 14, 1397. [Google Scholar] [CrossRef]
- Sundquist, W.I.; Krausslich, H.G. HIV-1 assembly, budding, and maturation. Cold Spring Harb. Perspect. Med. 2012, 2, a006924. [Google Scholar] [CrossRef]
- Shehu-Xhilaga, M.; Crowe, S.M.; Mak, J. Maintenance of the Gag/Gag-Pol ratio is important for human immunodeficiency virus type 1 RNA dimerization and viral infectivity. J. Virol. 2001, 75, 1834–1841. [Google Scholar] [CrossRef]
- Jurado, K.A.; Wang, H.; Slaughter, A.; Feng, L.; Kessl, J.J.; Koh, Y.; Wang, W.; Ballandras-Colas, A.; Patel, P.A.; Fuchs, J.R.; et al. Allosteric integrase inhibitor potency is determined through the inhibition of HIV-1 particle maturation. Proc. Natl. Acad. Sci. USA 2013, 110, 8690–8695. [Google Scholar] [CrossRef]
- Centore, R.C.; Sandoval, G.J.; Soares, L.M.M.; Kadoch, C.; Chan, H.M. Mammalian SWI/SNF Chromatin Remodeling Complexes: Emerging Mechanisms and Therapeutic Strategies. Trends Genet. 2020, 36, 936–950. [Google Scholar] [CrossRef]
- Reddy, D.; Bhattacharya, S.; Workman, J.L. (mis)-Targeting of SWI/SNF complex(es) in cancer. Cancer Metastasis Rev. 2023, 42, 455–470. [Google Scholar] [CrossRef]
- Wang, L.; Tang, J. SWI/SNF complexes and cancers. Gene 2023, 870, 147420. [Google Scholar] [CrossRef] [PubMed]
- Cooper, G.W.; Hong, A.L. SMARCB1-Deficient Cancers: Novel Molecular Insights and Therapeutic Vulnerabilities. Cancers 2022, 14, 3645. [Google Scholar] [CrossRef] [PubMed]
- Graf, M.; Interlandi, M.; Moreno, N.; Holdhof, D.; Gobel, C.; Melcher, V.; Mertins, J.; Albert, T.K.; Kastrati, D.; Alfert, A.; et al. Single-cell transcriptomics identifies potential cells of origin of MYC rhabdoid tumors. Nat. Commun. 2022, 13, 1544. [Google Scholar] [CrossRef] [PubMed]
- Sevenet, N.; Sheridan, E.; Amram, D.; Schneider, P.; Handgretinger, R.; Delattre, O. Constitutional mutations of the hSNF5/INI1 gene predispose to a variety of cancers. Am. J. Hum. Genet. 1999, 65, 1342–1348. [Google Scholar] [CrossRef]
- Bushman, F. Targeting retroviral integration. Science 1995, 267, 1443–1444. [Google Scholar] [CrossRef]
- Lesbats, P.; Botbol, Y.; Chevereau, G.; Vaillant, C.; Calmels, C.; Arneodo, A.; Andreola, M.L.; Lavigne, M.; Parissi, V. Functional coupling between HIV-1 integrase and the SWI/SNF chromatin remodeling complex for efficient in vitro integration into stable nucleosomes. PLoS Pathog. 2011, 7, e1001280. [Google Scholar] [CrossRef]
- Cheng, S.W.; Davies, K.P.; Yung, E.; Beltran, R.J.; Yu, J.; Kalpana, G.V. c-MYC interacts with INI1/hSNF5 and requires the SWI/SNF complex for transactivation function. Nat. Genet. 1999, 22, 102–105. [Google Scholar] [CrossRef]
- Craig, E.; Zhang, Z.K.; Davies, K.P.; Kalpana, G.V. A masked NES in INI1/hSNF5 mediates hCRM1-dependent nuclear export: Implications for tumorigenesis. EMBO J. 2002, 21, 31–42. [Google Scholar] [CrossRef]
