

High-Throughput Sequencing Reveals Apple Virome Diversity and Novel Viruses in the Czech Republic

 , , , and
, , , and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and HTS
2.2. Sequence and Recombination Analyses
2.3. Phylogenetic and Sequence Demarcation Analyses
3. Results
3.1. Virus Diversity and Co-Infections
3.2. Recombination Analysis
3.2.1. ACLSV
3.2.2. ASGV
3.2.3. ASPV and AGCaV
3.2.4. ApMV
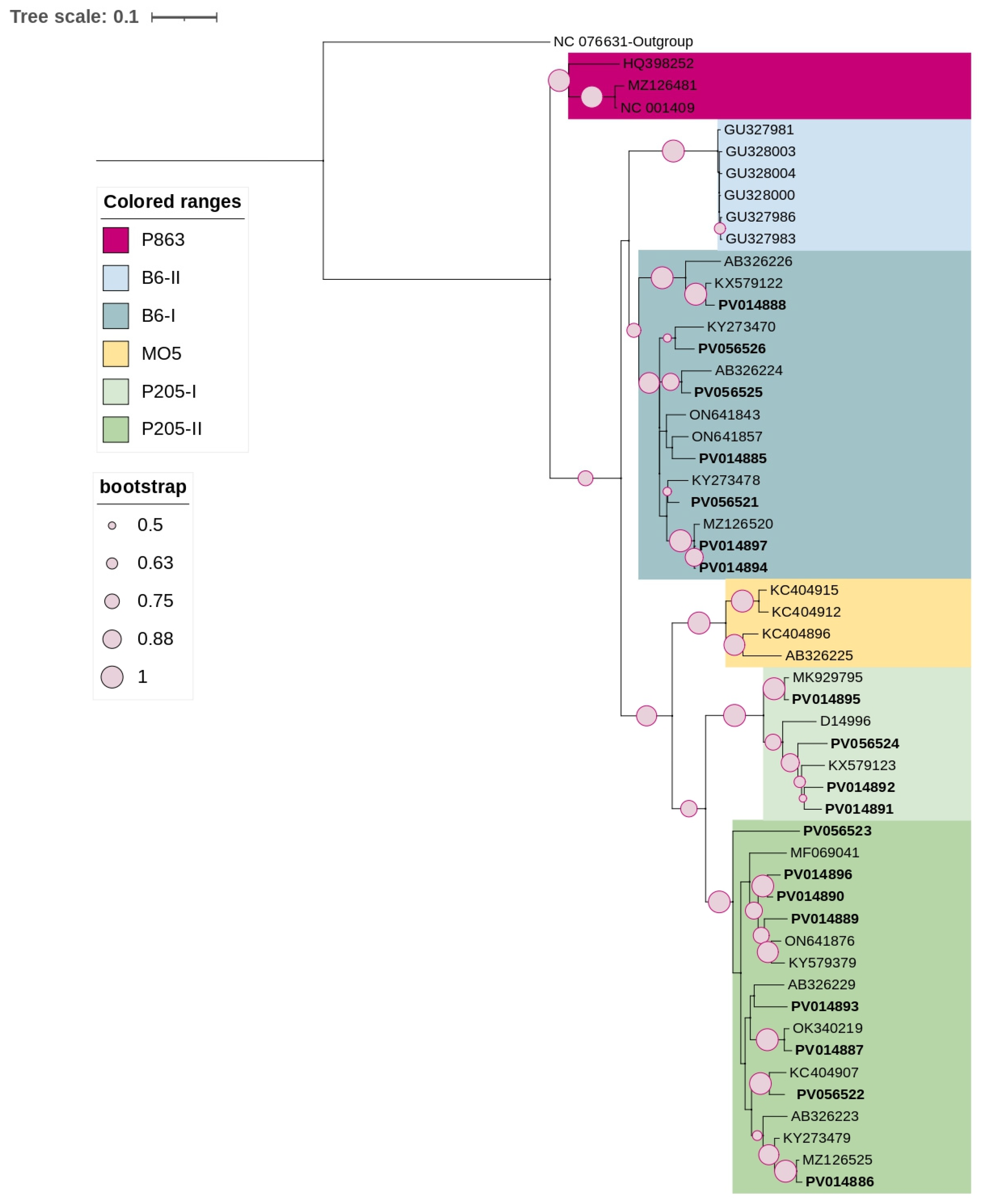
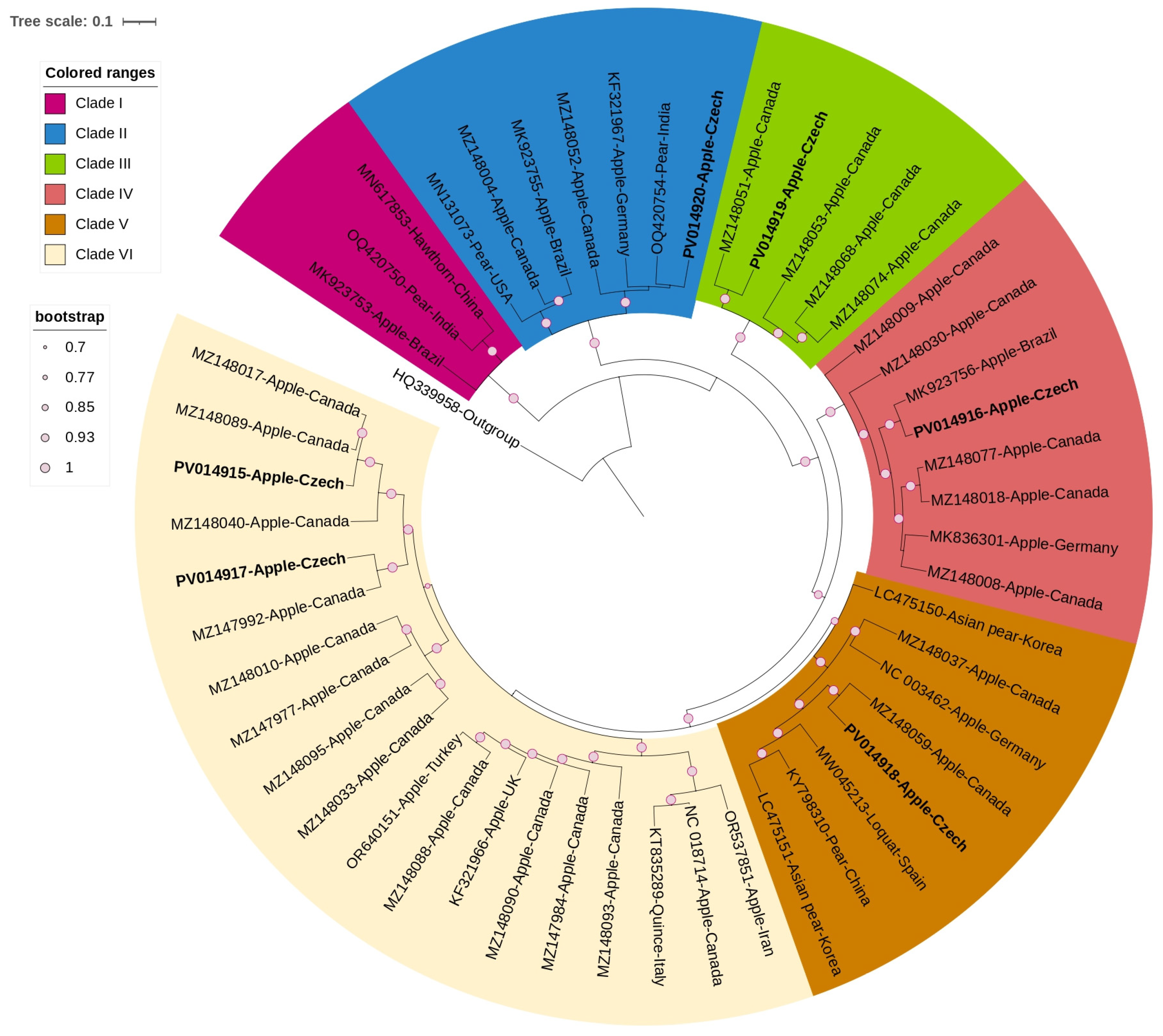
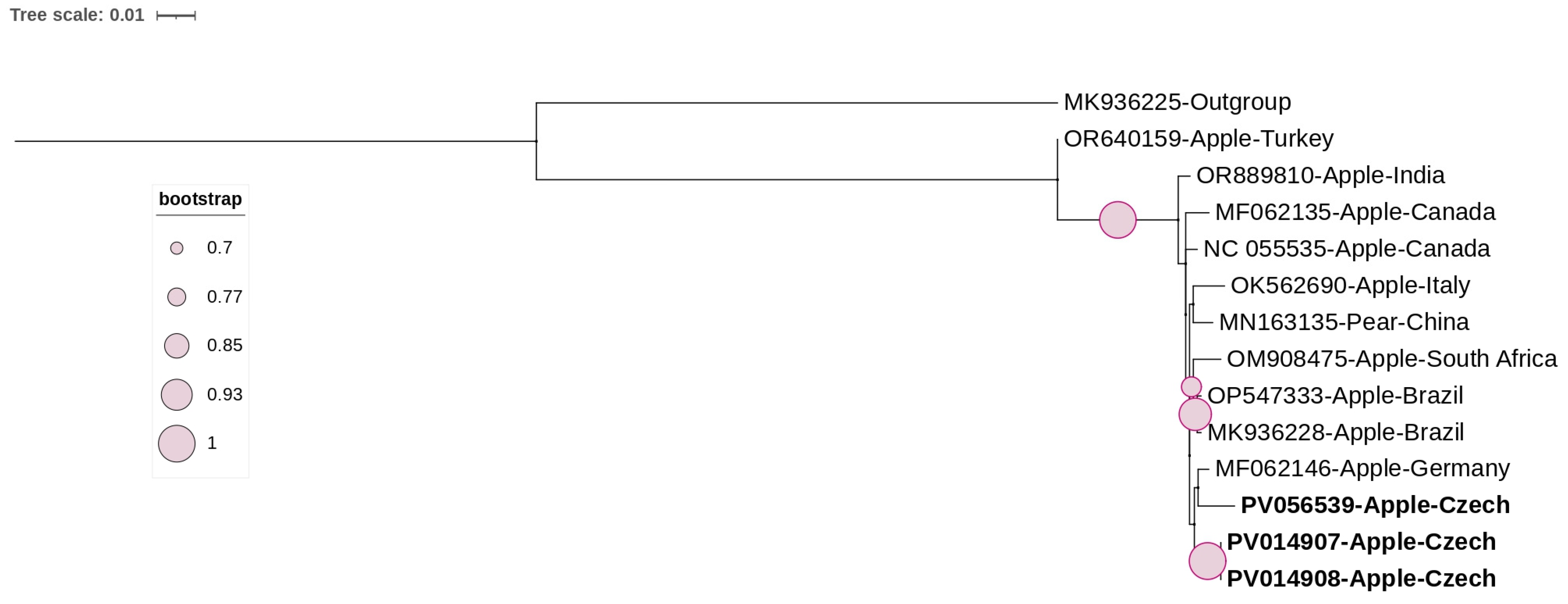
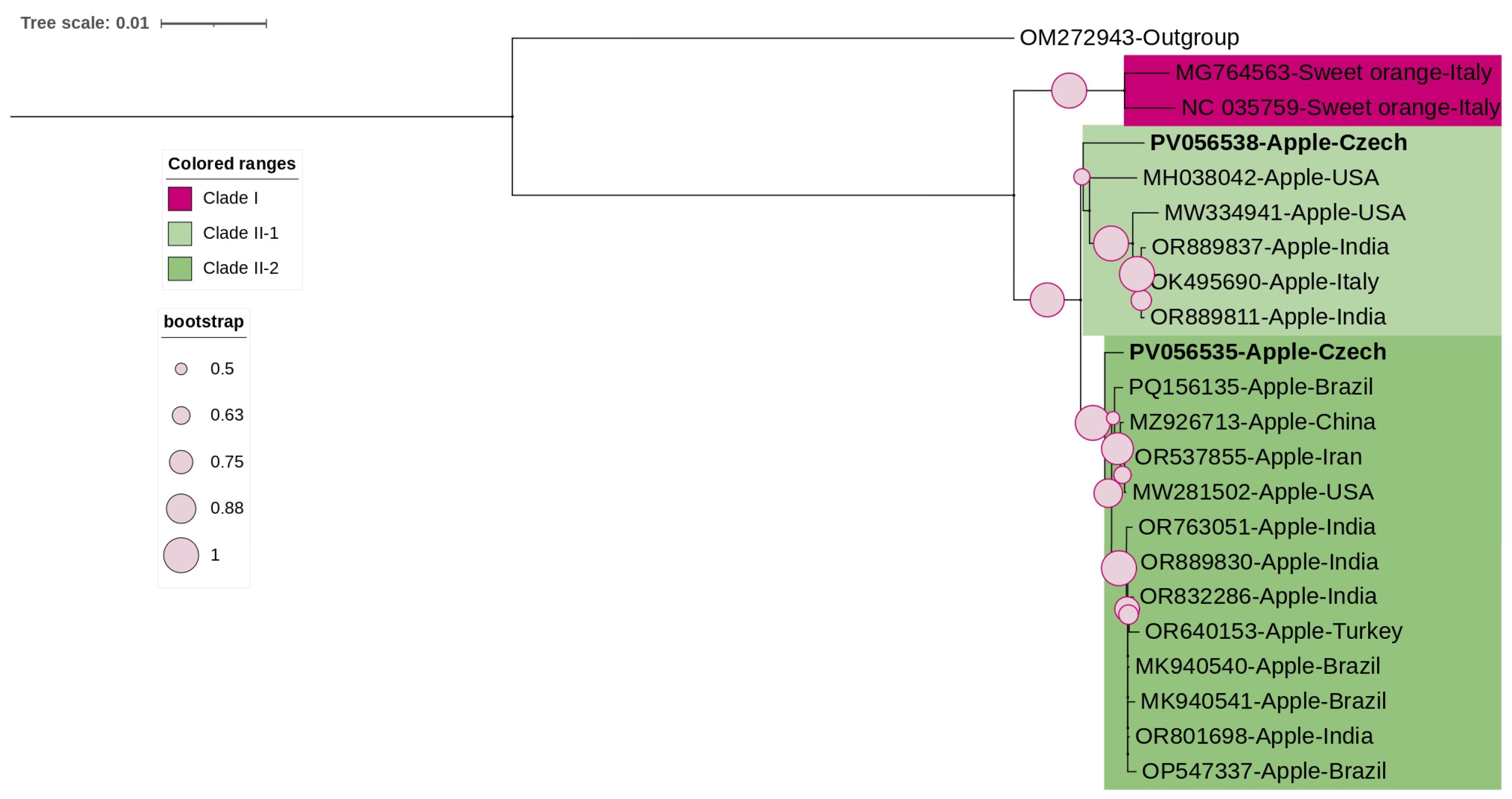
3.3. Phylogenetic and Sequence Demarcation Analyses
3.3.1. Betaflexiviridae
3.3.2. Bromoviridae
3.3.3. Phenuiviridae
4. Discussion
4.1. Application of HTS for Apple Virus Discovery
4.2. Unclear Symptom Association of ARWV1
4.3. Intra-Host and Mixed Viral Diversity
4.4. Frequency of Recombination Events
4.5. Observations on AGCaV and ASPV Relationship
4.6. Evidence of Potential Interspecific Recombination
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mansoor, S.; Ahmed, N.; Sharma, V.; Jan, S.; Nabi, S.U.; Mir, J.I.; Mir, M.A.; Masoodi, K.Z. Elucidating Genetic Variability and Population Structure in Venturia Inaequalis Associated with Apple Scab Diseaseusing SSR Markers. PLoS ONE 2019, 14, e0224300. [Google Scholar] [CrossRef]
- Menzel, W.; Zahn, V.; Maiss, E. Multiplex RT-PCR-ELISA Compared with Bioassay for the Detection of Four Apple Viruses. J. Virol. Methods 2003, 110, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Grimová, L.; Winkowska, L.; Zíka, L.; Ryšánek, P. Distribution of Viruses in Old Commercial and Abandoned Orchards and Wild Apple Trees. J. Plant Pathol. 2016, 98, 549–554. [Google Scholar] [CrossRef]
- Balík, J.; Rop, O.; Mlček, J.; Híc, P.; Horák, M.; Řezníček, V. Assessment of Nutritional Parameters of Native Apple Cultivars as New Gene Sources. Acta Univ. Agric. Silvic. Mendel. Brun. 2012, 60, 27–38. [Google Scholar] [CrossRef]
