
Genetic Profiles of Ten African Swine Fever Virus Strains from Outbreaks in Select Provinces of Luzon, Visayas, and Mindanao, Philippines, Between 2021 and 2023

 ,
,  , , ,
, , ,
Abstract
1. Introduction
2. Materials and Method
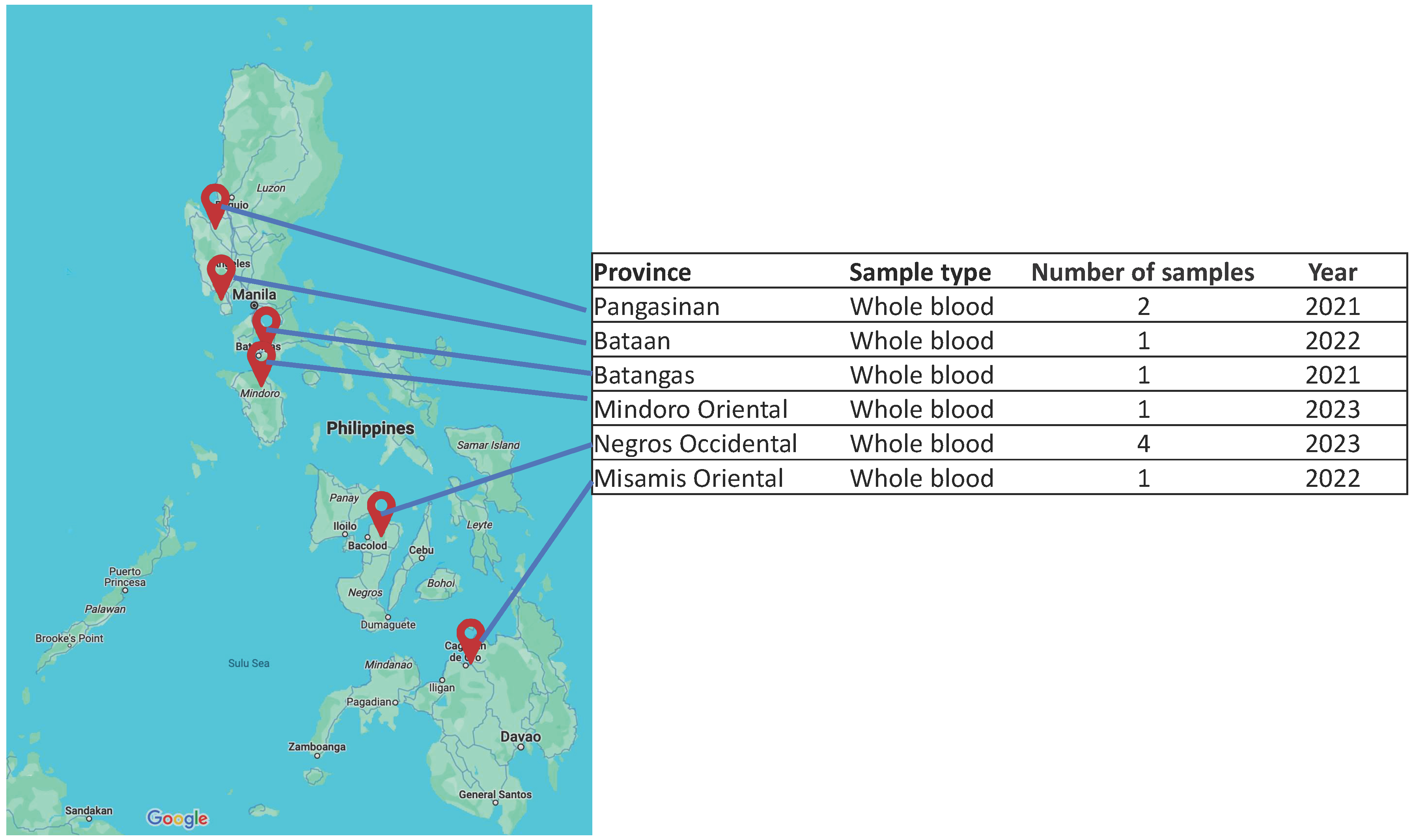
2.1. Sample Collection and Viral DNA Isolation
2.2. Library Preparation for Targeted Tiled Amplicon Sequencing
2.3. Genome Assembly and Annotation
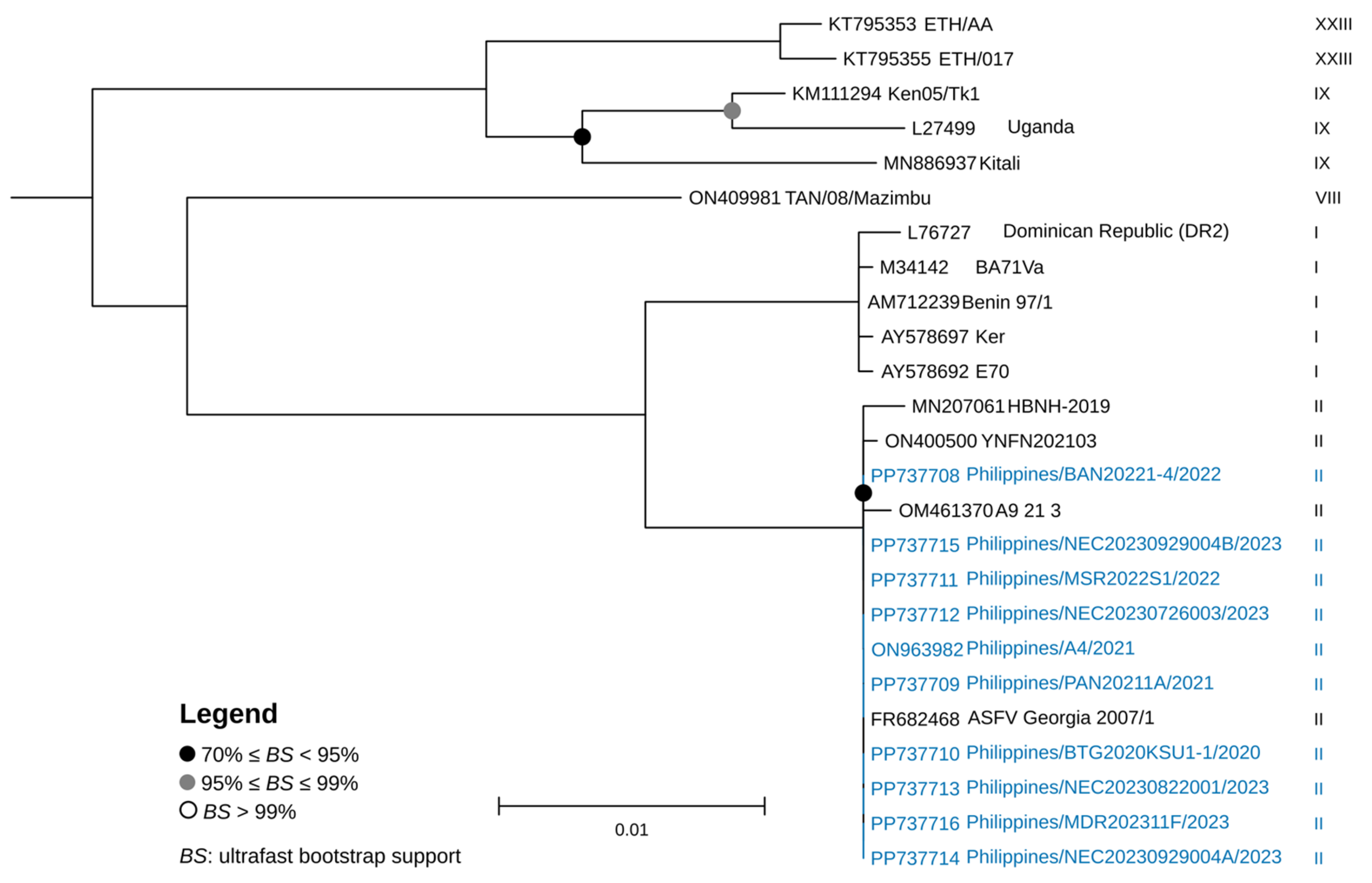
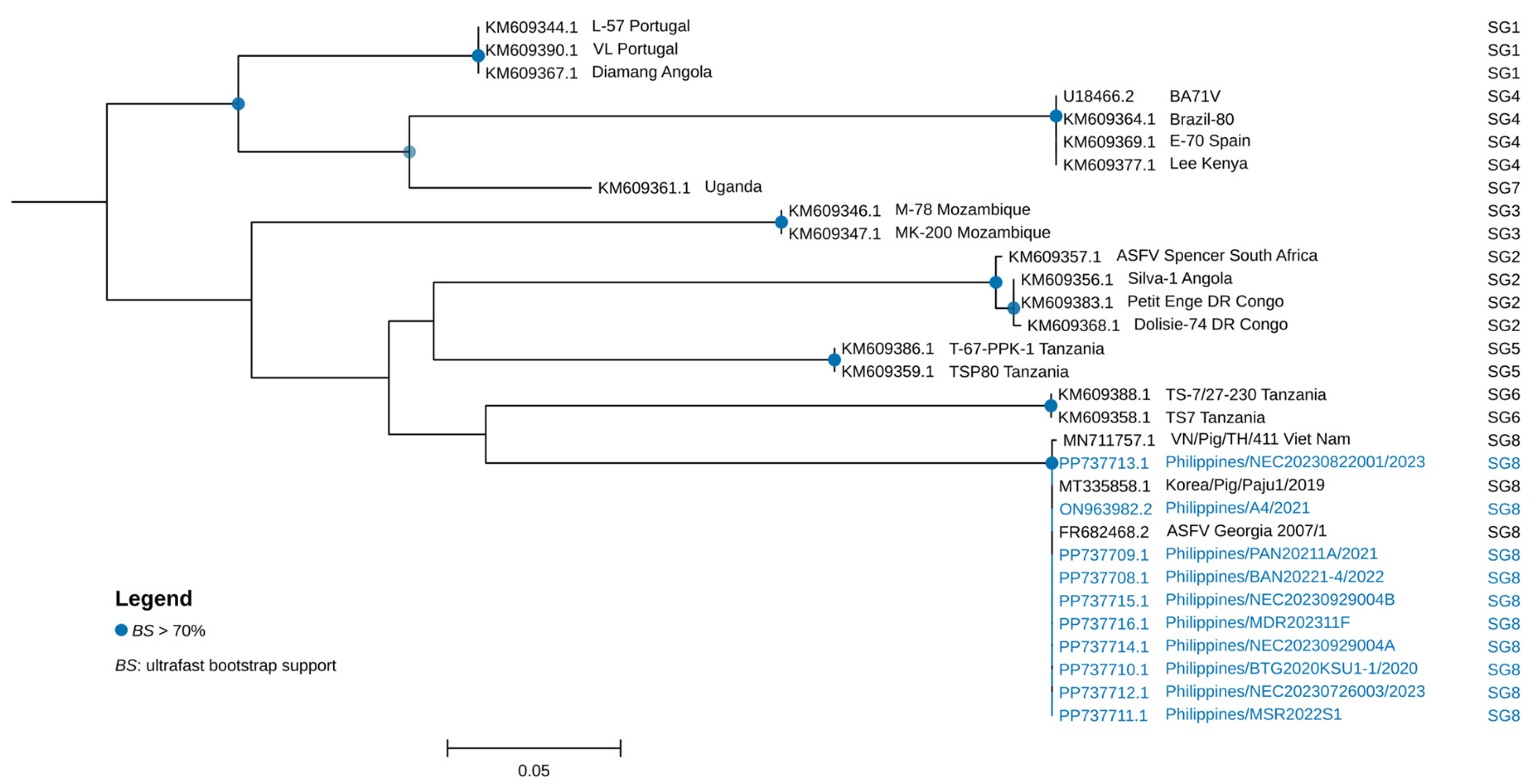
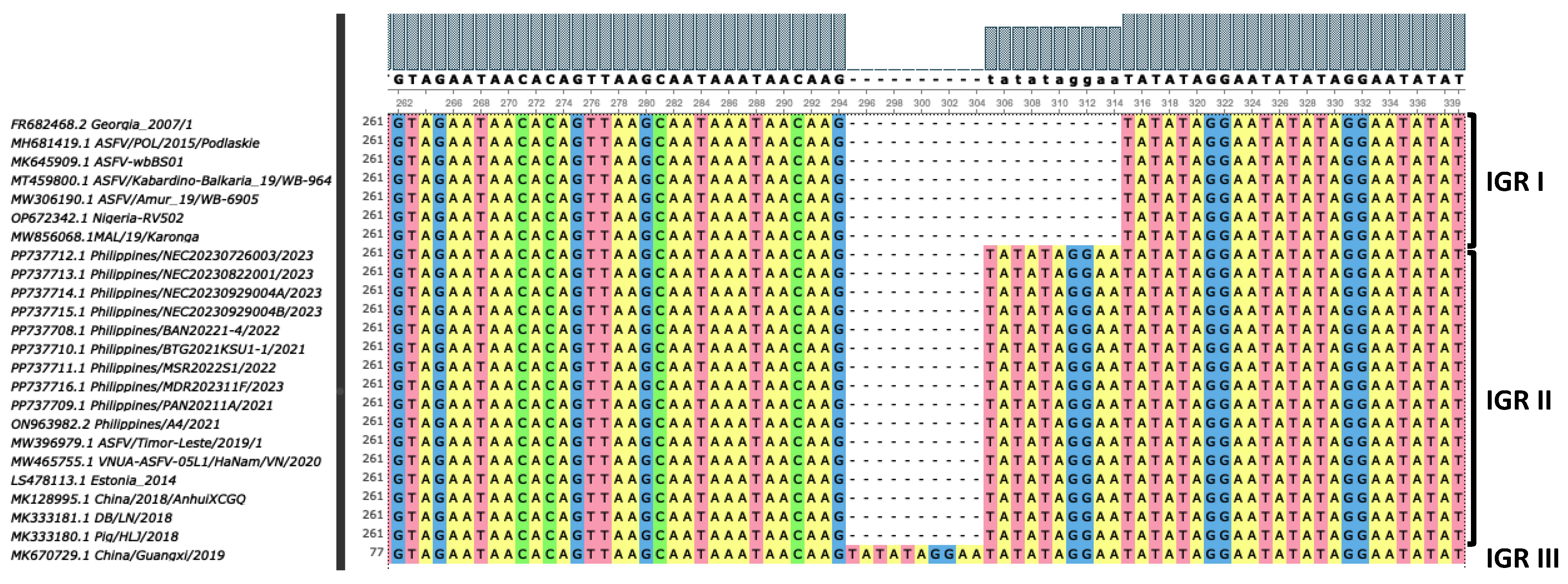
2.4. Genetic Characterization and Phylogenetic Analysis
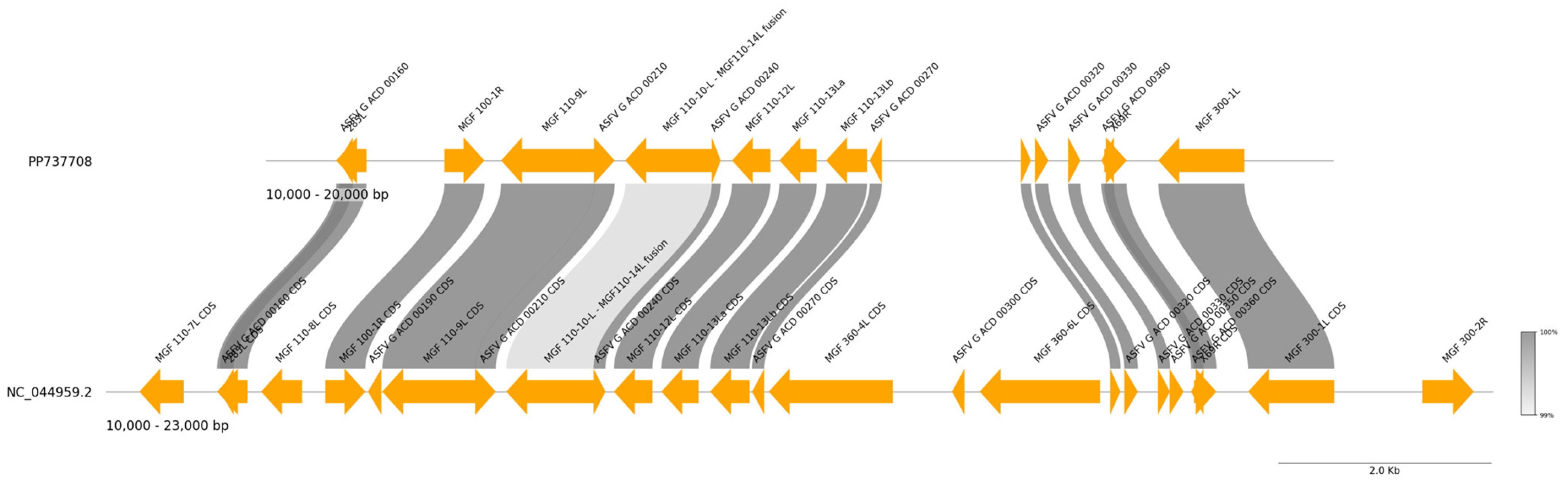
3. Results and Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lee, T.; Hansen, J. Southeast Asia’s Growing Meat Demand and Its Implications for Feedstuffs Imports. Amber Waves. United States Department of Agriculture, Economic Research Service, 1 April 2019. Available online: https://www.ers.usda.gov/amber-waves/2019/april/southeast-asia-s-growing-meat-demand-and-its-implications-for-feedstuffs-imports (accessed on 3 January 2025).
- Zsak, L.; Borca, M.; Risatti, G.; Zsak, A.; French, R.; Lu, Z.; Kutish, G.; Neilan, J.; Callahan, J.; Nelson, W. Preclinical Diagnosis of African Swine Fever in Contact-Exposed Swine by a Real-Time PCR Assay. J. Clin. Microbiol. 2005, 43, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Dixon, L.K.; Abrams, C.C.; Bowick, G.; Goatley, L.C.; Kay-Jackson, P.C.; Chapman, D.; Liverani, E.; Nix, R.; Silk, R.; Zhang, F. African Swine Fever Virus Proteins Involved in Evading Host Defence Systems. Vet. Immunol. Immunopathol. 2004, 100, 117–134. [Google Scholar] [CrossRef] [PubMed]
- Forth, J.H.; Forth, L.F.; King, J.; Groza, O.; Hübner, A.; Olesen, A.S.; Höper, D.; Dixon, L.K.; Netherton, C.L.; Rasmussen, T.B.; et al. A Deep-Sequencing Workflow for the Fast and Efficient Generation of High-Quality African Swine Fever Virus Whole-Genome Sequences. Viruses 2019, 11, 846. [Google Scholar] [CrossRef]
- de León, P.; Bustos, M.J.; Carrascosa, A.L. Laboratory Methods to Study African Swine Fever Virus. Virus Res. 2013, 173, 168–179. [Google Scholar] [CrossRef]
- Lu, G.; Pan, J.; Zhang, G. African Swine Fever Virus in Asia: Its Rapid Spread and Potential Threat to Unaffected Countries. J. Infect. 2020, 80, 350–371. [Google Scholar] [CrossRef]
