
Primary Cells from a CD46-Edited Bovine Heifer Have Reduced BVDV Susceptibility Despite Viral Adaptation to Heparan Sulfate

 , ,
, , 
Abstract
1. Introduction
2. Materials and Methods
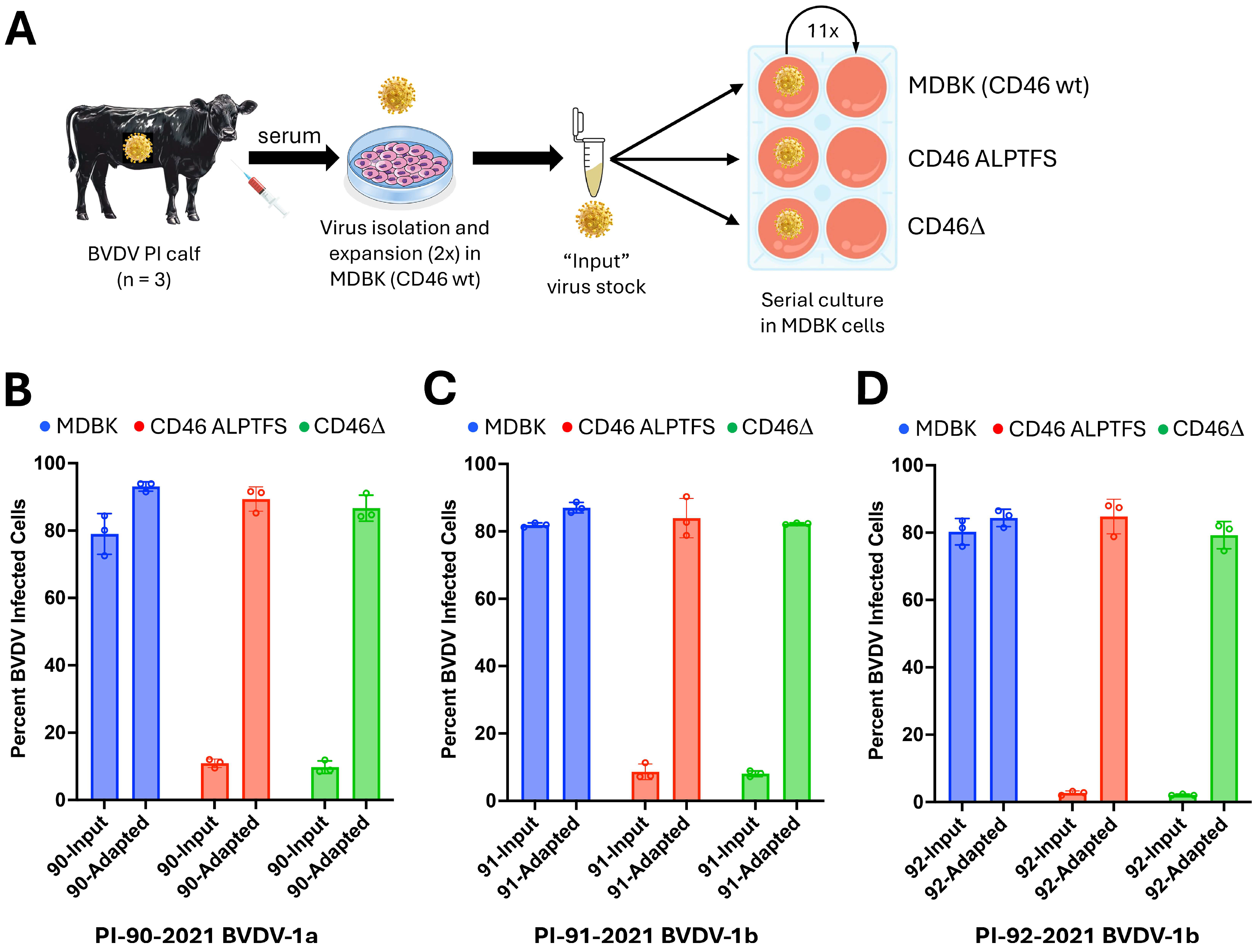
2.1. Study Population and Sample Collection
2.2. Cell Lines and Viruses
2.3. Passaging Virus Isolates on CD46-Edited MDBK Cells
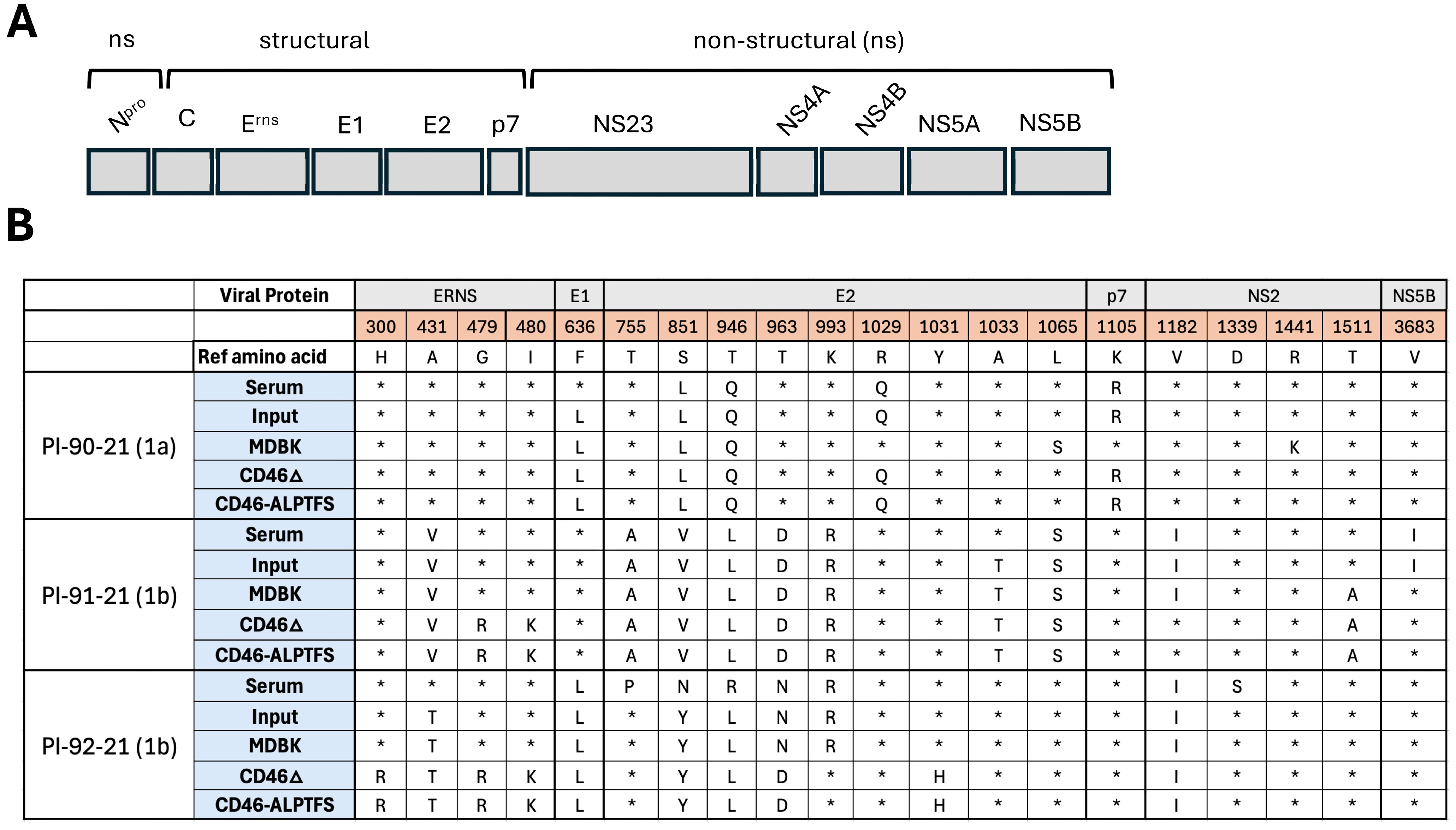
2.4. Whole Genome Sequencing, Assembly, and Comparisons of BVDV Isolates
2.5. Serum Virus Infections of CD46-Edited MDBK Cells
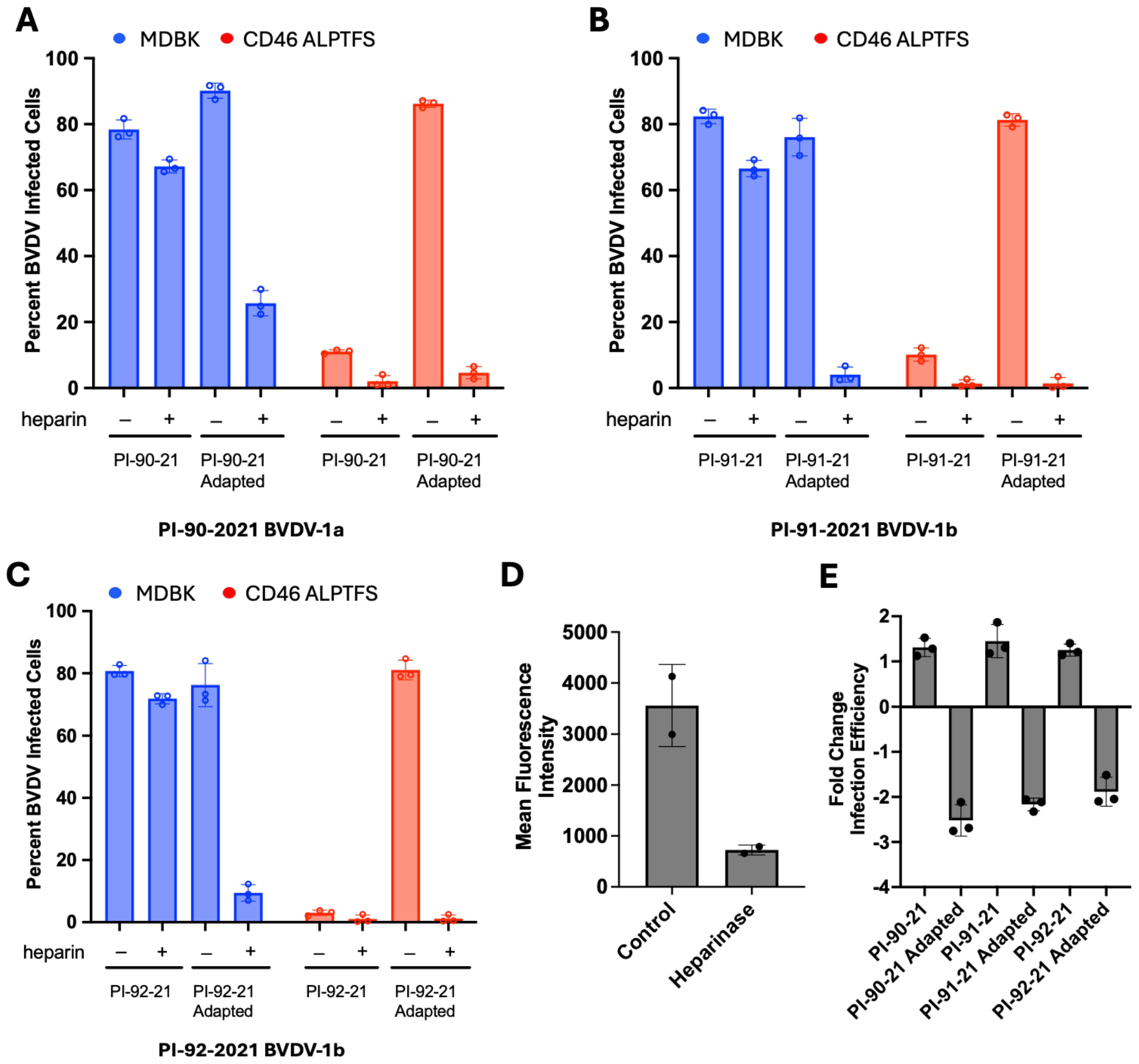
2.6. Infection of MDBK Cells with or Without Heparin Pre-Treatment
2.7. Heparinase Treatment and Infection of MDBK Cells
2.8. Flow Cytometric Detection of BVDV Antigen
2.9. Flow Cytometric Detection of Heparan Sulfate (HS)
2.10. Infection of Primary Skin Fibroblasts
2.11. Monocyte and Lymphocyte Isolation and Ex Vivo BVDV Challenge
2.12. Statistical Analyses
3. Results
3.1. Evolution of Three BVDV Field Strains After 11 Passages in CD46-Edited MDBK Cells
3.2. Disruption of HS-Mediated Entry Inhibits Adapted Virus Infection of MDBK-CD46 A82LPTFS Cells
3.3. A Virus Isolate from PI Yearling Calf Infects CD46-Edited MDBK Cells
3.4. Primary Cells from a CD46-Edited Heifer Have Reduced Susceptibility to the In Vitro- and In Vivo-Derived HS-Utilizing Viruses
3.5. Heparan Sulfate Expression Levels Correlate with Susceptibility to Adapted Viruses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BVDV | Bovine Viral Diarrhea Virus |
| CRISPRs | Clustered Regularly Interspaced Short Palindromic Repeats |
| HS | Heparan Sulfate |
| MDBK | Madin–Darby Bovine Kidney |
| PBMC | Peripheral Blood Mononuclear Cell |
| PI | Persistently Infected |
References
- Simmonds, P.; Becher, P.; Bukh, J.; Gould, E.A.; Meyers, G.; Monath, T.; Muerhoff, S.; Pletnev, A.; Rico-Hesse, R.; Smith, D.B.; et al. ICTV Virus Taxonomy Profile: Flaviviridae. J. Gen. Virol. 2017, 98, 2–3. [Google Scholar] [CrossRef] [PubMed]
- Olafson, P.; Maccallum, A.; Fox, F. An apparently new transmissible disease of cattle. Cornell Vet. 1946, 36, 205–213. [Google Scholar]