- Das, B.C.; Smith, M.E.; Kalpana, G.V. Design, synthesis of novel peptidomimetic derivatives of 4-HPR for rhabdoid tumors. Bioorg Med. Chem. Lett. 2008, 18, 4177–4180. [Google Scholar] [CrossRef]
- Das, B.C.; Smith, M.E.; Kalpana, G.V. Design and synthesis of 4-HPR derivatives for rhabdoid tumors. Bioorg Med. Chem. Lett. 2008, 18, 3805–3808. [Google Scholar] [CrossRef]
- Das, S.; Cano, J.; Kalpana, G.V. Multimerization and DNA binding properties of INI1/hSNF5 and its functional significance. J. Biol. Chem. 2009, 284, 19903–19914. [Google Scholar] [CrossRef] [PubMed]
- Morozov, A.; Yung, E.; Kalpana, G.V. Structure-function analysis of integrase interactor 1/hSNF5L1 reveals differential properties of two repeat motifs present in the highly conserved region. Proc. Natl. Acad. Sci. USA 1998, 95, 1120–1125. [Google Scholar] [CrossRef] [PubMed]
- Pathak, R.; Zin, F.; Thomas, C.; Bens, S.; Gayden, T.; Karamchandani, J.; Dudley, R.W.; Nemes, K.; Johann, P.D.; Oyen, F.; et al. Inhibition of nuclear export restores nuclear localization and residual tumor suppressor function of truncated SMARCB1/INI1 protein in a molecular subset of atypical teratoid/rhabdoid tumors. Acta Neuropathol. 2021, 142, 361–374. [Google Scholar] [CrossRef]
- Valencia, A.M.; Collings, C.K.; Dao, H.T.; St Pierre, R.; Cheng, Y.C.; Huang, J.; Sun, Z.Y.; Seo, H.S.; Mashtalir, N.; Comstock, D.E.; et al. Recurrent SMARCB1 Mutations Reveal a Nucleosome Acidic Patch Interaction Site That Potentiates mSWI/SNF Complex Chromatin Remodeling. Cell 2019, 179, 1342–1356.e23. [Google Scholar] [CrossRef]
- Allen, M.D.; Freund, S.M.; Zinzalla, G.; Bycroft, M. The SWI/SNF Subunit INI1 Contains an N-Terminal Winged Helix DNA Binding Domain that Is a Target for Mutations in Schwannomatosis. Structure 2015, 23, 1344–1349. [Google Scholar] [CrossRef]
- Morozov, A.; Lee, S.J.; Zhang, Z.K.; Cimica, V.; Zagzag, D.; Kalpana, G.V. INI1 induces interferon signaling and spindle checkpoint in rhabdoid tumors. Clin. Cancer Res. 2007, 13, 4721–4730. [Google Scholar] [CrossRef]
- Kohashi, K.; Oda, Y. Oncogenic roles of SMARCB1/INI1 and its deficient tumors. Cancer Sci. 2017, 108, 547–552. [Google Scholar] [CrossRef]
- Wu, D.Y.; Kalpana, G.V.; Goff, S.P.; Schubach, W.H. Epstein-Barr virus nuclear protein 2 (EBNA2) binds to a component of the human SNF-SWI complex, hSNF5/Ini1. J. Virol. 1996, 70, 6020–6028. [Google Scholar] [CrossRef]
- Qiu, L.; Bhutoria, S.; Kalpana, G.V.; Zou, X. Computational Modeling of IN-CTD/TAR Complex to Elucidate Additional Strategies to Inhibit HIV-1 Replication. Methods Mol. Biol. 2023, 2610, 75–84. [Google Scholar] [CrossRef]
- Xu, X.; Qiu, L.; Yan, C.; Ma, Z.; Grinter, S.Z.; Zou, X. Performance of MDockPP in CAPRI rounds 28-29 and 31-35 including the prediction of water-mediated interactions. Proteins 2017, 85, 424–434. [Google Scholar] [CrossRef]