- Nabi, S.U.; Mir, J.I.; Sharma, O.C.; Singh, D.B.; Zaffer, S.; Sheikh, M.A.; Masoodi, L.; Khan, K.A. Optimization of Tissue and Time for Rapid Serological and Molecular Detection of Apple Stem Pitting Virus and Apple Stem Grooving Virus in Apple’. Phytoparasitica 2018, 46, 705–713. [Google Scholar] [CrossRef]
- Cembali, T.; Folwell, R.J.; Wandschneider, P.; Eastwell, K.C.; Howell, W.E. Economic Implications of a Virus Prevention Program in Deciduous Tree Fruits in the USA. Crop Prot. 2003, 22, 1149–1156. [Google Scholar] [CrossRef]
- Xiao, H.; Hao, W.; Storoschuk, G.; MacDonald, J.L.; Sanfaçon, H. Characterizing the Virome of Apple Orchards Affected by Rapid Decline in the Okanagan and Similkameen Valleys of British Columbia (Canada). Pathogens 2022, 11, 1231. [Google Scholar] [CrossRef]
- Jiang, T.; Zhou, T. Unraveling the Mechanisms of Virus-Induced Symptom Development in Plants. Plants 2023, 12, 2830. [Google Scholar] [CrossRef]
- Takahashi, H.; Fukuhara, T.; Kitazawa, H.; Kormelink, R. Virus Latency and the Impact on Plants. Front. Microbiol. 2019, 10, 2764. [Google Scholar] [CrossRef]
- Simmonds, P.; Adriaenssens, E.; Lefkowitz, E.; Oksanen, H.; Siddell, S.; Zerbini, F.; Alfenas-Zerbini, P.; Aylward, F.; Dempsey, D.; Dutilh, B.; et al. Changes to Virus Taxonomy and the ICTV Statutes Ratified by the International Committee on Taxonomy of Viruses (2024). Arch. Virol. 2024, 169, 236. [Google Scholar] [CrossRef]
- Fuchs, M. Viruses and Apple Propagation: Why Should I Worry? Available online: http://www.hort.cornell.edu/expo/proceedings/2016/TreeFruit.%20Viruses%20and%20apple%20propagation%20why%20should%20I%20worry.%20Fuchs.pdf (accessed on 10 October 2024).
- Petri, J.L.; Hawerroth, F.J.; Fazio, G.; Francescatto, P.; Leite, G.B. Advances in Fruit Crop Propagation in Brazil and Worldwide—Apple Trees. Rev. Bras. Frutic. 2019, 41, e-004. [Google Scholar] [CrossRef]
- Hassan, M.; Polák, J.; Paprštein, F. Detection and Distribution of Four Pome Fruit Viruses in Germplasm Collection in the Czech Republic. Acta Hortic. 2008, 781, 113–118. [Google Scholar] [CrossRef]
