
Sequencing of One Unique Recombinant CRF85_BC/CRF01_AE Genome and Two Partial Genomes from Ningxia, China

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Source
2.2. RNA Extraction and Amplification
2.3. Sequence Analysis
3. Results
3.1. Demographic and Genetic Subtype Analysis
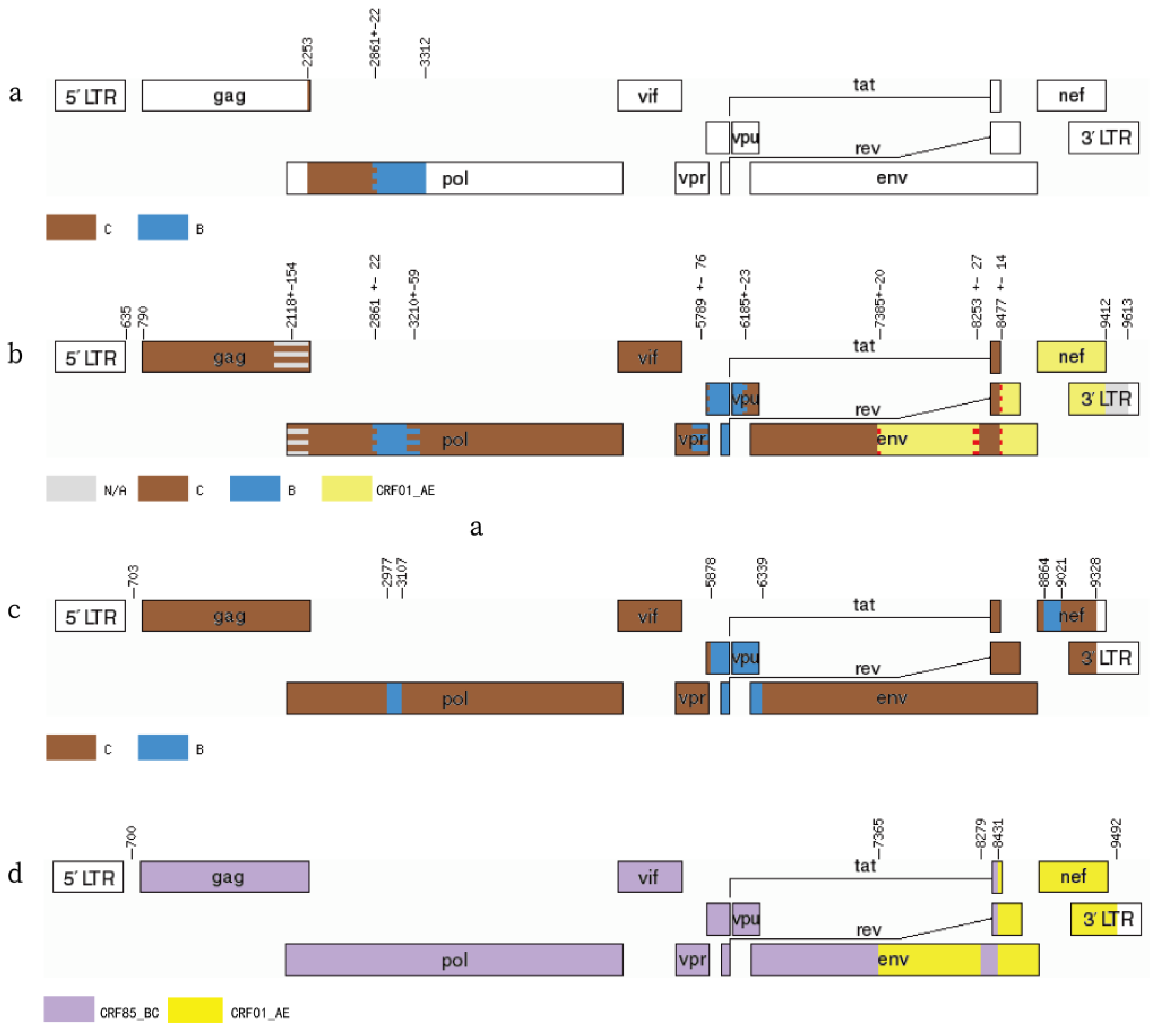
3.2. NFLG Recombination Patterns Analysis
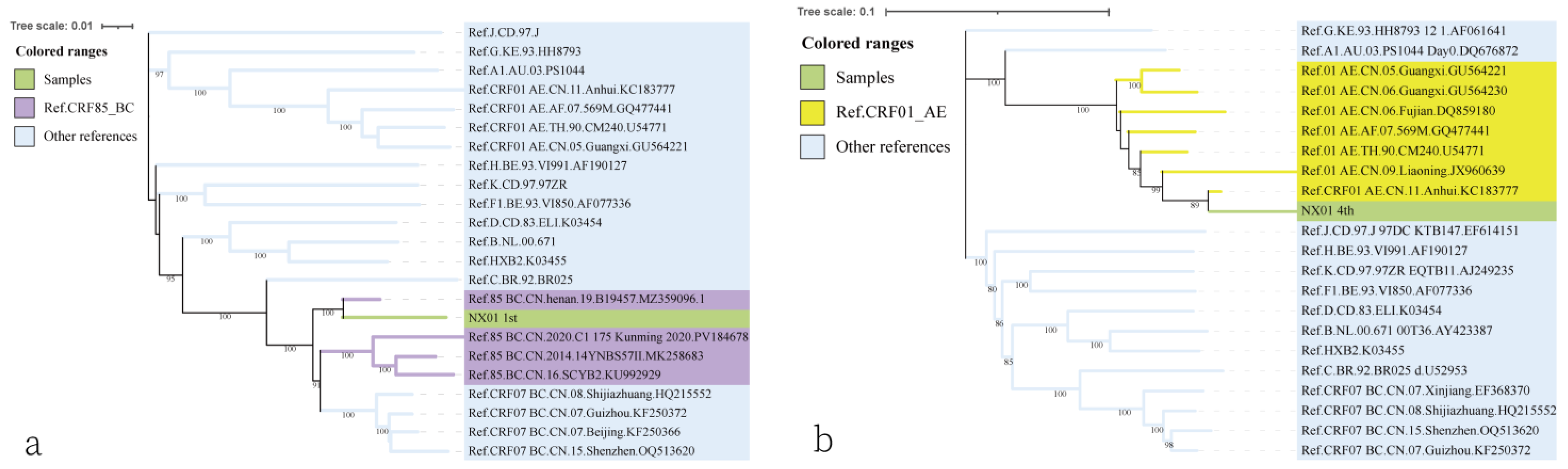
3.3. Traceability Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AIDS | Acquired Immune Deficiency Syndrome |
| HIV | Human Immunodeficiency Virus |
| CRFs | circulating recombinant forms |
| URFs | unique recombinant forms |
| NFLG | near full-length genome |
| MSM | men who have sex with men |
References
- HIV Sequence Database Main Page. Available online: https://www.hiv.lanl.gov/content/sequence/HIV/mainpage.html (accessed on 3 March 2025).
- Bolhassani, A. Editorial: The Global Phenotypic Diversity of HIV-1: Implications for Pathogenesis, Vaccine, and Cure. Front. Immunol. 2025, 16, 1572732. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Feng, Y.; Hao, J.; Hu, H.; Li, F.; Li, J.; Ruan, Y.; Liao, L.; Hu, J.; Song, C.; et al. National and Regional Molecular Epidemiology of HIV-1—China, 2004–2023. China CDC Wkly. 2024, 6, 1257–1263. [Google Scholar] [CrossRef] [PubMed]