- Walz, P.H.; Chamorro, M.F.; Falkenberg, S.M.; Passler, T.; van der Meer, F.; Woolums, A.R. Bovine viral diarrhea virus: An updated American College of Veterinary Internal Medicine consensus statement with focus on virus biology, hosts, immunosuppression, and vaccination. J. Vet. Intern. Med. 2020, 34, 1690–1706. [Google Scholar] [CrossRef] [PubMed]
- Mcclurkin, A.; Littledike, E.; Cutlip, R.; Frank, G.; Coria, M.; Bolin, S.R. Production of cattle immunotolerant to bovine viral diarrhea virus. Can. J. Comp. Med. 1984, 48, 156–161. [Google Scholar]
- Hessman, B.E.; Fulton, R.W.; Sjeklocha, D.B.; Murphy, T.A.; Ridpath, J.F.; Payton, M.E. Evaluation of economic effects and the health and performance of the general cattle population after exposure to cattle persistently infected with bovine viral diarrhea virus in a starter feedlot. Am. J. Vet. Res. 2009, 70, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, M.; Flick-Smith, H.; McCauley, J.W. Interactions of bovine viral diarrhoea virus glycoprotein E(rns) with cell surface glycosaminoglycans. J. Gen. Virol. 2000, 81 Pt 2, 451–459. [Google Scholar]
- Iqbal, M.; McCauley, J.W. McCauley, Identification of the glycosaminoglycan-binding site on the glycoprotein E(rns) of bovine viral diarrhoea virus by site-directed mutagenesis. J. Gen. Virol. 2002, 83 Pt 9, 2153–2159. [Google Scholar] [CrossRef]
- Krey, T.; Himmelreich, A.; Heimann, M.; Menge, C.; Thiel, H.-J.; Maurer, K.; Rumenapf, T. Function of bovine CD46 as a cellular receptor for bovine viral diarrhea virus is determined by complement control protein 1. J. Virol. 2006, 80, 3912–3922. [Google Scholar] [CrossRef]
- Maurer, K.; Krey, T.; Moennig, V.; Thiel, H.-J.; Rümenapf, T. CD46 is a cellular receptor for bovine viral diarrhea virus. J. Virol. 2004, 78, 1792–1799. [Google Scholar] [CrossRef]
- Chen, H.-W.; Huber, V.; Szakmary-Braendle, K.; Seitz, K.; Moetz, M.; Ruemenapf, T.; Riedel, C. Viral Traits and Cellular Knock-Out Genotype Affect Dependence of BVDV on Bovine CD46. Pathogens 2021, 10, 1620. [Google Scholar] [CrossRef]