- Huang, S.Y.; Yan, C.; Grinter, S.Z.; Chang, S.; Jiang, L.; Zou, X. Inclusion of the orientational entropic effect and low-resolution experimental information for protein-protein docking in Critical Assessment of PRedicted Interactions (CAPRI). Proteins 2013, 81, 2183–2191. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.Y.; Zou, X. A knowledge-based scoring function for protein-RNA interactions derived from a statistical mechanics-based iterative method. Nucleic Acids Res. 2014, 42, e55. [Google Scholar] [CrossRef]
- Lu, R.; Ghory, H.Z.; Engelman, A. Genetic analyses of conserved residues in the carboxyl-terminal domain of human immunodeficiency virus type 1 integrase. J. Virol. 2005, 79, 10356–10368. [Google Scholar] [CrossRef]
- Semenova, E.A.; Marchand, C.; Pommier, Y. HIV-1 integrase inhibitors: Update and perspectives. Adv. Pharmacol. 2008, 56, 199–228. [Google Scholar] [CrossRef]
- Wiskerchen, M.; Muesing, M.A. Human immunodeficiency virus type 1 integrase: Effects of mutations on viral ability to integrate, direct viral gene expression from unintegrated viral DNA templates, and sustain viral propagation in primary cells. J. Virol. 1995, 69, 376–386. [Google Scholar] [CrossRef]
- Ao, Z.; Huang, G.; Yao, H.; Xu, Z.; Labine, M.; Cochrane, A.W.; Yao, X. Interaction of human immunodeficiency virus type 1 integrase with cellular nuclear import receptor importin 7 and its impact on viral replication. J. Biol. Chem. 2007, 282, 13456–13467. [Google Scholar] [CrossRef]
- Shema Mugisha, C.; Dinh, T.; Kumar, A.; Tenneti, K.; Eschbach, J.E.; Davis, K.; Gifford, R.; Kvaratskhelia, M.; Kutluay, S.B. Emergence of Compensatory Mutations Reveals the Importance of Electrostatic Interactions between HIV-1 Integrase and Genomic RNA. mBio 2022, 13, e0043122. [Google Scholar] [CrossRef]
- Cannon, P.M.; Byles, E.D.; Kingsman, S.M.; Kingsman, A.J. Conserved sequences in the carboxyl terminus of integrase that are essential for human immunodeficiency virus type 1 replication. J. Virol. 1996, 70, 651–657. [Google Scholar] [CrossRef]
- Charpentier, C.; Descamps, D. Resistance to HIV Integrase Inhibitors: About R263K and E157Q Mutations. Viruses 2018, 10, 41. [Google Scholar] [CrossRef]
- De Houwer, S.; Demeulemeester, J.; Thys, W.; Rocha, S.; Dirix, L.; Gijsbers, R.; Christ, F.; Debyser, Z. The HIV-1 integrase mutant R263A/K264A is 2-fold defective for TRN-SR2 binding and viral nuclear import. J. Biol. Chem. 2014, 289, 25351–25361. [Google Scholar] [CrossRef]
- Rozina, A.; Anisenko, A.; Kikhai, T.; Silkina, M.; Gottikh, M. Complex Relationships between HIV-1 Integrase and Its Cellular Partners. Int. J. Mol. Sci. 2022, 23, 12341. [Google Scholar] [CrossRef] [PubMed]
- Cereseto, A.; Manganaro, L.; Gutierrez, M.I.; Terreni, M.; Fittipaldi, A.; Lusic, M.; Marcello, A.; Giacca, M. Acetylation of HIV-1 integrase by p300 regulates viral integration. EMBO J. 2005, 24, 3070–3081. [Google Scholar] [CrossRef] [PubMed]