- Pei, J.; Wu, Z.; Si, B.; Ma, C.; Liu, Y.; Ma, X.; Kuai, W.; Zhang, Y.; Li, Y. HIV-1 Drug Resistance and Genetic Transmission Networks among Patients with Sexually Transmitted HIV in Ningxia, China. Front. Public Health 2024, 12, 1485516. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Pei, J.; Ma, X.; Si, B.; Ma, C.; Li, Y.; Zhu, X.; Yang, D.; Wu, Z. Characterization of HIV-1 drug resistance and molecular transmission network in Ningxia, 2023. Virol. Sin. 2024, 40, 1384–1390. [Google Scholar] [CrossRef]
- Lv, R.; Xu, C.; Fu, S.; Li, S.; Jiang, B. Occurrence of genotypic drug resistance in HIV-infected/AIDS patients with virologic failure of antiretroviral therapy and factors influencing it. Guangxi Med. J. 2019, 41, 1238–1242. [Google Scholar]
- Zazzi, M.; Romano, L.; Catucci, M.; Venturi, G.; De Milito, A.; Valensin, P.E. Clinical Evaluation of an In-House Reverse Transcription-Competitive PCR for Quantitation of Human Immunodeficiency Virus Type 1 RNA in Plasma. J. Clin. Microbiol. 1999, 37, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Global HIV & AIDS Statistics—Fact Sheet. Available online: https://www.unaids.org/en/resources/fact-sheet (accessed on 3 March 2025).