- Kundu, J.K. Detection, Distribution and Genetic Diversities of Apple Stem Pitting Virus and Apple Stem Grooving Virus in the Czech Republic. Acta Hortic. 2008, 781, 135–142. [Google Scholar] [CrossRef]
- Adams, I.P.; Glover, R.H.; Monger, W.A.; Mumford, R.; Jackeviciene, E.; Navalinskiene, M.; Samuitiene, M.; Boonham, N. Next-Generation Sequencing and Metagenomic Analysis: A Universal Diagnostic Tool in Plant Virology. Mol. Plant Pathol. 2009, 10, 537–545. [Google Scholar] [CrossRef]
- Várallyay, E.; Přibylová, J.; Galbacs, Z.N.; Jahan, A.; Varga, T.; Špak, J.; Lenz, O.; Fránová, J.; Sedlák, J.; Koloniuk, I. Detection of Apple Hammerhead Viroid, Apple Luteovirus 1 and Citrus Concave Gum-Associated Virus in Apple Propagation Materials and Orchards in the Czech Republic and Hungary. Viruses 2022, 14, 2347. [Google Scholar] [CrossRef] [PubMed]
- Koloniuk, I.; Přibylová, J.; Fránová, J.; Špak, J. Genomic Characterization of Malus Domestica Virus A (MdoVA), a Novel Velarivirus Infecting Apple. Arch. Virol. 2020, 165, 479–482. [Google Scholar] [CrossRef]
- Koloniuk, I.; Přibylová, J.; Špak, J. Complete Genome Sequence of a Mite-Associated Virus Obtained by High-Throughput Sequencing Analysis of an Apple Leaf Sample. Arch. Virol. 2020, 165, 1501–1504. [Google Scholar] [CrossRef] [PubMed]
- Komínková, M.; Ben Mansour, K.; Komínek, P.; Brožová, J.; Střalková, R. Multiple Infections with Viruses of the Family Tymoviridae in Czech Grapevines. Viruses 2024, 16, 343. [Google Scholar] [CrossRef]
- Koloniuk, I.; Matyášová, A.; Brázdová, S.; Veselá, J.; Přibylová, J.; Várallyay, E.; Fránová, J. Analysis of Virus-Derived SiRNAs in Strawberry Plants Co-Infected with Multiple Viruses and Their Genotypes. Plants 2023, 12, 2564. [Google Scholar] [CrossRef]