- Blome, S.; Franzke, K.; Beer, M. African Swine Fever—A Review of Current Knowledge. Virus Res. 2020, 287, 198099. [Google Scholar] [CrossRef]
- Dixon, L.K.; Stahl, K.; Jori, F.; Vial, L.; Pfeiffer, D.U. African Swine Fever Epidemiology and Control. Annu. Rev. Anim. Biosci. 2020, 8, 221–246. [Google Scholar] [CrossRef]
- Costard, S.; Mur, L.; Lubroth, J.; Sanchez-Vizcaino, J.M.; Pfeiffer, D.U. Epidemiology of African Swine Fever Virus. Virus Res. 2013, 173, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Rowlands, R.J.; Michaud, V.; Heath, L.; Hutchings, G.; Oura, C.; Vosloo, W.; Dwarka, R.; Onashvili, T.; Albina, E.; Dixon, L.K. African Swine Fever Virus Isolate, Georgia, 2007. Emerg. Infect. Dis. 2008, 14, 1870–1874. [Google Scholar] [CrossRef]
- Ge, S.; Li, J.; Fan, X.; Liu, F.; Li, L.; Wang, Q.; Ren, W.; Bao, J.; Liu, C.; Wang, H.; et al. Molecular Characterization of African Swine Fever Virus, China, 2018. Emerg. Infect. Dis. 2018, 24, 2131–2133. [Google Scholar] [CrossRef]
- Ito, S.; Kawaguchi, N.; Bosch, J.; Aguilar-Vega, C.; Sánchez-Vizcaíno, J.M. What Can We Learn from the Five-Year African Swine Fever Epidemic in Asia? Front. Vet. Sci. 2023, 10, 1273417. [Google Scholar] [CrossRef]
- Kim, H.-J.; Cho, K.-H.; Ryu, J.-H.; Jang, M.-K.; Chae, H.-G.; Choi, J.-D.; Nah, J.-J.; Kim, Y.-J.; Kang, H.-E. Isolation and Genetic Characterization of African Swine Fever Virus from Domestic Pig Farms in South Korea, 2019. Viruses 2020, 12, 1237. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.-H.; Yoo, D.-S.; Hong, S.-K.; Kim, D.-Y.; Jang, M.-K.; Kang, H.-E.; Kim, Y.-H. Genetic Profile of African Swine Fever Viruses Circulating at Pig Farms in South Korea during the Outbreaks between 2022 and April 2023. Viruses 2023, 15, 1552. [Google Scholar] [CrossRef]
- Kwon, O.-K.; Kim, D.-W.; Heo, J.-H.; Kim, J.-Y.; Nah, J.-J.; Choi, J.-D.; Lee, D.-W.; Cho, K.-H.; Hong, S.-K.; Kim, Y.-H.; et al. Genomic Epidemiology of African Swine Fever Virus Identified in Domestic Pig Farms in South Korea during 2019–2021. Transbound. Emerg. Dis. 2024, 2024, 9077791. [Google Scholar] [CrossRef]
- Fernandez-Colorado, C.P.; Kim, W.H.; Flores, R.A.; Min, W. African Swine Fever in the Philippines: A Review on Surveillance, Prevention, and Control Strategies. Animals 2024, 14, 1816. [Google Scholar] [CrossRef]