- Madison, M.K.; Lawson, D.Q.; Elliott, J.; Ozanturk, A.N.; Koneru, P.C.; Townsend, D.; Errando, M.; Kvaratskhelia, M.; Kutluay, S.B. Allosteric HIV-1 Integrase Inhibitors Lead to Premature Degradation of the Viral RNA Genome and Integrase in Target Cells. J. Virol. 2017, 91, e00821-17. [Google Scholar] [CrossRef] [PubMed]
- Brockman, M.A.; Chopera, D.R.; Olvera, A.; Brumme, C.J.; Sela, J.; Markle, T.J.; Martin, E.; Carlson, J.M.; Le, A.Q.; McGovern, R.; et al. Uncommon pathways of immune escape attenuate HIV-1 integrase replication capacity. J. Virol. 2012, 86, 6913–6923. [Google Scholar] [CrossRef]
- Nomaguchi, M.; Miyake, A.; Doi, N.; Fujiwara, S.; Miyazaki, Y.; Tsunetsugu-Yokota, Y.; Yokoyama, M.; Sato, H.; Masuda, T.; Adachi, A. Natural single-nucleotide polymorphisms in the 3′ region of the HIV-1 pol gene modulate viral replication ability. J. Virol. 2014, 88, 4145–4160. [Google Scholar] [CrossRef]
- Li, M.; Craigie, R. Processing of viral DNA ends channels the HIV-1 integration reaction to concerted integration. J. Biol. Chem. 2005, 280, 29334–29339. [Google Scholar] [CrossRef]
- Ghasabi, F.; Hashempour, A.; Khodadad, N.; Bemani, S.; Keshani, P.; Shekiba, M.J.; Hasanshahi, Z. First report of computational protein-ligand docking to evaluate susceptibility to HIV integrase inhibitors in HIV-infected Iranian patients. Biochem. Biophys. Rep. 2022, 30, 101254. [Google Scholar] [CrossRef]
- Rogers, L.; Obasa, A.E.; Jacobs, G.B.; Sarafianos, S.G.; Sonnerborg, A.; Neogi, U.; Singh, K. Structural Implications of Genotypic Variations in HIV-1 Integrase From Diverse Subtypes. Front. Microbiol. 2018, 9, 1754. [Google Scholar] [CrossRef]
- Katz, A.; Solden, L.; Zou, S.B.; Navarre, W.W.; Ibba, M. Molecular evolution of protein-RNA mimicry as a mechanism for translational control. Nucleic Acids Res. 2014, 42, 3261–3271. [Google Scholar] [CrossRef]
- Tsonis, P.A.; Dwivedi, B. Molecular mimicry: Structural camouflage of proteins and nucleic acids. Biochim. Biophys. Acta 2008, 1783, 177–187. [Google Scholar] [CrossRef]
- Walbott, H.; Machado-Pinilla, R.; Liger, D.; Blaud, M.; Rety, S.; Grozdanov, P.N.; Godin, K.; van Tilbeurgh, H.; Varani, G.; Meier, U.T.; et al. The H/ACA RNP assembly factor SHQ1 functions as an RNA mimic. Genes. Dev. 2011, 25, 2398–2408. [Google Scholar] [CrossRef] [PubMed]
- Kutluay, S.B.; Zang, T.; Blanco-Melo, D.; Powell, C.; Jannain, D.; Errando, M.; Bieniasz, P.D. Global changes in the RNA binding specificity of HIV-1 gag regulate virion genesis. Cell 2014, 159, 1096–1109. [Google Scholar] [CrossRef] [PubMed]
- Passos, D.O.; Li, M.; Craigie, R.; Lyumkis, D. Retroviral integrase: Structure, mechanism, and inhibition. Enzymes 2021, 50, 249–300. [Google Scholar] [CrossRef]
- Passos, D.O.; Li, M.; Yang, R.; Rebensburg, S.V.; Ghirlando, R.; Jeon, Y.; Shkriabai, N.; Kvaratskhelia, M.; Craigie, R.; Lyumkis, D. Cryo-EM structures and atomic model of the HIV-1 strand transfer complex intasome. Science 2017, 355, 89–92. [Google Scholar] [CrossRef]
