
Genotypic Characterization of Human Respiratory Syncytial Viruses Detected in Mexico Between 2021 and 2024

 , , , , , , , and
, , , , , , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Samples
2.2. HRSV-A and HRSV-B Genotyping
2.3. Global HRSV Sequences
2.4. Recombination Analysis
2.5. Phylogenetic Analysis
2.6. Lineage Assignment
2.7. Analysis of Amino Acid Sequences and Amino Acid-Based Lineage Definition
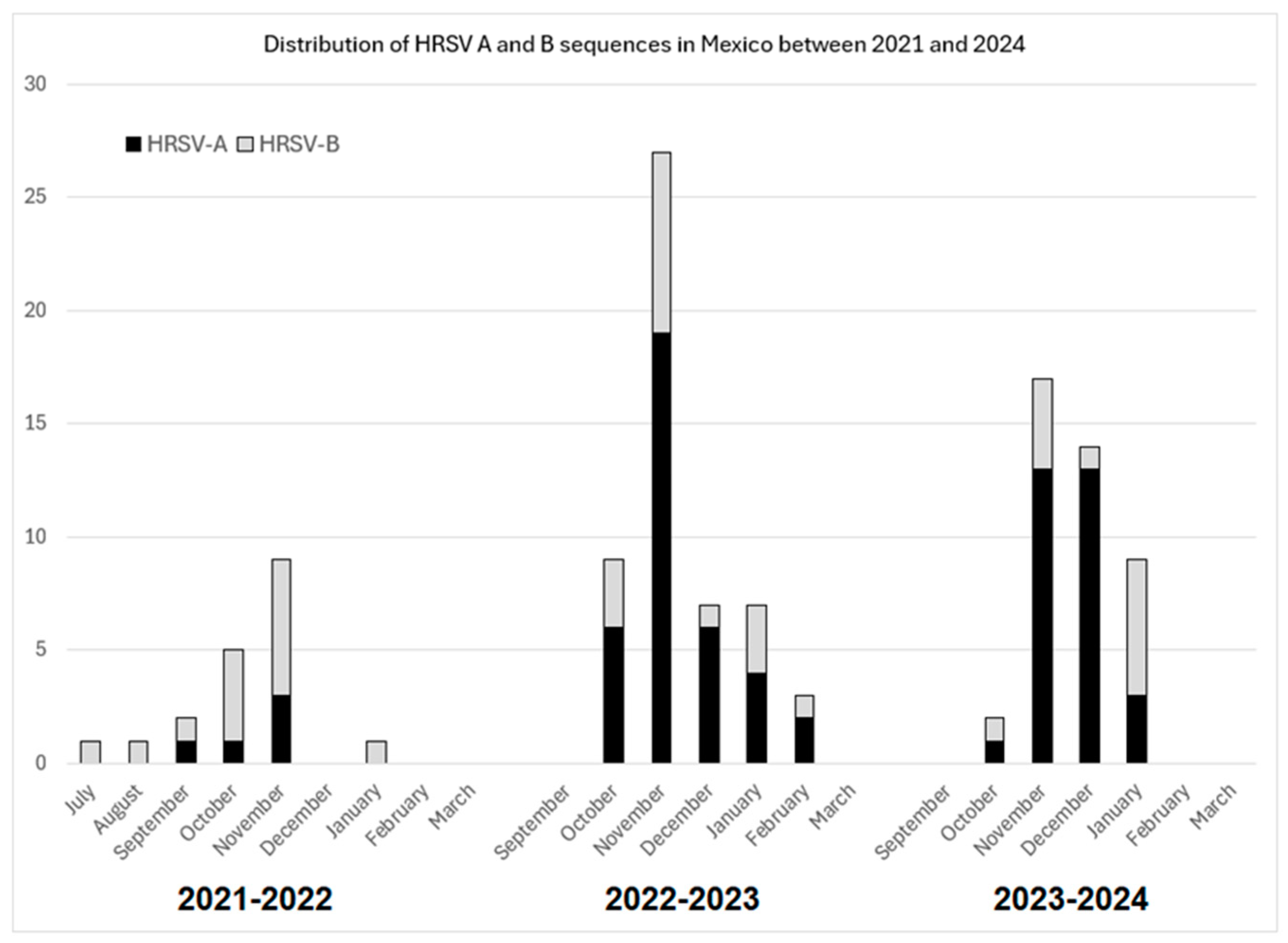
3. Results
3.1. Recombination Analysis
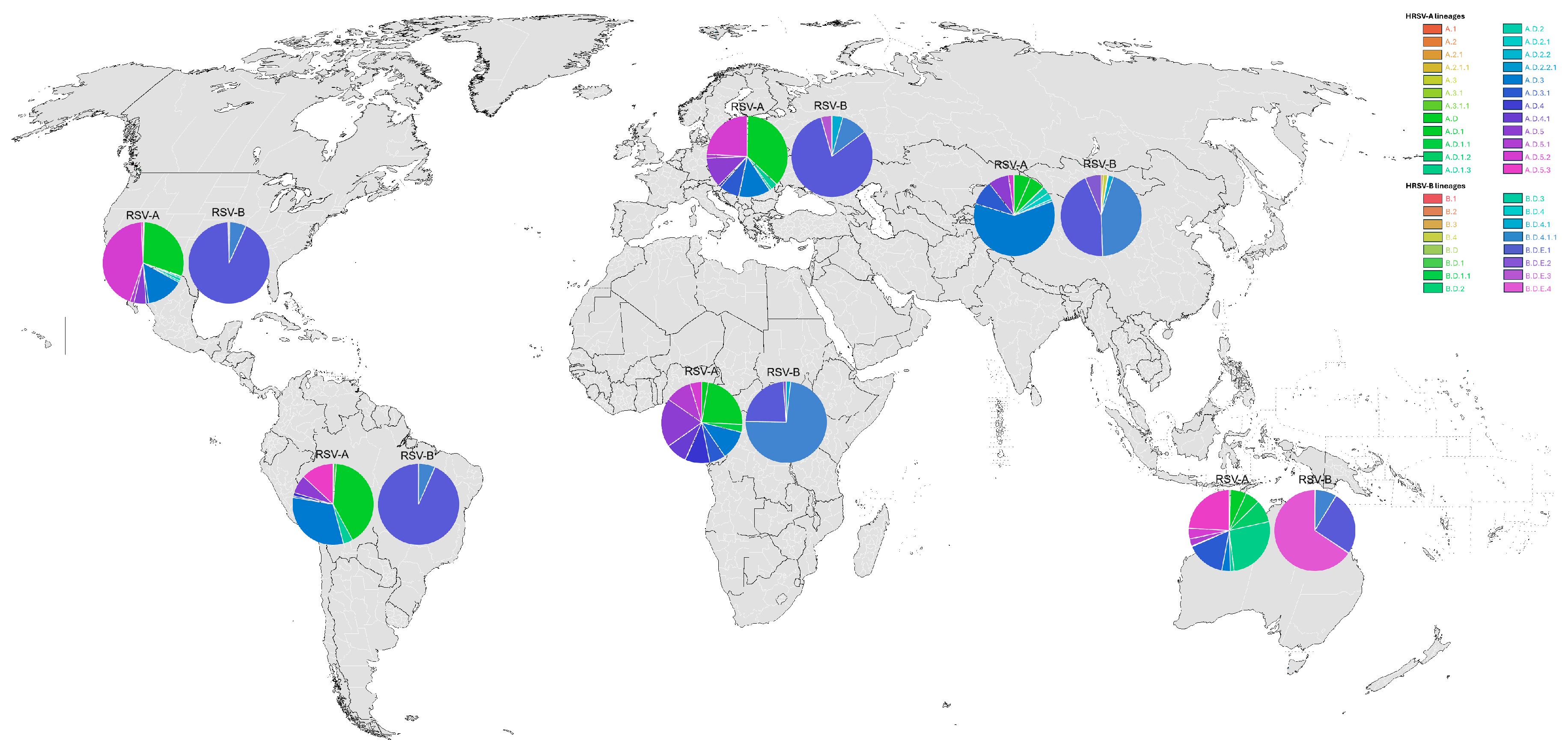
3.2. Global HRSV Sequences
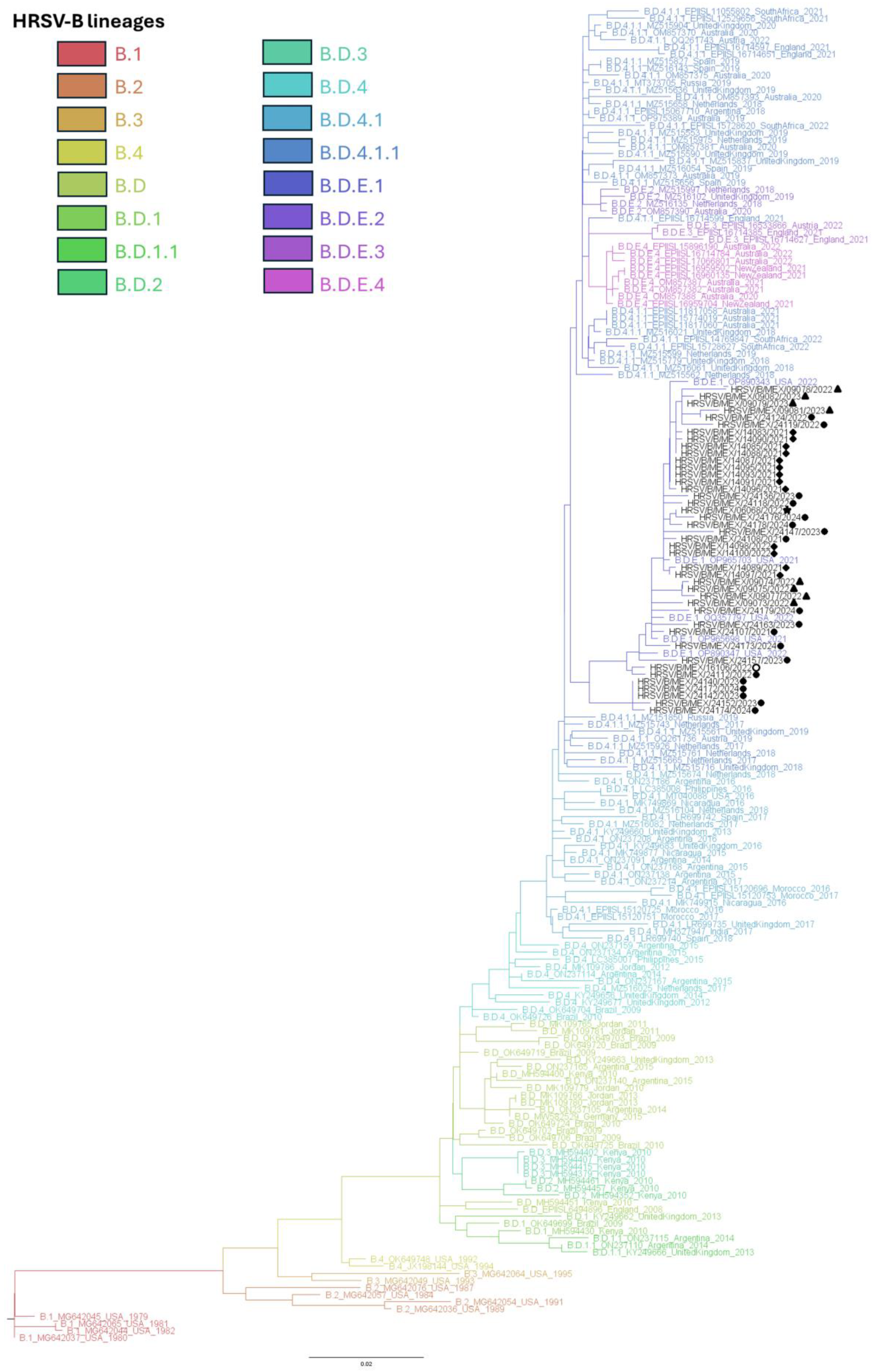
3.3. Phylogenetic Inference and Lineage Assignment
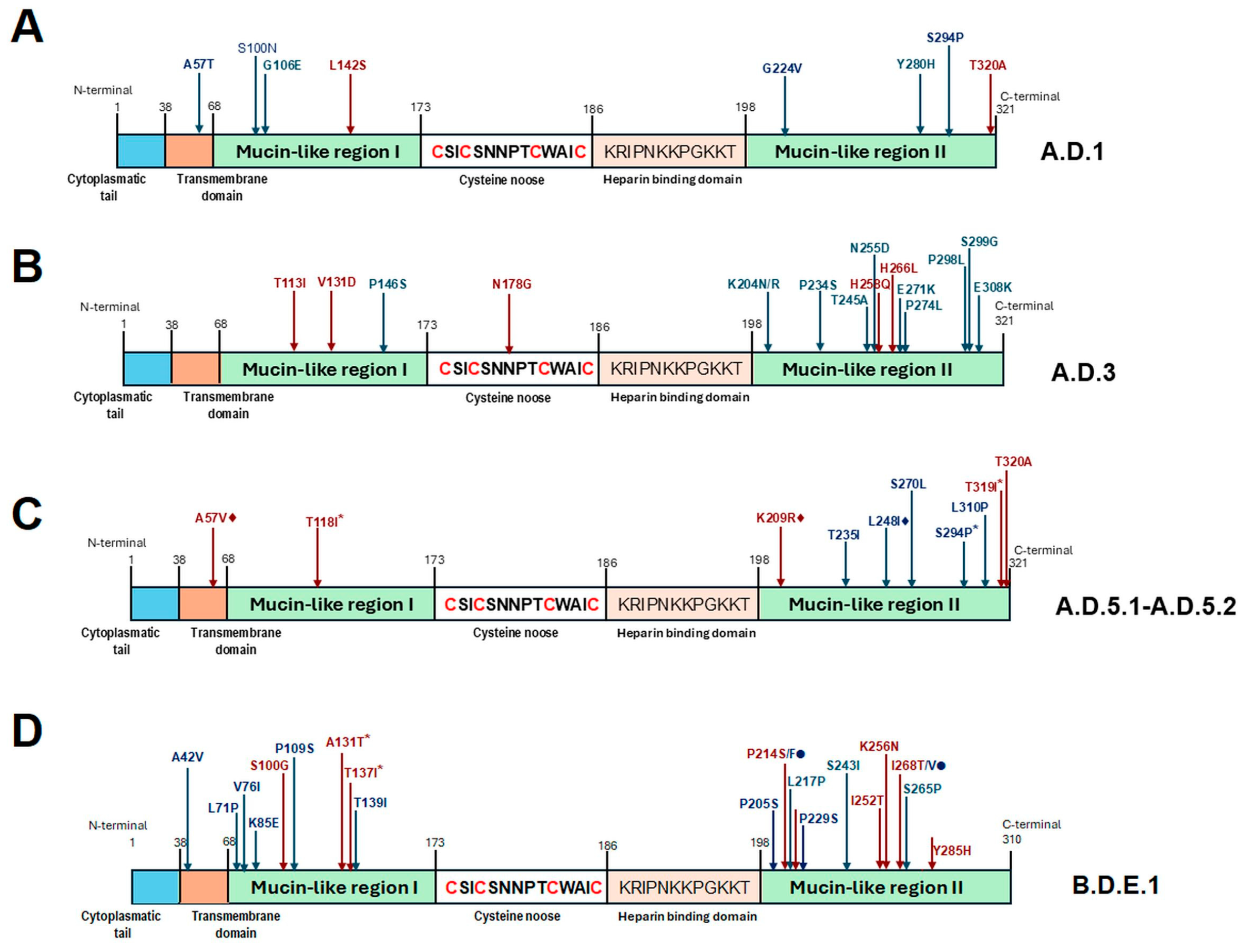
3.4. Analysis of Amino Acid Sequences and Amino Acid-Based Lineage Definition
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chadha, M.; Hirve, S.; Bancej, C.; Barr, I.; Baumeister, E.; Caetano, B.; Chittaganpitch, M.; Darmaa, B.; Ellis, J.; Fasce, R.; et al. Human respiratory syncytial virus and influenza seasonality patterns-Early findings from the WHO global respiratory syncytial virus surveillance. Influenza Other Respir. Viruses 2020, 14, 638–646. [Google Scholar] [CrossRef] [PubMed]
- Reeves, R.M.; van Wijhe, M.; Tong, S.; Lehtonen, T.; Stona, L.; Teirlinck, A.C.; Fernandez, L.V.; Li, Y.; Giaquinto, C.; Fischer, T.K.; et al. Respiratory syncytial virus-associated hospital admissions in children younger than 5 years in 7 European countries using coutinely Collected datasets. J. Infect. Dis. 2020, 222 (Suppl. S7), S599–S605. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Reeves, R.M.; Wang, X.; Bassat, Q.; Brooks, W.A.; Cohen, C.; Moore, D.P.; Nunes, M.; Rath, B.; Campbell, H.; et al. Global patterns in monthly activity of influenza virus, respiratory syncytial virus, parainfluenza virus, and metapneumovirus: A systematic analysis. Lancet Glob. Health 2019, 7, e1031–e1045. [Google Scholar] [CrossRef]
- Cai, W.; Buda, S.; Schuler, E.; Hirve, S.; Zhang, W.; Haas, W. Risk factors for hospitalized respiratory syncytial virus disease and its severe outcomes. Influenza Other Respir. Viruses 2020, 14, 658–670. [Google Scholar] [CrossRef]
- Shmueli, E.; Goldberg, O.; Mei-Zahav, M.; Stafler, P.; Bar-On, O.; Levine, H.; Steuer, G.; Mussaffi, H.; Gendler, Y.; Blau, H.; et al. Risk factors for respiratory syncytial virus bronchiolitis hospitalizations in children with chronic diseases. Pediatr. Pulmonol. 2021, 56, 2204–2211. [Google Scholar] [CrossRef] [PubMed]