- Li, X.; Li, W.; Zhong, P.; Fang, K.; Zhu, K.; Musa, T.H.; Song, Y.; Du, G.; Gao, R.; Guo, Y.; et al. Nationwide Trends in Molecular Epidemiology of HIV-1 in China. AIDS Res. Hum. Retroviruses 2016, 32, 851–859. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, D.; Hu, J.; Song, C.; Liao, L.; Feng, Y.; Li, D.; Xing, H.; Ruan, Y. Changes in HIV-1 Subtypes/Sub-Subtypes, and Transmitted Drug Resistance Among ART-Naïve HIV-Infected Individuals—China, 2004–2022. China CDC Wkly. 2023, 5, 664–671. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Yang, D.; Li, Z.; Li, L.; Pei, J.; Wu, Z. Analysis of Genetic Subtypes and Drug Resistance of HIV-1 Infected Patients in Yinchuan City, 2020. Virol. Sin. 2022, 38, 1160–1165. [Google Scholar] [CrossRef]
- Si, B.; Ma, C.; Wu, Y.; Li, Z.; Pei, J.; Yang, D.; Wu, Z.; Zhang, Y. Characterization of HIV-1 genotype subtypes and drug resistance in heterosexually transmitted populations in Ningxia, 2021. Virol. Sin. 2023, 39, 783–790. [Google Scholar] [CrossRef]
- Cheng, H.; Zhang, J.; Capizzi, J.; Young, N.L.; Mastro, T.D. HIV-1 Subtype E in Yunnan, China. Lancet 1994, 344, 953–954. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Young, N.L.; Subbarao, S.; Warachit, P.; Saguanwongse, S.; Wongsheree, S.; Jayavasu, C.; Luo, C.C.; Mastro, T.D. HIV Type 1 Subtypes in Guangxi Province, China, 1996. AIDS Res. Hum. Retroviruses 1999, 15, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Liao, L.; Feng, Y.; Zhang, J.; Yan, J.; He, C.; Xu, W.; Ruan, Y.; Xing, H.; Shao, Y. Trends of HIV Subtypes and Phylogenetic Dynamics among Young Men Who Have Sex with Men in China, 2009–2014. Sci. Rep. 2015, 5, 16708. [Google Scholar] [CrossRef] [PubMed]
- Steain, M.C.; Wang, B.; Dwyer, D.E.; Saksena, N.K. HIV-1 Co-Infection, Superinfection and Recombination. Sex Health 2004, 1, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Su, L.; Wei, D.; Yang, H.; Zeng, Y.; Hu, Y.; Yuan, D.; Feng, L.; Ruan, Y.; Qin, G.; Liang, S. Identification of a Novel HIV-1 Circulating Recombinant Form (CRF85_BC) in Sichuan, China. AIDS Res. Hum. Retroviruses 2016, 32, 895–899. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Wang, Y.; Tian, F. Analysis of HIV Sentinel Surveillance Results of Male Mobile Population in Dongcheng District, Beijing, 2016–2020. Occup. Health 2022, 38, 2499–2502. [Google Scholar] [CrossRef]
- Wang, H.; Li, S.; Li, F.; Liu, H.; Guo, S.; Hou, Z. Analysis of the results of HIV sentinel surveillance of the mobile population at construction sites in Changping District, Beijing, China, 2018–2021. Cap. J. Public Health 2023, 17, 184–187. [Google Scholar] [CrossRef]
- Chen, Y.; Wu, Y. Advances in the drug resistance genotype detection of HIV-1. J. Mol. Diagn. Ther. 2017, 2, 142–146. [Google Scholar]
- Jiao, Y.; Huang, Y.; Li, S.; Li, Z.; Wang, Y.; Yin, Q.; Ma, L. Replication kinetic properties of HIV-1 CRF_BC novel drug resistance associated mutations. Chin. J. Prev. Med. 2015, 49, 436–440. [Google Scholar]
- Liu, M.; Xing, H.; Zhang, Y.; Li, Y.; Wang, Y.; An, N.; Lu, X. Study on the molecular transmission network of HIV-1 drug-resistant strains in the pre-treatment population in Hebei Province, China. Chin. J. Zoonoses 2020, 36, 746–751. [Google Scholar]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Pei, J.; Zhu, X.; Liu, Y.; Ma, X.; Yang, D.; Wu, Z. Sequencing of One Unique Recombinant CRF85_BC/CRF01_AE Genome and Two Partial Genomes from Ningxia, China. Viruses 2025, 17, 655. https://doi.org/10.3390/v17050655
Li Y, Pei J, Zhu X, Liu Y, Ma X, Yang D, Wu Z. Sequencing of One Unique Recombinant CRF85_BC/CRF01_AE Genome and Two Partial Genomes from Ningxia, China. Viruses. 2025; 17(5):655. https://doi.org/10.3390/v17050655
Chicago/Turabian StyleLi, Yufeng, Jianxin Pei, Xiaohong Zhu, Yichang Liu, Xiaofa Ma, Dongzhi Yang, and Zhonglan Wu. 2025. "Sequencing of One Unique Recombinant CRF85_BC/CRF01_AE Genome and Two Partial Genomes from Ningxia, China" Viruses 17, no. 5: 655. https://doi.org/10.3390/v17050655
APA StyleLi, Y., Pei, J., Zhu, X., Liu, Y., Ma, X., Yang, D., & Wu, Z. (2025). Sequencing of One Unique Recombinant CRF85_BC/CRF01_AE Genome and Two Partial Genomes from Ningxia, China. Viruses, 17(5), 655. https://doi.org/10.3390/v17050655