- Jing, T.; Shan, Z.; Dinh, T.; Biswas, A.; Jang, S.; Greenwood, J.; Li, M.; Zhang, Z.; Gray, G.; Shin, H.J.; et al. Oligomeric HIV-1 Integrase Structures Reveal Functional Plasticity for Intasome Assembly and RNA Binding. bioRxiv 2024. [Google Scholar] [CrossRef]
- Harrison, J.; Passos, D.O.; Bruhn, J.F.; Bauman, J.D.; Tuberty, L.; DeStefano, J.J.; Ruiz, F.X.; Lyumkis, D.; Arnold, E. Cryo-EM structure of the HIV-1 Pol polyprotein provides insights into virion maturation. Sci. Adv. 2022, 8, eabn9874. [Google Scholar] [CrossRef]
- Angelov, D.; Charra, M.; Seve, M.; Cote, J.; Khochbin, S.; Dimitrov, S. Differential remodeling of the HIV-1 nucleosome upon transcription activators and SWI/SNF complex binding. J. Mol. Biol. 2000, 302, 315–326. [Google Scholar] [CrossRef]
- Ariumi, Y.; Serhan, F.; Turelli, P.; Telenti, A.; Trono, D. The integrase interactor 1 (INI1) proteins facilitate Tat-mediated human immunodeficiency virus type 1 transcription. Retrovirology 2006, 3, 47. [Google Scholar] [CrossRef]
- Rafati, H.; Parra, M.; Hakre, S.; Moshkin, Y.; Verdin, E.; Mahmoudi, T. Repressive LTR nucleosome positioning by the BAF complex is required for HIV latency. PLoS Biol. 2011, 9, e1001206, Erratum in PLoS Biol. 2015, 13, e1002302. [Google Scholar] [CrossRef]
- Treand, C.; du Chene, I.; Bres, V.; Kiernan, R.; Benarous, R.; Benkirane, M.; Emiliani, S. Requirement for SWI/SNF chromatin-remodeling complex in Tat-mediated activation of the HIV-1 promoter. EMBO J. 2006, 25, 1690–1699. [Google Scholar] [CrossRef]
- Maillot, B.; Levy, N.; Eiler, S.; Crucifix, C.; Granger, F.; Richert, L.; Didier, P.; Godet, J.; Pradeau-Aubreton, K.; Emiliani, S.; et al. Structural and functional role of INI1 and LEDGF in the HIV-1 preintegration complex. PLoS ONE 2013, 8, e60734. [Google Scholar] [CrossRef] [PubMed]
- Maroun, M.; Delelis, O.; Coadou, G.; Bader, T.; Segeral, E.; Mbemba, G.; Petit, C.; Sonigo, P.; Rain, J.C.; Mouscadet, J.F.; et al. Inhibition of early steps of HIV-1 replication by SNF5/Ini1. J. Biol. Chem. 2006, 281, 22736–22743. [Google Scholar] [CrossRef]
- Ali, A.M.; Atmaj, J.; Van Oosterwijk, N.; Groves, M.R.; Domling, A. Stapled Peptides Inhibitors: A New Window for Target Drug Discovery. Comput. Struct. Biotechnol. J. 2019, 17, 263–281. [Google Scholar] [CrossRef] [PubMed]
- Moiola, M.; Memeo, M.G.; Quadrelli, P. Stapled Peptides-A Useful Improvement for Peptide-Based Drugs. Molecules 2019, 24, 3654. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| IN Residues | IN Mutations *, ** | IN-INI1 Interaction | IN-RNA Interaction | Infection | Capsid Morphology | Reference |
|---|---|---|---|---|---|---|
| Charged | ||||||
| R228 | R228A | Defective | Defective | Defective | Defective | [11,32,64] |