- Abascal, F.; Zardoya, R.; Telford, M.J. TranslatorX: Multiple Alignment of Nucleotide Sequences Guided by Amino Acid Translations. Nucleic Acids Res. 2010, 38, W7–W13. [Google Scholar] [CrossRef]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT Online Service: Multiple Sequence Alignment, Interactive Sequence Choice and Visualization. Brief. Bioinform. 2018, 20, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.A. BioEdit: A User-Friendly Biological Sequence Alignment Editor and Analysis Program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and Analysis of Recombination Patterns in Virus Genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Muhire, B.M.; Varsani, A.; Martin, D.P. SDT: A Virus Classification Tool Based on Pairwise Sequence Alignment and Identity Calculation. PLoS ONE 2014, 9, e108277. [Google Scholar] [CrossRef]
- Zuļģe, N.; Gospodaryk, A.; Moročko–Bičevska, I. Genetic Diversity and Phylogenetic Relationships of Apple Chlorotic Leaf Spot Virus Isolates from Malus, Pyrus and Prunus Hosts in Latvia. Plant Pathol. 2023, 72, 900–911. [Google Scholar] [CrossRef]
- Rollin, J.; Bester, R.; Brostaux, Y.; Caglayan, K.; De Jonghe, K.; Eichmeier, A.; Foucart, Y.; Haegeman, A.; Koloniuk, I.; Kominek, P.; et al. Detection of Single Nucleotide Polymorphisms in Virus Genomes Assembled from High-Throughput Sequencing Data: Large-Scale Performance Testing of Sequence Analysis Strategies. PeerJ 2023, 11, e15816. [Google Scholar] [CrossRef] [PubMed]
- Grimová, L.; Winkowska, L.; Konrady, M.; Ryšánek, P. Apple Mosaic Virus. Phytopathol. Mediterr. 2016, 55, 1–19. [Google Scholar] [CrossRef]
- Herath, V.; Romay, G.; Urrutia, C.D.; Verchot, J. Family Level Phylogenies Reveal Relationships of Plant Viruses within the Order Bunyavirales. Viruses 2020, 12, 1010. [Google Scholar] [CrossRef]
- Ben Mansour, K.; Komínek, P.; Komínková, M.; Brožová, J. Characterization of Prunus Necrotic Ringspot Virus and Cherry Virus A Infecting Myrobalan Rootstock. Viruses 2023, 15, 1723. [Google Scholar] [CrossRef]
- Rott, M.E.; Kesanakurti, P.; Berwarth, C.; Rast, H.; Boyes, I.; Phelan, J.; Jelkmann, W. Discovery of Negative-Sense RNA Viruses in Trees Infected with Apple Rubbery Wood Disease by next-Generation Sequencing. Plant Dis. 2018, 102, 1254–1263. [Google Scholar] [CrossRef] [PubMed]