- Workman, A.M.; Heaton, M.P.; Webster, D.A.; Harhay, G.P.; Kalbfleisch, T.S.; Smith, T.P.L.; Falkenberg, S.M.; Carlson, D.F.; Sonstegard, T.S. Evaluating Large Spontaneous Deletions in a Bovine Cell Line Selected for Bovine Viral Diarrhea Virus Resistance. Viruses 2021, 13, 2147. [Google Scholar] [CrossRef] [PubMed]
- Workman, A.M.; Heaton, M.P.; Vander Ley, B.L.; Webster, D.A.; Sherry, L.; Bostrom, J.R.; Larson, S.; Kalbfleisch, T.S.; Harhay, G.P.; Jobman, E.E.; et al. First gene-edited calf with reduced susceptibility to a major viral pathogen. PNAS Nexus 2023, 2, pgad125. [Google Scholar] [CrossRef]
- Leveringhaus, E.; Cagatay, G.N.; Hardt, J.; Becher, P.; Postel, A. Different impact of bovine complement regulatory protein 46 (CD46(bov)) as a cellular receptor for members of the species Pestivirus H and Pestivirus G. Emerg. Microbes Infect. 2022, 11, 60–72. [Google Scholar] [CrossRef]
- Szillat, K.P.; Koethe, S.; Wernike, K.; Höper, D.; Beer, M. A CRISPR/Cas9 Generated Bovine CD46-knockout Cell Line-A Tool to Elucidate the Adaptability of Bovine Viral Diarrhea Viruses (BVDV). Viruses 2020, 12, 859. [Google Scholar] [CrossRef] [PubMed]
- Mahlum, C.E.; Haugerud, S.; Shivers, J.L.; Rossow, K.D.; Goyal, S.M.; Collins, J.E.; Faaberg, K.S. Detection of bovine viral diarrhea virus by TaqMan reverse transcription polymerase chain reaction. J. Vet. Diagn. Investig. 2002, 14, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Workman, A.M.; Heaton, M.P.; Harhay, G.P.; Smith, T.P.; Grotelueschen, D.M.; Sjeklocha, D.; Brodersen, B.; Petersen, J.L.; Chitko-McKown, C.G. Resolving Bovine viral diarrhea virus subtypes from persistently infected U.S. beef calves with complete genome sequence. J. Vet. Diagn. Investig. 2016, 28, 519–528. [Google Scholar] [CrossRef]
- Meneghetti, M.C.Z.; Hughes, A.J.; Rudd, T.R.; Nader, H.B.; Powell, A.K.; Yates, E.A.; Lima, M.A. Heparan sulfate and heparin interactions with proteins. J. R. Soc. Interface 2015, 12, 0589. [Google Scholar] [CrossRef]