- Hsu, C.-H.; Montenegro, M.; Perez, A. Space–Time Dynamics of African Swine Fever Spread in the Philippines. Microorganisms 2023, 11, 1492. [Google Scholar] [CrossRef]
- Mazur-Panasiuk, N.; Woźniakowski, G.; Niemczuk, K. The First Complete Genomic Sequences of African Swine Fever Virus Isolated in Poland. Sci. Rep. 2019, 9, 4556. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Sun, Y.; Qiu, H.-J. African Swine Fever: An Unprecedented Disaster and Challenge to China. Infect. Dis. Poverty 2018, 7, 111. [Google Scholar] [CrossRef]
- Montecillo, A.D.; Baybay, Z.; Cabug, R.C.; Cariaso, W.; Jose, J.P.; Mantaring, S.; Briones, A.; Warr, A.; Burkard, C.; Villegas, L.; et al. Coding-Complete Genome Sequence of an African Swine Fever Virus from an Outbreak in 2021 among Domestic Pigs in Pangasinan, Philippines. Microbiol. Resour. Announc. 2022, 11, e0071922. [Google Scholar] [CrossRef]
- Warr, A.; Newman, C.; Craig, N.; Vendelė, I.; Pilare, R.; Cruz, L.C.; Barangan, T.G.; Morales, R.G.; Opriessnig, T.; Venturina, V.M.; et al. No Part Gets Left behind: Tiled Nanopore Sequencing of Whole ASFV Genomes Stitched Together Using Lilo. bioRxiv 2021, 1–35. [Google Scholar] [CrossRef]
- Mazur-Panasiuk, N.; Walczak, M.; Juszkiewicz, M.; Woźniakowski, G. The Spillover of African Swine Fever in Western Poland Revealed Its Estimated Origin on the Basis of O174L, K145R, MGF 505-5R and IGR I73R/I329L Genomic Sequences. Viruses 2020, 12, 1094. [Google Scholar] [CrossRef] [PubMed]
- Nix, R.J.; Gallardo, C.; Hutchings, G.; Blanco, E.; Dixon, L.K. Molecular Epidemiology of African Swine Fever Virus Studied by Analysis of Four Variable Genome Regions. Arch. Virol. 2006, 151, 2475–2494. [Google Scholar] [CrossRef] [PubMed]
- Gallardo, C.; Casado, N.; Soler, A.; Djadjovski, I.; Krivko, L.; Madueño, E.; Nieto, R.; Perez, C.; Simon, A.; Ivanova, E.; et al. A Multi Gene-Approach Genotyping Method Identifies 24 Genetic Clusters within the Genotype II-European African Swine Fever Viruses Circulating from 2007 to 2022. Front. Vet. Sci. 2023, 10, 1112850. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A Tool to Design Target-Specific Primers for Polymerase Chain Reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise Alignment for Nucleotide Sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Smolka, M.; Paulin, L.F.; Grochowski, C.M.; Horner, D.W.; Mahmoud, M.; Behera, S.; Kalef-Ezra, E.; Gandhi, M.; Hong, K.; Pehlivan, D.; et al. Detection of Mosaic and Population-Level Structural Variants with Sniffles2. Nat. Biotechnol. 2024, 42, 1571–1580. [Google Scholar] [CrossRef]