- Wright, A.A.; Szostek, S.A.; Beaver-Kanuya, E.; Harper, S.J. Diversity of Three Bunya-like Viruses Infecting Apple. Arch. Virol. 2018, 163, 3339–3343. [Google Scholar] [CrossRef] [PubMed]
- Nickel, O.; Fajardo, T.V.M.; Candresse, T. First Report on Detection of Three Bunya-like Viruses in Apples in Brazil. Plant Dis. 2020, 104, 3088. [Google Scholar] [CrossRef]
- Hu, G.; Dong, Y.; Zhang, Z.; Fan, X.; Ren, F.; Lu, X. First Report of Apple Rubbery Wood Virus 1 in Apple in China. Plant Dis. 2021, 105, 3324–3735. [Google Scholar] [CrossRef]
- Hu, G.J.; Dong, Y.F.; Zhang, Z.P.; Fan, X.D.; Ren, F.; Lu, X.K. First Report of Apple Rubbery Wood Virus 2 Infection of Apples in China. Plant Dis. 2021, 105, 519. [Google Scholar] [CrossRef]
- Minutolo, M.; Cinque, M.; Di Serio, F.; Navarro, B.; Alioto, D. Occurrence of Apple Rubbery Wood Virus 1 and Apple Rubbery Wood Virus 2 in Pear and Apple in Campania (Southern Italy) and Development of Degenerate Primers for the Rapid Detection of Rubodviruses. J. Plant Pathol. 2023, 105, 567–572. [Google Scholar] [CrossRef]
- Lim, S.; Baek, D.; Moon, J.; Cho, I.; Choi, G.; Do, Y.; Lee, D.; Lee, S.H. First Report of Apple Luteovirus 1 and Apple Rubbery Wood Virus 1 on Apple Tree Rootstocks in Korea. Plant Dis. 2019, 103, 591. [Google Scholar] [CrossRef]
- Bester, R.; Bougard, K.; Maree, H. First Report of Apple Rubodvirus 2 Infecting Apples (Malus domestica) in South Africa. J. Plant Pathol. 2022, 104, 1199–1200. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, G.P.; Hong, N.; Wang, Y.X.; Yang, Z.K.; Guo, J.S.; Zhang, Z.; Li, L.; Li, Y.J.; Li, Q.Y.; et al. First Report of Apple Rubbery Wood Virus 2 Infecting Pear (Pyrus spp.) in China. Plant Dis. 2019, 103, 3293. [Google Scholar] [CrossRef]