- Sarrazin, S.; Lamanna, W.C.; Esko, J.D. Heparan sulfate proteoglycans. Cold Spring Harb. Perspect. Biol. 2011, 3, a004952. [Google Scholar] [CrossRef] [PubMed]
- Cagno, V.; Tseligka, E.D.; Jones, S.T.; Tapparel, C. Heparan Sulfate Proteoglycans and Viral Attachment: True Receptors or Adaptation Bias? Viruses 2019, 11, 596. [Google Scholar] [CrossRef]
- Hulst, M.M.; van Gennip, H.G.; Moormann, R.J. Passage of classical swine fever virus in cultured swine kidney cells selects virus variants that bind to heparan sulfate due to a single amino acid change in envelope protein E(rns). J. Virol. 2000, 74, 9553–9561. [Google Scholar] [CrossRef]
- Hulst, M.M.; van Gennip, H.G.P.; Vlot, A.C.; Schooten, E.; de Smit, A.J.; Moormann, R.J.M. Interaction of classical swine fever virus with membrane-associated heparan sulfate: Role for virus replication in vivo and virulence. J. Virol. 2001, 75, 9585–9595. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.; von Freyburg, M.; Elbers, K.; Meyers, G. Recovery of virulent and RNase-negative attenuated type 2 bovine viral diarrhea viruses from infectious cDNA clones. J. Virol. 2002, 76, 8494–8503. [Google Scholar] [CrossRef]
- Meyers, G.; Saalmuller, A.; Buttner, M. Mutations abrogating the RNase activity in glycoprotein E(rns) of the pestivirus classical swine fever virus lead to virus attenuation. J. Virol. 1999, 73, 10224–10235. [Google Scholar] [CrossRef]
- Reimann, I.; Depner, K.; Trapp, S.; Beer, M. An avirulent chimeric Pestivirus with altered cell tropism protects pigs against lethal infection with classical swine fever virus. Virology 2004, 322, 143–157. [Google Scholar] [CrossRef]
- Dalrymple, N.; Mackow, E.R. Productive dengue virus infection of human endothelial cells is directed by heparan sulfate-containing proteoglycan receptors. J. Virol. 2011, 85, 9478–9485. [Google Scholar] [CrossRef] [PubMed]
- Mandl, C.W.; Kroschewski, H.; Allison, S.L.; Kofler, R.; Holzmann, H.; Meixner, T.; Heinz, F.X. Adaptation of tick-borne encephalitis virus to BHK-21 cells results in the formation of multiple heparan sulfate binding sites in the envelope protein and attenuation in vivo. J. Virol. 2001, 75, 5627–5637. [Google Scholar] [CrossRef] [PubMed]