- Zilberbeg, M.D.; Khan, I.; Shorr, A.F. Respiratory viruses in nosocomial pneumonia: An evolving paradigm. Viruses 2023, 15, 1676. [Google Scholar] [CrossRef]
- Rima, B.; Collins, P.; Easton, A.; Fouchier, R.; Kurath, G.; Lamb, R.A.; Lee, B.; Maisner, A.; Rota, P.; Wang, L.; et al. ICTV virus taxonomy profile: Pneumoviridae. J. Gen. Virol. 2017, 98, 2912–2913. [Google Scholar] [CrossRef]
- Melero, J.A.; García-Barreno, B.; Martínez, I.; Pringle, C.R.; Cane, P.A. Antigenic structure, evolution and immunobiology of human respiratory syncytial virus attachment (G) protein. J. Gen. Virol. 1997, 78 Pt 10, 2411–2418. [Google Scholar] [CrossRef]
- Cantu-Flores, K.; Rivera-Alfaro, G.; Muñoz-Escalante, J.C.; Noyola, D.E. Global distribution of respiratory syncytial virus A and B infections: A systematic review. Pathog. Glob. Health 2022, 116, 398–409. [Google Scholar] [CrossRef]
- Muñoz-Escalante, J.C.; Comas-García, A.; Bernal-Silva, S.; Robles-Espinoza, C.D.; Gómez-Leal, G.; Noyola, D.E. Respiratory syncytial virus A genotype classification based on systematic intergenotypic and intragenotypic sequence analysis. Sci. Rep. 2019, 9, 20097. [Google Scholar] [CrossRef]
- Muñoz-Escalante, J.C.; Comas-García, A.; Bernal-Silva, S.; Noyola, D.E. Respiratory syncytial virus B sequence analysis reveals a novel early genotype. Sci. Rep. 2021, 11, 3452. [Google Scholar] [CrossRef] [PubMed]
- Amjad, M.N.; Wang, J.; Ashraf, M.A.; Shen, B.; Din, G.U.; Raza, M.A.; Shoaib, M.; Yue, L.; Chen, L.; Xu, H.; et al. Evolutionary trends of respiratory syncytial viruses: Insights from large-scale surveillance and molecular dynamics of G glycoprotein. Heliyon 2024, 10, e30886. [Google Scholar] [CrossRef] [PubMed]
- Tramuto, F.; Maida, C.M.; Mazzucco, W.; Costantino, C.; Amodio, E.; Sferlazza, G.; Previti, A.; Immordino, P.; Vitale, F. Molecular epidemiology and genetic diversity of human respiratory syncytial virus in Sicily during pre- and post-COVID-19 surveillance seasons. Pathogens 2023, 12, 1099. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, S.; Phyu, W.W.; Wagatsuma, K.; Nagai, T.; Sano, Y.; Taniguchi, K.; Nagata, N.; Tomimoto, K.; Sato, I.; Kaji, H.; et al. Molecular epidemiology of respiratory syncytial virus during 2019–2022 and surviving genotypes after the COVID-19 pandemic in Japan. Viruses 2023, 15, 2382. [Google Scholar] [CrossRef]