- Bougard, K.; Maree, H.; Pietersen, G.; Meitz-Hopkins, J.; Bester, R. First Report of Apple Rubodvirus 2 Infecting Pear (Pyrus communis) in South Africa. Plant Dis. 2022, 106, 1535. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Y.; Wang, G.; Li, Q.; Zhang, Z.; Li, L.; Lv, Y.; Yang, Z.; Guo, J.; Hong, N. Molecular Characteristics and Incidence of Apple Rubbery Wood Virus 2 and Citrus Virus A Infecting Pear Trees in China. Viruses 2022, 14, 576. [Google Scholar] [CrossRef] [PubMed]
- Cho, I.S.; Igori, D.; Lim, S.; Choi, G.S.; Hammond, J.; Lim, H.S.; Moon, J.S. Deep Sequencing Analysis of Apple Infecting Viruses in Korea. Plant Pathol. J. 2016, 32, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Dhir, S.; Mathioudakis, M.M.; Hasiów-Jaroszewska, B.; Hallan, V. Serological and Molecular Analysis Indicates the Presence of Distinct Viral Genotypes of Apple Stem Pitting Virus in India. 3 Biotech 2021, 11, 278. [Google Scholar] [CrossRef] [PubMed]
- Wright, A.A.; Cross, A.R.; Harper, S.J. A Bushel of Viruses: Identification of Seventeen Novel Putative Viruses by RNA-Seq in Six Apple Trees. PLoS ONE 2020, 15, e0227669. [Google Scholar] [CrossRef]
- Jia, X.J.; Zhang, Z.P.; Fan, X.D.; Ren, F.; Dong, Y.F.; Hu, G.J. Detection and Genetic Variation Analysis of Apple Chlorotic Leaf Spot Virus and Apple Stem Grooving Virus in Wild Apple (Malus sieversii) from Xinjiang, China. For. Pathol. 2022, 52, e12771. [Google Scholar] [CrossRef]
- Canales, C.; Morán, F.; Olmos, A.; Ruiz-García, A.B. First Detection and Molecular Characterization of Apple Stem Grooving Virus, Apple Chlorotic Leaf Spot Virus, and Apple Hammerhead Viroid in Loquat in Spain. Plants 2021, 10, 2293. [Google Scholar] [CrossRef]
- Hajizadeh, M.; Ben Mansour, K.; Gibbs, A.J. A Genetic Study of Spillovers in the Bean Common Mosaic Subgroup of Potyviruses. Viruses 2024, 16, 1351. [Google Scholar] [CrossRef]
- Morelli, M.; D’Attoma, G.; Saldarelli, P.; Minafra, A. The Evolution of Wisteria Vein Mosaic Virus: A Case Study Approach to Track the Emergence of New Potyvirus Threats. Pathogens 2024, 13, 1001. [Google Scholar] [CrossRef]