- Su, C.-M.; Liao, C.-L.; Lin, Y.-L. Highly sulfated forms of heparin sulfate are involved in japanese encephalitis virus infection. Virology 2001, 286, 206–215. [Google Scholar] [CrossRef]
- Chen, Y.; Maguire, T.; Hileman, R.E.; Fromm, J.R.; Esko, J.D.; Linhardt, R.J.; Marks, R.M. Dengue virus infectivity depends on envelope protein binding to target cell heparan sulfate. Nat. Med. 1997, 3, 866–871. [Google Scholar] [CrossRef]
- Lee, E.; Hall, R.A.; Lobigs, M. Common E protein determinants for attenuation of glycosaminoglycan-binding variants of Japanese encephalitis and West Nile viruses. J. Virol. 2004, 78, 8271–8280. [Google Scholar] [CrossRef]
- Drager, C.; Beer, M.; Blome, S. Porcine complement regulatory protein CD46 and heparan sulfates are the major factors for classical swine fever virus attachment in vitro. Arch. Virol. 2015, 160, 739–746. [Google Scholar] [CrossRef]
- Lee, E.; Lobigs, M. Mechanism of virulence attenuation of glycosaminoglycan-binding variants of Japanese encephalitis virus and Murray valley encephalitis virus. J. Virol. 2002, 76, 4901–4911. [Google Scholar] [CrossRef] [PubMed]
- Hulst, M.M.; Moormann, R.J. Inhibition of pestivirus infection in cell culture by envelope proteins E(rns) and E2 of classical swine fever virus: E(rns) and E2 interact with different receptors. J. Gen. Virol. 1997, 78 Pt 11, 2779–2787. [Google Scholar] [CrossRef] [PubMed]
- Colitti, B.; Nogarol, C.; Giacobini, M.; Capucchio, M.T.; Biasato, I.; Rosati, S.; Bertolotti, L. Compartmentalized evolution of Bovine Viral Diarrhoea Virus type 2 in an immunotolerant persistently infected cow. Sci. Rep. 2019, 9, 15460. [Google Scholar] [CrossRef] [PubMed]
- Dow, N.; Chernick, A.; Orsel, K.; van Marle, G.; van der Meer, F. Genetic Variability of Bovine Viral Diarrhea Virus and Evidence for a Possible Genetic Bottleneck during Vertical Transmission in Persistently Infected Cattle. PLoS ONE 2015, 10, e0131972. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PI-90-21 | Serum | INPUT | p11 MDBK | p11 CD46Δ | p11 CD46 ALPTFS | |