- Vila, J.; Lera, E.; Peremiquel-Trillas, P.; Andrés, C.; Martínez, L.; Barceló, I.; Carsi, A.; Balcells, J.; Rodrigo-Pendás, J.Á.; Soler-Palacín, P.; et al. Increased RSV-A bronchiolitis severity in RSV-infected children admitted to a reference center in Catalonia (Spain) between 2014 and 2018. J. Pediatr. Infect. Dis. Soc. 2023, 12, 180–183. [Google Scholar] [CrossRef]
- Cai, W.; Köndgen, S.; Tolksdorf, K.; Dürrwald, R.; Schuler, E.; Biere, B.; Schweiger, B.; Goerlitz, L.; Haas, W.; Wolff, T.; et al. Atypical age distribution and high disease severity in children with RSV infections during two irregular epidemic seasons throughout the COVID-19 pandemic, Germany, 2021 to 2023. Eurosurveillance 2024, 29, 2300465. [Google Scholar] [CrossRef]
- Piñana, M.; González-Sánchez, A.; Andrés, C.; Vila, J.; Creus-Costa, A.; Prats-Méndez, I.; Arnedo-Muñoz, M.; Saubi, N.; Esperalba, J.; Rando, A.; et al. Genomic evolution of human respiratory syncytial virus during a decade (2013–2023): Bridging the path to monoclonal antibody surveillance. J. Infect. 2024, 88, 106153. [Google Scholar] [CrossRef]
- Walker, G.J.; Foster, C.S.; Sevendal, A.; Domazetovska, A.; Kamalakkannan, A.; Williams, P.C.; Kim, K.W.; Condylios, A.; Stelzer-Braid, S.; Bartlett, A.W.; et al. Clinical, Genomic, and Immunological Characterization of RSV Surge in Sydney, Australia, 2022. Pediatrics 2024, 153, e2023063667. [Google Scholar] [CrossRef]
- Bermudez Barrezueta, L.; Del Pozo, V.M.; López-Casillas, P.; Raposo, M.B.; Zamorano, M.G.; Vázquez, M.d.l.A.P. Variation in the seasonality of the respiratory syncytial virus during the COVID-19 pandemic. Infection 2022, 50, 1001–1005. [Google Scholar] [CrossRef]
- Eden, J.S.; Sikazwe, C.; Xie, R.; Deng, Y.-M.; Sullivan, S.G.; Michie, A.; Levy, A.; Cutmore, E.; Blyth, C.C.; Britton, P.N.; et al. Off-season RSV epidemics in Australia after easing of COVID-19 restrictions. Nat. Commun. 2022, 13, 2884. [Google Scholar] [CrossRef]
- Jia, R.; Lu, L.; Su, L.; Lin, Z.; Gao, D.; Lv, H.; Xu, M.; Liu, P.; Cao, L.; Xu, J. Resurgence of respiratory syncytial virus infection during COVID-19 pandemic among children in Shanghai, China. Front. Microbiol. 2022, 13, 938372. [Google Scholar] [CrossRef]
- Leija-Martinez, J.J.; Esparza-Miranda, L.A.; Rivera-Alfaro, G.; Noyola, D.E. Impact of nonpharmaceutical interventions during the COVID-19 pandemic on the prevalence of respiratory syncytial virus in hospitalized children with lower respiratory tract infections: A systematic review and meta-analysis. Viruses 2024, 16, 429. [Google Scholar] [CrossRef]