- Ji, D.; Aboukhalil, R.; Moshiri, N. ViralWasm: A Client-Side User-Friendly Web Application Suite for Viral Genomics. Bioinformatics 2024, 40, btae018. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A Flexible Suite of Utilities for Comparing Genomic Features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Tcherepanov, V.; Ehlers, A.; Upton, C. Genome Annotation Transfer Utility (GATU): Rapid Annotation of Viral Genomes Using a Closely Related Reference Genome. BMC Genom. 2006, 7, 150. [Google Scholar] [CrossRef]
- Gallardo, C.; Mwaengo, D.M.; Macharia, J.M.; Arias, M.; Taracha, E.A.; Soler, A.; Okoth, E.; Martín, E.; Kasiti, J.; Bishop, R.P. Enhanced Discrimination of African Swine Fever Virus Isolates through Nucleotide Sequencing of the P54, P72, and pB602L (CVR) Genes. Virus Genes 2009, 38, 85–95. [Google Scholar] [CrossRef]
- Gallardo, C.; Fernández-Pinero, J.; Pelayo, V.; Gazaev, I.; Markowska-Daniel, I.; Pridotkas, G.; Nieto, R.; Fernández-Pacheco, P.; Bokhan, S.; Nevolko, O.; et al. Genetic Variation among African Swine Fever Genotype II Viruses, Eastern and Central Europe. Emerg. Infect. Dis. 2014, 20, 1544–1547. [Google Scholar] [CrossRef] [PubMed]
- Tran, H.T.T.; Truong, A.D.; Dang, A.K.; Ly, D.V.; Chu, N.T.; Van Hoang, T.; Nguyen, H.T.; Netherton, C.L.; Dang, H.V. Novel Method for Sub-Grouping of Genotype II African Swine Fever Viruses Based on the Intergenic Region between the A179L and A137R Genes. Vet. Med. Sci. 2022, 8, 607–609. [Google Scholar] [CrossRef] [PubMed]
- Sanna, G.; Dei Giudici, S.; Bacciu, D.; Angioi, P.P.; Giammarioli, M.; De Mia, G.M.; Oggiano, A. Improved Strategy for Molecular Characterization of African Swine Fever Viruses from Sardinia, Based on Analysis of P30, CD2V and I73R/I329L Variable Regions. Transbound. Emerg. Dis. 2017, 64, 1280–1286. [Google Scholar] [CrossRef]
- Dinhobl, M.; Spinard, E.; Birtley, H.; Tesler, N.; Borca, M.V.; Gladue, D.P. African Swine Fever Virus P72 Genotyping Tool. Microbiol. Resour. Announc. 2024, 13, e0089123. [Google Scholar] [CrossRef] [PubMed]