| Accession | PQ613788 | PQ613786 | PQ613787 | PQ613785 | PQ613784 | |
| Read Depth | 1374 | 144,875 | 75,534 | 116,976 | 130,942 | |
| PI-91-21 | Serum | INPUT | p11 MDBK | p11 CD46Δ | p11 CD46 ALPTFS | |
| Accession | PQ613793 | PQ613791 | PQ613792 | PQ613790 | PQ613789 | |
| Read depth | 831 | 162,529 | 54,832 | 83,482 | 126,951 | |
| PI-92-21 | Serum | INPUT | p11 MDBK | p11 CD46Δ | p11 CD46 ALPTFS | |
| Accession | PQ613798 | PQ613796 | PQ613797 | PQ613795 | PQ613794 | |
| Read depth | 559 | 210,742 | 84,011 | 89,169 | 159,937 | |
| Amino acid, position | Virus strain | Serum | INPUT | p11 MDBK | p11 CD46Δ | p11 CD46 ALPTFS |
| PI-90-21 | * | * | * | * | * | |
| Histidine, 300 | PI-91-21 | Leucine (1.6%) | * | * | * | * |
| PI-92-21 | Leucine (0.7%) | * | Arginine (23%) | Arginine (83.0%) | Arginine (81.7%) | |
| PI-90-21 | * | * | * | * | * | |
| Cysteine, 441 | PI-91-21 | * | * | * | * | * |
| PI-92-21 | * | * | * | * | * | |
| PI-90-21 | * | * | * | * | * | |
| Glycine, 479 | PI-91-21 | Arginine (2%) | * | Arginine (43.5%) | Arginine (94.9%) | Arginine (90.2%) |
| PI-92-21 | * | * | Arginine (25.8%) | Arginine (98.1%) | Arginine (98.0%) | |
| PI-90-21 | Valine (1.7%) | Valine (0.8%) | Threonine (0.5%) | Valine (0.5%) | * | |
| Leucine (0.5%) | ||||||
| Isoleucine, 480 | PI-91-21 | Lysine (1.8%) | * | Lysine (27.8%) | Lysine (76.1%) | Lysine (69.8%) |
| Arginine (0.9%) | Arginine (15.8%) | Arginine (19.2%) | Arginine (21.1%) | |||
| PI-92-21 | Threonine (0.9%) | * | Lysine (25%) | Lysine (82.7%) | Lysine (83.8%) | |
| Leucine (0.5%) |
| PI-86-21 | Serum | 72 h MDBK | 72 h MDBK A82LPTFS | INPUT | |
| Accession | PQ613779 | PQ613778 | |||
| Read Depth | 1967 | 2361 | |||
| PI-86-22 | Serum | 72 h MDBK | 72 h MDBK A82LPTFS | INPUT | |
| Accession | PQ613783 | PQ613781 | PQ613780 | PQ613782 | |
| Read depth | 5377 | 108,859 | 206,498 | 1060 | |