- Haapanen, M.; Renko, M.; Artama, M.; Kuitunen, I. The impact of the lockdown and the re-opening of schools and day cares on the epidemiology of SARS-CoV-2 and other respiratory infections in children—A nationwide register study in Finland. eClinicalMedicine 2021, 34, 100807. [Google Scholar] [CrossRef]
- Honemann, M.; Thiem, S.; Bergs, S.; Berthold, T.; Propach, C.; Siekmeyer, M.; Frille, A.; Wallborn, T.; Maier, M.; Pietsch, C. In-depth analysis of the re-emergence of respiratory syncytial virus at a tertiary care hospital in Germany in the summer of 2021 after the alleviation of non-pharmaceutical interventions due to the SARS-CoV-2 pandemic. Viruses 2023, 15, 877. [Google Scholar] [CrossRef]
- Foley, D.A.; Yeoh, D.K.; Minney-Smith, C.A.; Martin, A.C.; Mace, A.O.; Sikazwe, C.T.; Le, H.; Levy, A.; Moore, H.C.; Blyth, C.C. The interseasonal resurgence of respiratory syncytial virus in Australian children following the reduction of coronavirus disease 2019-related public health measures. Clin. Infect. Dis. 2021, 73, e2829–e2830. [Google Scholar] [CrossRef]
- Frohlich, G.C.; Gregianini, T.S.; Pinheiro, F.G.; Nascimento, R.; Cezar, T.M.; Pscheidt, V.M.; Selayaran, T.; Martins, L.G.; Gomes, M.F.d.C.; Salvato, R.S.; et al. Resurgence of human respiratory syncytial virus during COVID-19 pandemic in Southern Brazil. J. Med. Virol. 2024, 96, e29551. [Google Scholar] [CrossRef]
- Pierangeli, A.; Piralla, A.; Renteria, S.U.; Giacomel, G.; Lunghi, G.; Pagani, E.; Giacobazzi, E.; Vian, E.; Biscaro, V.; Piccirilli, G.; et al. Multicenter epidemiological investigation and genetic characterization of respiratory syncytial virus and metapneumovirus infections in the pre-pandemic 2018–2019 season in northern and central Italy. Clin. Exp. Med. 2023, 23, 2725–2737. [Google Scholar] [CrossRef]
- Goya, S.; Sereewit, J.; Pfalmer, D.; Nguyen, T.V.; Bakhash, S.A.M.; Sobolik, E.B.; Greninger, A.L. Genomic characterization of respiratory syncytial virus during 2022–23 outbreak, Washington, USA. Emerg. Infect. Dis. 2023, 29, 865–868. [Google Scholar] [CrossRef]
- Holland, L.A.; Holland, S.C.; Smith, M.F.; Leonard, V.R.; Murugan, V.; Nordstrom, L.; Mulrow, M.; Salgado, R.; White, M.; Lim, E.S. Genomic sequencing surveillance to identify respiratory syncytial virus mutations, Arizona, USA. Emerg. Infect. Dis. 2023, 29, 2380–2382. [Google Scholar] [CrossRef]