- Ben Mansour, K.; Gibbs, A.J.; Komínková, M.; Komínek, P.; Brožová, J.; Kazda, J.; Zouhar, M.; Ryšánek, P. Watermelon Mosaic Virus in the Czech Republic, Its Recent and Historical Origins. Plamt Pathol. 2023, 72, 1528–1538. [Google Scholar] [CrossRef]
- Gibbs, A.J.; Hajizadeh, M.; Ohshima, K.; Jones, R.A.C. The Potyviruses: An Evolutionary Synthesis Is Emerging. Viruses 2020, 12, 132. [Google Scholar] [CrossRef]
- Suman, R.; Rani, A.; Rishi, N.; Dhir, S.; Hallan, V.; Chandel, V. First Report of Apple Stem Grooving Virus Infection in Loquat from India. VirusDisease 2022, 33, 334–337. [Google Scholar] [CrossRef] [PubMed]
- Mathioudakis, M.M.; Maliogka, V.I.; Candresse, T.; Nickel, O.; Fajardo, T.V.M.; Budzyńska, D.; Hasiów-Jaroszewska, B.; Katis, N.I. Molecular Characterization of the Coat Protein Gene of Greek Apple Stem Pitting Virus Isolates: Evolution through Deletions, Insertions, and Recombination Events. Plants 2021, 10, 917. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Yaegashi, H.; Kishigami, R.; Kawakubo, A.; Yamagishi, N.; Ito, T.; Yoshikawa, N. Apple Russet Ring and Apple Green Crinkle Diseases: Fulfillment of Koch’s Postulates by Virome Analysis, Amplification of Full-Length CDNA of Viral Genomes, in Vitro Transcription of Infectious Viral RNAs, and Reproduction of Symptoms on Fruits of Apple T. Front. Microbiol. 2020, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- James, D.; Varga, A.; Jesperson, G.D.; Navratil, M.; Safarova, D.; Constable, F.; Horner, M.; Eastwell, K.; Jelkmann, W. Identification and Complete Genome Analysis of a Virus Variant or Putative New Foveavirus Associated with Apple Green Crinkle Disease. Arch. Virol. 2013, 158, 1877–1887. [Google Scholar] [CrossRef]
- Çelik, A.; Morca, A.F.; Coşkan, S.; Santosa, A.I. Global Population Structure of Apple Mosaic Virus (ApMV, Genus Ilarvirus). Viruses 2023, 15, 1221. [Google Scholar] [CrossRef]
- Noorani, M.S.; Baig, M.S.; Khan, J.A.; Pravej, A. Whole Genome Characterization and Diagnostics of Prunus Necrotic Ringspot Virus (PNRSV) Infecting Apricot in India. Sci. Rep. 2023, 13, 4393. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Apple Plant | Cultivar | Origin | Locality |