- Dinhobl, M.; Spinard, E.; Tesler, N.; Birtley, H.; Signore, A.; Ambagala, A.; Masembe, C.; Borca, M.V.; Gladue, D.P. Reclassification of ASFV into 7 Biotypes Using Unsupervised Machine Learning. Viruses 2024, 16, 67. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A Novel Method for Rapid Multiple Sequence Alignment Based on Fast Fourier Transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Okonechnikov, K.; Golosova, O.; Fursov, M.; the UGENE team. Unipro UGENE: A Unified Bioinformatics Toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Bianchini, G.; Sánchez-Baracaldo, P. TreeViewer: Flexible, Modular Software to Visualise and Manipulate Phylogenetic Trees. Ecol. Evol. 2024, 14, e10873. [Google Scholar] [CrossRef]
- Garcia, G.; Munar, M.; Salinas, M.B.; Gregorio, G.; Mananggit, M. Molecular Characterization and Identification of African Swine Fever Virus Isolates from Emerging Cases of Infection in Central Luzon, Philippines. Vet. Integr. Sci. 2024, 22, 111–119. [Google Scholar] [CrossRef]
- Serdeña, A.P.R.; Bernardo, J.M.G.; Pangga, G.M.V.; Salamat, S.E.A.; Agulto, T.N.; Desamero, M.J.M.; Atienza, C.P.G.; Calumpang, G.J.A.; Canlas, R.M.P.; Castillo, M.S.M.; et al. Molecular Detection of African Swine Fever Virus in Pork and Pork Products and Associated Risk Factors in the Philippines. J. Vet. Med. Sci. 2025, 87, 13–27. [Google Scholar] [CrossRef]
- Nguyen, V.T.; Cho, K.-H.; Mai, N.T.A.; Park, J.-Y.; Trinh, T.B.N.; Jang, M.-K.; Nguyen, T.T.H.; Vu, X.D.; Nguyen, T.L.; Nguyen, V.D.; et al. Multiple Variants of African Swine Fever Virus Circulating in Vietnam. Arch. Virol. 2022, 167, 1137–1140. [Google Scholar] [CrossRef]
- Shi, K.; Liu, H.; Yin, Y.; Si, H.; Long, F.; Feng, S. Molecular Characterization of African Swine Fever Virus From 2019-2020 Outbreaks in Guangxi Province, Southern China. Front. Vet. Sci. 2022, 9, 912224. [Google Scholar] [CrossRef] [PubMed]
- Elsukova, A.; Shevchenki, I.; Varentsova, A.; Zinyakov, N.; Igolkin, A.; Vlasova, N. African Swine Fever (ASF), Intergenic Region, 9R/10R, NGS, Tandem Repeat Sequences in the Intergenic Region MGF 505 9R/10R Is a New Marker of the Genetic Variability among ASF Genotype II Viruses. In Proceedings of the EPIZONE, 10 th Annual Meeting 2016, Madrid, Spain, 27–29 April 2016; Volume 9, p. 78. [Google Scholar]