- Rios-Guzman, E.; Simons, L.M.; Dean, T.J.; Agnes, F.; Pawlowski, A.; Alisoltanidehkordi, A.; Nam, H.H.; Ison, M.G.; Ozer, E.A.; Lorenzo-Redondo, R.; et al. Deviations in RSV epidemiological patterns and population structures in the United States following the COVID-19 pandemic. Nat. Commun. 2024, 15, 3374. [Google Scholar] [CrossRef]
- Gamino-Arroyo, A.E.; Moreno-Espinosa, S.; Llamosas-Gallardo, B.; Ortiz-Hernández, A.A.; Guerrero, M.L.; Galindo-Fraga, A.; Galán-Herrera, J.F.; Prado-Galbarro, F.J.; Beigel, J.H.; Ruiz-Palacios, G.M.; et al. Epidemiology and clinical characteristics of respiratory syncytial virus infections among children and adults in Mexico. Influenza Other Respir. Viruses 2017, 11, 48–56. [Google Scholar] [CrossRef]
- Wong-Chew, R.M.; García-León, M.L.; Noyola, D.E.; Gonzalez, L.F.P.; Meza, J.G.; Vilaseñor-Sierra, A.; Martinez-Aguilar, G.; Rivera-Nuñez, V.H.; Newton-Sánchez, O.A.; Firo-Reyes, V.; et al. Respiratory viruses detected in Mexican children younger than 5 years old with community-acquired pneumonia: A national multicenter study. Int. J. Infect. Dis. 2017, 62, 32–38. [Google Scholar] [CrossRef]
- Noyola, D.E.; Hunsberger, S.; Powers, J.H.; Galindo-Fraga, A.; Ramirez-Venegas, A.; Moreno-Espinosa, S.; Llamosas-Gallardo, B.; Guerrero, M.L.; Beigel, J.H.; Ruiz-Palacios, G.; et al. Comparison of rates of hospitalization between single and dual virus detection in a Mexican cohort of children and adults with influenza-like illness. Open Forum Infect. Dis. 2019, 6, ofz424. [Google Scholar] [CrossRef]
- Benitez-Guerra, D.; Piña-Flores, C.; Zamora-López, M.; Escalante-Padrón, F.; Lima-Rogel, V.; González-Ortiz, A.M.; Guevara-Tovar, M.; Bernal-Silva, S.; Benito-Cruz, B.; Castillo-Martínez, F.; et al. Respiratory syncytial virus acute respiratory infection-associated hospitalizations in preterm Mexican infants: A cohort study. Influenza Other Respir. Viruses 2020, 14, 182–188. [Google Scholar] [CrossRef]
- Nieto-Rivera, B.; Saldaña-Ahuactzi, Z.; Parra-Ortega, I.; Flores-Alanis, A.; Carbajal-Franco, E.; Cruz-Rangel, A.; Galaviz-Hernández, S.; Romero-Navarro, B.; de la Rosa-Zamboni, D.; Salazar-García, M.; et al. Frequency of respiratory virus-associated infection among children and adolescents from a tertiary-care hospital in Mexico City. Sci. Rep. 2023, 13, 19763. [Google Scholar] [CrossRef]