|---|---|---|---|
| A9 | Rubín | CARC | Prague–Ruzyně |
| G4 | Spartan | CARC | Prague–Ruzyně |
| H8 | Spartan | CARC | Prague–Ruzyně |
| S1 | Táborita | RBIP | Holovousy |
| S4 | Golden Delicious | RBIP | Holovousy |
| S7 | Red Boskoop | RBIP | Holovousy |
| S10 | Golden Delicious | RBIP | Holovousy |
| S16 | Red Boskoop | RBIP | Holovousy |
| Plant/Virus | ACLSV | ASGV | ASPV | AGCaV | ApMV | ARWV1 | ARWV2 | CCGaV | AHVd |
|---|---|---|---|---|---|---|---|---|---|
| A9 | + | + | + | + | + | ||||
| G4 | + | + | + | + | + | + | + | + | |
| H8 | + | + | + | + | + | + | + | + | |
| S1 | + | + | + | + | + | + | + | ||
| S4 | + | + | + | + | + | ||||
| S7 | + | ||||||||
| S10 | + | ||||||||
| S16 | + |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ben Mansour, K.; Koloniuk, I.; Brožová, J.; Komínková, M.; Přibylová, J.; Sarkisova, T.; Sedlák, J.; Špak, J.; Komínek, P. High-Throughput Sequencing Reveals Apple Virome Diversity and Novel Viruses in the Czech Republic. Viruses 2025, 17, 650. https://doi.org/10.3390/v17050650
Ben Mansour K, Koloniuk I, Brožová J, Komínková M, Přibylová J, Sarkisova T, Sedlák J, Špak J, Komínek P. High-Throughput Sequencing Reveals Apple Virome Diversity and Novel Viruses in the Czech Republic. Viruses. 2025; 17(5):650. https://doi.org/10.3390/v17050650
Chicago/Turabian StyleBen Mansour, Karima, Igor Koloniuk, Jana Brožová, Marcela Komínková, Jaroslava Přibylová, Tatiana Sarkisova, Jiří Sedlák, Josef Špak, and Petr Komínek. 2025. "High-Throughput Sequencing Reveals Apple Virome Diversity and Novel Viruses in the Czech Republic" Viruses 17, no. 5: 650. https://doi.org/10.3390/v17050650
APA StyleBen Mansour, K., Koloniuk, I., Brožová, J., Komínková, M., Přibylová, J., Sarkisova, T., Sedlák, J., Špak, J., & Komínek, P. (2025). High-Throughput Sequencing Reveals Apple Virome Diversity and Novel Viruses in the Czech Republic. Viruses, 17(5), 650. https://doi.org/10.3390/v17050650