- Mazloum, A.; van Schalkwyk, A.; Chernyshev, R.; Igolkin, A.; Heath, L.; Sprygin, A. A Guide to Molecular Characterization of Genotype II African Swine Fever Virus: Essential and Alternative Genome Markers. Microorganisms 2023, 11, 642. [Google Scholar] [CrossRef] [PubMed]
- Afonso, C.L.; Piccone, M.E.; Zaffuto, K.M.; Neilan, J.; Kutish, G.F.; Lu, Z.; Balinsky, C.A.; Gibb, T.R.; Bean, T.J.; Zsak, L.; et al. African Swine Fever Virus Multigene Family 360 and 530 Genes Affect Host Interferon Response. J. Virol. 2004, 78, 1858–1864. [Google Scholar] [CrossRef]
- Zsak, L.; Lu, Z.; Burrage, T.G.; Neilan, J.G.; Kutish, G.F.; Moore, D.M.; Rock, D.L. African Swine Fever Virus Multigene Family 360 and 530 Genes Are Novel Macrophage Host Range Determinants. J. Virol. 2001, 75, 3066–3076. [Google Scholar] [CrossRef]
- Chapman, D.A.G.; Tcherepanov, V.; Upton, C.; Dixon, L.K. Comparison of the Genome Sequences of Non-Pathogenic and Pathogenic African Swine Fever Virus Isolates. J. Gen. Virol. 2008, 89, 397–408. [Google Scholar] [CrossRef]
- Igolkin, A.; Mazloum, A.; Zinyakov, N.; Chernyshev, R.; Schalkwyk, A.V.; Shotin, A.; Lavrentiev, I.; Gruzdev, K.; Chvala, I. Detection of the First Recombinant African Swine Fever Virus (Genotypes I and II) in Domestic Pigs in Russia. Mol. Biol. Rep. 2024, 51, 1011. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Sun, E.; Huang, L.; Ding, L.; Zhu, Y.; Zhang, J.; Shen, D.; Zhang, X.; Zhang, Z.; Ren, T.; et al. Highly Lethal Genotype I and II Recombinant African Swine Fever Viruses Detected in Pigs. Nat. Commun. 2023, 14, 3096. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Vu, T.T.H.; Yeom, M.; Nguyen, V.D.; Than, T.T.; Nguyen, V.T.; Jeong, D.G.; Ambagala, A.; Le, V.P.; Song, D. Molecular Characterization of Emerging Recombinant African Swine Fever Virus of Genotype I and II in Vietnam, 2023. Emerg. Microbes Infect. 2024, 13, 2404156. [Google Scholar] [CrossRef] [PubMed]
- Le, V.P.; Nguyen, V.T.; Le, T.B.; Mai, N.T.A.; Nguyen, V.D.; Than, T.T.; Lai, T.N.H.; Cho, K.H.; Hong, S.-K.; Kim, Y.H.; et al. Detection of Recombinant African Swine Fever Virus Strains of P72 Genotypes I and II in Domestic Pigs, Vietnam. Emerg. Infect. Dis. 2023, 30, 991–994. [Google Scholar] [CrossRef]