- Bose, M.E.; He, J.; Shrivastava, S.; Nelson, M.I.; Bera, J.; Halpin, R.A.; Town, C.D.; Lorenzi, H.A.; Noyola, D.E.; Falcone, V.; et al. Sequencing and analysis of globally obtained human respiratory syncytial virus A and B genomes. PLoS ONE 2015, 10, e0120098. [Google Scholar] [CrossRef]
- Comas-Garcia, A.; Noyola, D.E.; Cadena-Mota, S.; Rico-Hernández, M.; Bernal-Silva, S. Respiratory syncytial virus-A ON1 genotype emergence in central Mexico in 2009 and evidence of multiple duplication events. J. Infect. Dis. 2018, 217, 1089–1098. [Google Scholar] [CrossRef]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Window 98/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Goya, S.; Ruis, C.; Neher, R.A.; Meijer, A.; Aziz, A.; Hinrichs, A.S.; von Gottberg, A.; Roemer, C.; Amoako, D.G.; Acuña, D.; et al. Standardized phylogenetic classification of human respiratory syncytial virus below the subgroup level. Emerg. Infect. Dis. 2024, 30, 1631–1641. [Google Scholar] [CrossRef]
- Martin, D.P.; Varsani, A.; Roumagnac, P.; Botha, G.; Maslamoney, S.; Schwab, T.; Kelz, Z.; Kumar, V.; Murrell, B. RDP5: A computer program for analyzing recombination in, and removing signals of recombination from, nucleotide sequence datasets. Virus Evol. 2020, 7, veaa087. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; Von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the ultrafast nootstrap approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Adams, G.; Moreno, G.K.; Petros, B.A.; Uddin, R.; Levine, Z.; Kotzen, B.; Messer, K.S.; Dobbins, S.T.; DeRuff, K.C.; Loreth, C.M.; et al. Viral lineages in the 2022 RSV surge in the United States. N. Engl. J. Med. 2023, 388, 1335–1337. [Google Scholar] [CrossRef]
- Ono, T.; Hashimoto, K.; Kume, Y.; Chishiki, M.; Okabe, H.; Sato, M.; Norito, S.; Aso, J.; Sada, M.; Mochizuki, I.; et al. Molecular diversity of human respiratory syncytial virus before and during the COVID-19 pandemic in two neighboring Japanese cities. Microbiol. Spectr. 2023, 11, e0260622. [Google Scholar] [CrossRef]
- Goya, S.; Galiano, M.; Nauwelaers, I.; Trento, A.; Openshaw, P.J.; Mistchenko, A.S.; Zambon, M.; Viegas, M. Toward unified molecular surveillance of RSV: A proposal for genotype definition. Influenza Other Respir. Viruses 2020, 14, 274–285. [Google Scholar] [CrossRef]