| Amino acid, position | Virus strain | Serum | 72 h MDBK | 72 h MDBK A82LPTFS | INPUT |
| PI-86-21 | * | * | |||
| Histidine, 300 | PI-86-22 | Tyrosine (0.5%) | Arginine (0.5%) | Tyrosine (0.7%) | * |
| Arginine (0.6%) | |||||
| Leucine (0.6%) | |||||
| PI-86-21 | * | * | |||
| Cysteine, 441 | PI-86-22 | * | * | * | * |
| PI-86-21 | Arginine (2.6%) | Arginine (1%) | |||
| Glycine, 479 | P8-86-22 | Arginine (21.2%) | Lysine (51.4%) | Arginine (55.5%) | Lysine (79.6%) |
| Lysine (19.1%) | Arginine (25.7%) | Lysine (42.6%) | Arginine (11.2%) | ||
| PI-86-21 | Lysine (1.9%) | * | |||
| Arginine (0.6%) | |||||
| Threonine (0.5%) | |||||
| Isoleucine, 480 | PI-86-22 | Lysine (21.8%) | Lysine (24%) | Lysine (58.1%) | Lysine (11.1%) |
| Methionine (2.5%) | Methionine (7.4%) | Methionine (8.3%) | |||
| Valine (1.0%) | Threonine (3.6%) | Valine (1.8%) | |||
| Threonine (0.7%) | Arginine (0.5%) | ||||
| Arginine (0.6%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krueger, A.C.; Vander Ley, B.L.; Heaton, M.P.; Sonstegard, T.S.; Workman, A.M. Primary Cells from a CD46-Edited Bovine Heifer Have Reduced BVDV Susceptibility Despite Viral Adaptation to Heparan Sulfate. Viruses 2025, 17, 634. https://doi.org/10.3390/v17050634
Krueger AC, Vander Ley BL, Heaton MP, Sonstegard TS, Workman AM. Primary Cells from a CD46-Edited Bovine Heifer Have Reduced BVDV Susceptibility Despite Viral Adaptation to Heparan Sulfate. Viruses. 2025; 17(5):634. https://doi.org/10.3390/v17050634
Chicago/Turabian StyleKrueger, Alexandria C., Brian L. Vander Ley, Michael P. Heaton, Tad S. Sonstegard, and Aspen M. Workman. 2025. "Primary Cells from a CD46-Edited Bovine Heifer Have Reduced BVDV Susceptibility Despite Viral Adaptation to Heparan Sulfate" Viruses 17, no. 5: 634. https://doi.org/10.3390/v17050634
APA StyleKrueger, A. C., Vander Ley, B. L., Heaton, M. P., Sonstegard, T. S., & Workman, A. M. (2025). Primary Cells from a CD46-Edited Bovine Heifer Have Reduced BVDV Susceptibility Despite Viral Adaptation to Heparan Sulfate. Viruses, 17(5), 634. https://doi.org/10.3390/v17050634