- Van Nguyen, D.; Thi Nguyen, N.; Van Nguyen, D.; Thi Bui, T.; Ngoc Tran, T. Genome Sequences of Recombinant African Swine Fever Virus Isolated from Domestic Pigs in Vietnam. Microbiol. Resour. Announc. 2024, 13, e0082124. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Province | Cq Value | Assembly Length (bp) | %GC | Mean Coverage | Predicted ORFs | NCBI Accession No. |
|---|---|---|---|---|---|---|---|
| ASFV Philippines/BAN20221-4/2022 | Bataan | 17.6 | 187,609 | 38.5 | 4729x | 183 | PP737708 |
| ASFV Philippines/PAN20211A/2021 | Pangasinan | 17.9 | 189,514 | 38.4 | 3183x | 187 | PP737709 |
| ASFV Philippines/BTG2021KSU1-1/2021 | Batangas | 20.2 | 189,540 | 38.4 | 4985x | 184 | PP737710 |
| ASFV Philippines/MSR2022S1/2022 | Misamis Oriental | 13 | 189,514 | 38.4 | 5576x | 175 | PP737711 |
| ASFV Philippines/NEC20230726003/2023 | Negros Occidental | 19.2 | 189,537 | 38.4 | 5092x | 188 | PP737712 |
| ASFV Philippines/NEC20230822001/2023 | Negros Occidental | 18.7 | 189,528 | 38.4 | 2673x | 188 | PP737713 |
| ASFV Philippines/NEC20230929004A/2023 | Negros Occidental | 19.9 | 189,539 | 38.4 | 2905x | 186 | PP737714 |
| ASFV Philippines/NEC20230929004B/2023 | Negros Occidental | 20.3 | 189,519 | 38.4 | 3283x | 187 | PP737715 |
| ASFV Philippines/MDR202311F/2023 | Mindoro Oriental | 19.1 | 189,501 | 38.4 | 1022x | 187 | PP737716 |
| ASFV Philippines/Pangasinan/A4/2021 | Pangasinan | 21 | 192,265 | 38.3 | 21x | 187 | ON963982.2 |
| Strain | p72 Genotype | CD2v Serogroup | CVR | IGRI73R-I329L | IGRA179L-A137R | IGRMGF 505 9R/10R | ECO2 | O174L | K145R | MGF 505-5R | Bt/Sj | CP204L | J268L |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Philippines/BAN20221-4/2022 | II | 8 | CVR1 | II | No deletion | MGF-1 | ECO2-I | I-with deletion | I | I | 100% | 100% | 100% |
| Philippines/PAN20211A/2021 | II | 8 | CVR1 | II | No deletion | MGF-1 | ECO2-I | I-with deletion | I | I | 100% | 100% | 100% |
| Philippines/BTG2021KSU1-1/2021 | II | 8 | CVR1 | II | No deletion | MGF-1 | ECO2-I | I-with deletion | I | I | 100% | One base substitution (A-to-G) | One base substitution (G-to-A) |
| Philippines/MSR2022S1/2022 | II | 8 | CVR1 | II | No deletion | MGF-1 | ECO2-I | I-with deletion | I | I | 100% | 100% | 100% |
| Philippines/NEC20230726003/2023 | II | 8 | CVR1 | II | No deletion; with SNPs | MGF-1 | ECO2-I | I-with deletion | I | I | 100% | 100% | 100% |