- Ramaekers, K.; Rector, A.; Cuypers, L.; Lemey, P.; Keyaerts, E.; Van Ranst, M. Towards a unified classification for human respiratory syncytial virus genotypes. Virus Evol. 2020, 6, veaa052. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Collection Site | Number of Samples in Which HRSV Was Tested | HRSV-Positive Diagnosis | HRSV-A Specific PCR Positive | HRSV-A Sequences | HRSV-B Specific PCR Positive | HRSV-B Sequences |
|---|---|---|---|---|---|---|
| Hospital General de Colima | 41 | 6 | 3 | 3 | 1 | 1 |
| Hospital General de Mexico | 92 | 16 | 3 | 2 | 6 | 6 |
| Hospital Pediátrico de Coyoacán | 146 | 28 | 2 | 2 | 2 | 2 |
| Hospital Civil de Guadalajara | 161 | 35 | 10 | 10 | 15 | 14 |
| Hospital Central-SLP | 152 | 35 | 14 | 13 | 2 | 2 |
| Hospital del Niño y la Mujer | 390 | 160 | 34 | 30 | 14 | 12 |
| IPICYT | 50 | 5 | 0 | 0 | 1 | 1 |
| UASLP | 284 | 28 | 12 | 12 | 4 | 4 |
| Total | 1316 | 216 | 78 | 72 | 45 | 42 |
| Lineage | 1956–2019 | 1956–2019 | 2020 | 2020 | 2021 | 2021 | 2022 | 2022 | 2023 | 2023 | 2024 | 2024 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Global * | Mexico ** | Global | Mexico | Global | Mexico | Global | Mexico | Global | Mexico | Global | Mexico | |
| A.1 | 21 | |||||||||||
| A.2 | 30 | |||||||||||
| A.2.1 | 323 | |||||||||||
| A.2.1.1 | 99 | 1 | ||||||||||
| A.3 | 81 | 2 | ||||||||||
| A.3.1 | 794 | 1 | ||||||||||
| A.3.1.1 | 343 | |||||||||||
| A.D | 2776 | 5 | 119 | 23 | 26 | 8 | 1 | |||||
| A.D.1 | 1114 | 358 | 242 | 3 | 459 | 22 | 786 | 16 | 303 | 2 | ||
| A.D.1.1 | 43 | 26 | 51 | 19 | 1 | 1 | ||||||
| A.D.1.2 | 141 | 80 | ||||||||||
| A.D.1.3 | 3 | 266 | 215 | 25 | 1 | |||||||
| A.D.1.4 | 3 | 5 | 17 | 24 | 64 | 10 | ||||||
| A.D.2 | 35 | 5 | 14 | 27 | 14 | 2 | ||||||
| A.D.2.1 | 54 | 19 | 1 | 9 | 2 | 1 | ||||||
| A.D.2.2 | 667 | 87 | 2 | 3 | ||||||||
| A.D.2.2.1 | 124 | 6 | 2 | |||||||||
| A.D.3 | 520 | 207 | 329 | 2 | 307 | 3 | 277 | 5 | 128 | 1 | ||
| A.D.3.1 | 11 | 2 | 94 | 223 | 131 | 17 | ||||||
| A.D.4 | 276 | 2 | 15 | 12 | 44 | 6 | ||||||
| A.D.4.1 | 131 | 7 | 31 | 8 | 3 | |||||||
| A.D.5 | 319 | 76 | 218 | 142 | 184 | 47 | ||||||
| A.D.5.1 | 10 | 8 | 61 | 69 | 1 | 10 | ||||||
| A.D.5.2 | 6 | 3 | 256 | 784 | 6 | 423 | 11 | 143 | ||||
| A.D.5.3 | 6 | 6 | 219 | 7 | 5 | |||||||
| Total | 7789 | 7 | 1335 | 2 | 1816 | 5 | 2135 | 31 | 2014 | 33 | 669 | 3 |
| Lineage | 1956–2019 | 1956–2019 | 2020 | 2020 | 2021 | 2021 | 2022 | 2022 | 2023 | 2023 | 2024 | 2024 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Global * | Mexico ** | Global | Mexico | Global | Mexico | Global | Mexico | Global | Mexico | Global | Mexico | |