| K244 | K244A | Defective | Defective | Defective | ND | [32,64] |
| K244E | ND | ND | Defective | ND | [65] | |
| K244A/E246A | ND | ND | Defective | ND | [66] | |
| K240A, K244A/R263A, K264A | ND | ND | Defective | ND | [67] | |
| R262 | R262A | ND | ND | Not Defective | ND | [64] |
| R262A/R263A | ND | Defective | Defective | Defective | [11,64,68] | |
| R262A/K264A | ND | ND | Defective | ND | [64] | |
| R262I/K264T | ND | ND | Defective | ND | [69] | |
| R262D/R263V/K264E | ND | ND | Defective | ND | [65] | |
| R263 | R263A | ND | ND | Less Defective | ND | [64] |
| R263K | ND | ND | Not Defective | ND | [70] | |
| R263L | ND | ND | Not Defective | ND | [65] | |
| R263S | ND | ND | Not Defective | ND | [69] | |
| R263A/K264A | ND | ND | Defective | ND | [71] | |
| K264 | K264A | ND | ND | Not Defective | ND | [64] |
| K264E | ND | ND | Defective | ND | [64] | |
| K264R | ND | ND | Not Defective | ND | [72] | |
| K264A/K266A | Defective | Defective | Defective | Defective | [17,32] | |
| K264R/K266R/K273R | ND | ND | Defective | ND | [73] | |
| R269 | R269A | ND | ND | Reduced and delayed | ND | [64,65] |
| R269A/D270A | ND | ND | Reduced | ND | [64,65] | |
| R269A/K273A | Defective | Defective | Defective | Defective | [17,32,74] | |
| Hydrophobic | ||||||
| I220 | I220L | ND | ND | Slightly Reduced | ND | [75] |
| F223 | F223A | ND | ND | Defective | ND | [76] |
| F223E | ND | ND | Defective | ND | [76] | |
| F223G | ND | ND | Defective | ND | [76] | |
| F223H | ND | ND | Less Defective | ND | [76] | |
| F223K | ND | ND | Defective | ND | [76] | |
| F223S | ND | ND | Defective | ND | [76] | |
| F223Y | ND | ND | Not Defective | ND | [76] | |
| W235 | W235A | Defective | Defective | Defective | ND | [32,34,77] |
| W235E | Defective | Defective | Defective | Defective | [32,34,77] | |
| W235K | Defective | Defective | Defective | ND | [32,34,77] | |
| W235F | Not Defective | Not Defective | Not Defective | ND | [32,34,77] | |
| A265 | A265T | ND | ND | Not Defective | ND | [78] |
| A265V | ND | ND | Not Defective | ND | [78,79] | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalpana, G.V.; Ernst, E.; Haldar, S. TAR RNA Mimicry of INI1 and Its Influence on Non-Integration Function of HIV-1 Integrase. Viruses 2025, 17, 693. https://doi.org/10.3390/v17050693
Kalpana GV, Ernst E, Haldar S. TAR RNA Mimicry of INI1 and Its Influence on Non-Integration Function of HIV-1 Integrase. Viruses. 2025; 17(5):693. https://doi.org/10.3390/v17050693
Chicago/Turabian StyleKalpana, Ganjam V., Emilie Ernst, and Swati Haldar. 2025. "TAR RNA Mimicry of INI1 and Its Influence on Non-Integration Function of HIV-1 Integrase" Viruses 17, no. 5: 693. https://doi.org/10.3390/v17050693
APA StyleKalpana, G. V., Ernst, E., & Haldar, S. (2025). TAR RNA Mimicry of INI1 and Its Influence on Non-Integration Function of HIV-1 Integrase. Viruses, 17(5), 693. https://doi.org/10.3390/v17050693