| Philippines/NEC20230822001/2023 | II | 8 | CVR1 | II | No deletion; with SNPs | MGF-1 | ECO2-I | I-with deletion | I | I | 100% | 100% | 100% |
| Philippines/NEC20230929004A/2023 | II | 8 | CVR1 | II | No deletion; with SNPs | MGF-1 | ECO2-I | I-with deletion | I | I | 100% | 100% | 100% |
| Philippines/NEC20230929004B/2023 | II | 8 | CVR1 | II | No deletion; with SNPs | MGF-1 | ECO2-I | I-with deletion | I | I | 100% | 100% | 100% |
| Philippines/MDR202311F/2023 | II | 8 | CVR1 | II | No deletion; with SNPs | MGF-1 | ECO2-I | I-with deletion | I | I | 100% | 100% | 100% |
| Philippines/A4/2021 | II | 8 | CVR1 | II | No deletion | MGF-1 | ECO2-I | I-with deletion | I | I | 100% | 100% | 100% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Montecillo, A.D.; Baybay, Z.K.; Ferrer, J.B.C.; Cariaso, W.; Pantua, A.; Jose, J.P.; Madera, R.; Shi, J.; Doysabas, K.C.; Dargantes, A.; et al. Genetic Profiles of Ten African Swine Fever Virus Strains from Outbreaks in Select Provinces of Luzon, Visayas, and Mindanao, Philippines, Between 2021 and 2023. Viruses 2025, 17, 588. https://doi.org/10.3390/v17040588
Montecillo AD, Baybay ZK, Ferrer JBC, Cariaso W, Pantua A, Jose JP, Madera R, Shi J, Doysabas KC, Dargantes A, et al. Genetic Profiles of Ten African Swine Fever Virus Strains from Outbreaks in Select Provinces of Luzon, Visayas, and Mindanao, Philippines, Between 2021 and 2023. Viruses. 2025; 17(4):588. https://doi.org/10.3390/v17040588
Chicago/Turabian StyleMontecillo, Andrew D., Zyne K. Baybay, Jimwel Bryan Christopher Ferrer, Wreahlen Cariaso, Airish Pantua, John Paulo Jose, Rachel Madera, Jishu Shi, Karla Cristine Doysabas, Alan Dargantes, and et al. 2025. "Genetic Profiles of Ten African Swine Fever Virus Strains from Outbreaks in Select Provinces of Luzon, Visayas, and Mindanao, Philippines, Between 2021 and 2023" Viruses 17, no. 4: 588. https://doi.org/10.3390/v17040588
APA StyleMontecillo, A. D., Baybay, Z. K., Ferrer, J. B. C., Cariaso, W., Pantua, A., Jose, J. P., Madera, R., Shi, J., Doysabas, K. C., Dargantes, A., Dargantes, K. A. T., Boongaling, A. R. A., Manglicmot, A. P., Villegas, L. C., & Pantua, H. D. (2025). Genetic Profiles of Ten African Swine Fever Virus Strains from Outbreaks in Select Provinces of Luzon, Visayas, and Mindanao, Philippines, Between 2021 and 2023. Viruses, 17(4), 588. https://doi.org/10.3390/v17040588