| B.1 | 32 | |||||||||||
| B.2 | 63 | |||||||||||
| B.3 | 85 | |||||||||||
| B.4 | 363 | 3 | ||||||||||
| B.D | 1042 | 1 | 1 | |||||||||
| B.D.1 | 147 | |||||||||||
| B.D.1.1 | 22 | |||||||||||
| B.D.2 | 47 | |||||||||||
| B.D.3 | 99 | |||||||||||
| B.D.4 | 524 | |||||||||||
| B.D.4.1 | 837 | 14 | 2 | 68 | 43 | |||||||
| B.D.4.1.1 | 3311 | 408 | 9 | 285 | 211 | 118 | 15 | |||||
| B.D.E.1 | 15 | 6 | 778 | 13 | 1448 | 13 | 977 | 10 | 183 | 6 | ||
| B.D.E.2 | 79 | 29 | 15 | 5 | 5 | |||||||
| B.D.E.3 | 4 | 83 | 10 | 4 | ||||||||
| B.D.E.4 | 3 | 23 | 304 | 309 | 8 | |||||||
| Not assignable | 76 | 2 | ||||||||||
| Total | 6745 | 4 | 484 | 11 | 1535 | 13 | 2026 | 13 | 1113 | 10 | 198 | 6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez-Marrero, N.; Muñoz-Escalante, J.C.; Wong-Chew, R.M.; Torres-González, P.; García-León, M.L.; Bautista-Carbajal, P.; Martínez-Arce, P.A.; Espinosa-Sotero, M.d.C.; Tabla-Orozco, V.; Rojas-Larios, F.; et al. Genotypic Characterization of Human Respiratory Syncytial Viruses Detected in Mexico Between 2021 and 2024. Viruses 2025, 17, 651. https://doi.org/10.3390/v17050651
Martínez-Marrero N, Muñoz-Escalante JC, Wong-Chew RM, Torres-González P, García-León ML, Bautista-Carbajal P, Martínez-Arce PA, Espinosa-Sotero MdC, Tabla-Orozco V, Rojas-Larios F, et al. Genotypic Characterization of Human Respiratory Syncytial Viruses Detected in Mexico Between 2021 and 2024. Viruses. 2025; 17(5):651. https://doi.org/10.3390/v17050651
Chicago/Turabian StyleMartínez-Marrero, Nadia, Juan Carlos Muñoz-Escalante, Rosa Maria Wong-Chew, Pedro Torres-González, Miguel Leonardo García-León, Patricia Bautista-Carbajal, Pedro Antonio Martínez-Arce, María del Carmen Espinosa-Sotero, Verónica Tabla-Orozco, Fabian Rojas-Larios, and et al. 2025. "Genotypic Characterization of Human Respiratory Syncytial Viruses Detected in Mexico Between 2021 and 2024" Viruses 17, no. 5: 651. https://doi.org/10.3390/v17050651
APA StyleMartínez-Marrero, N., Muñoz-Escalante, J. C., Wong-Chew, R. M., Torres-González, P., García-León, M. L., Bautista-Carbajal, P., Martínez-Arce, P. A., Espinosa-Sotero, M. d. C., Tabla-Orozco, V., Rojas-Larios, F., Juárez-Tobías, S., González-Ortiz, A. M., Alpuche-Solís, Á. G., & Noyola, D. E. (2025). Genotypic Characterization of Human Respiratory Syncytial Viruses Detected in Mexico Between 2021 and 2024. Viruses, 17(5), 651. https://doi.org/10.3390/v17050651