
Impact of SARS-CoV-2 Wuhan and Omicron Variant Proteins on Type I Interferon Response


Abstract
1. Introduction
2. Materials and Methods
2.1. Cells
2.2. Expression Constructs
2.3. Dual-Luciferase Reporter Assay
2.4. Cell Viability Assay
2.5. Production of Lentiviruses Encoding Single SARS-CoV-2 Viral Proteins
2.6. Transduction of HUVEC and IFN Stimulation
2.7. RT-qPCR
2.8. Figure Generation
3. Results
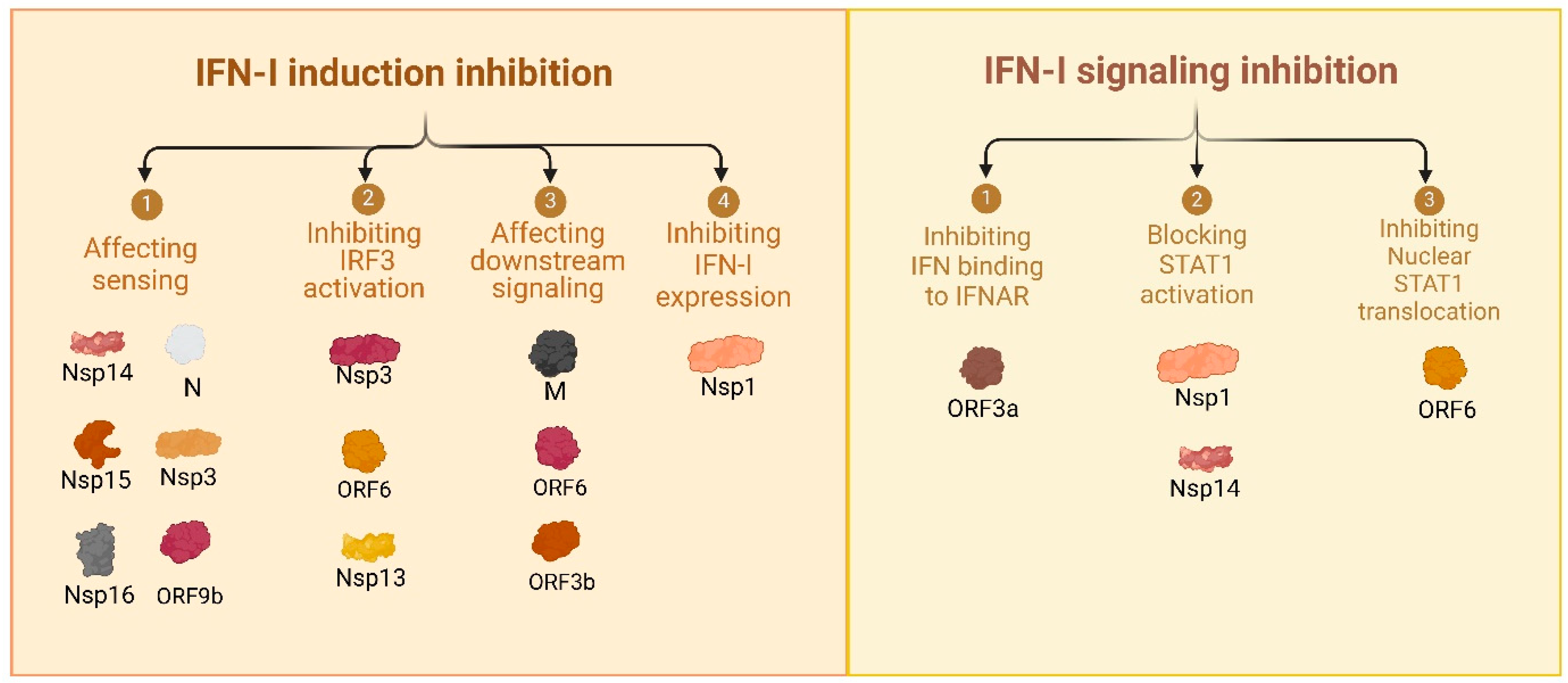
3.1. Comprehensive Review of SARS-CoV-2 Proteins and Their Immune-Modulatory Effects

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Effect on Host |
|---|---|
| Spike (S) | |
| Nucleocapsid (N) | |
| Membrane (M) | |
| Envelope (E) |
|
| Nsp1 | |
| Nsp2 |
|
| Nsp3 | |
| Nsp4 | |
| Nsp5 | |
| Nsp6 | |
| Nsp7 |
|
| Nsp8 | |
| Nsp9 |
|
| Nsp10 |
|
| Nsp11 |
|
| Nsp12 | |
| Nsp13 |
|
| Nsp14 | |
| Nsp15 |
|
| Nsp16 |
|
| ORF3a | |
| ORF6 | |
| ORF7a |
|
| ORF7b | |
| ORF8 |
|
| ORF9b |
|
| ORF10 |
| Strategy | Mechanism of Action |
|---|---|
| Hijacking protein synthesis machinery |
|
| Protecting viral mRNA |
|
| Protecting viral proteins |
|
| Safe Release of virions |
|
| Immune modulation |
|
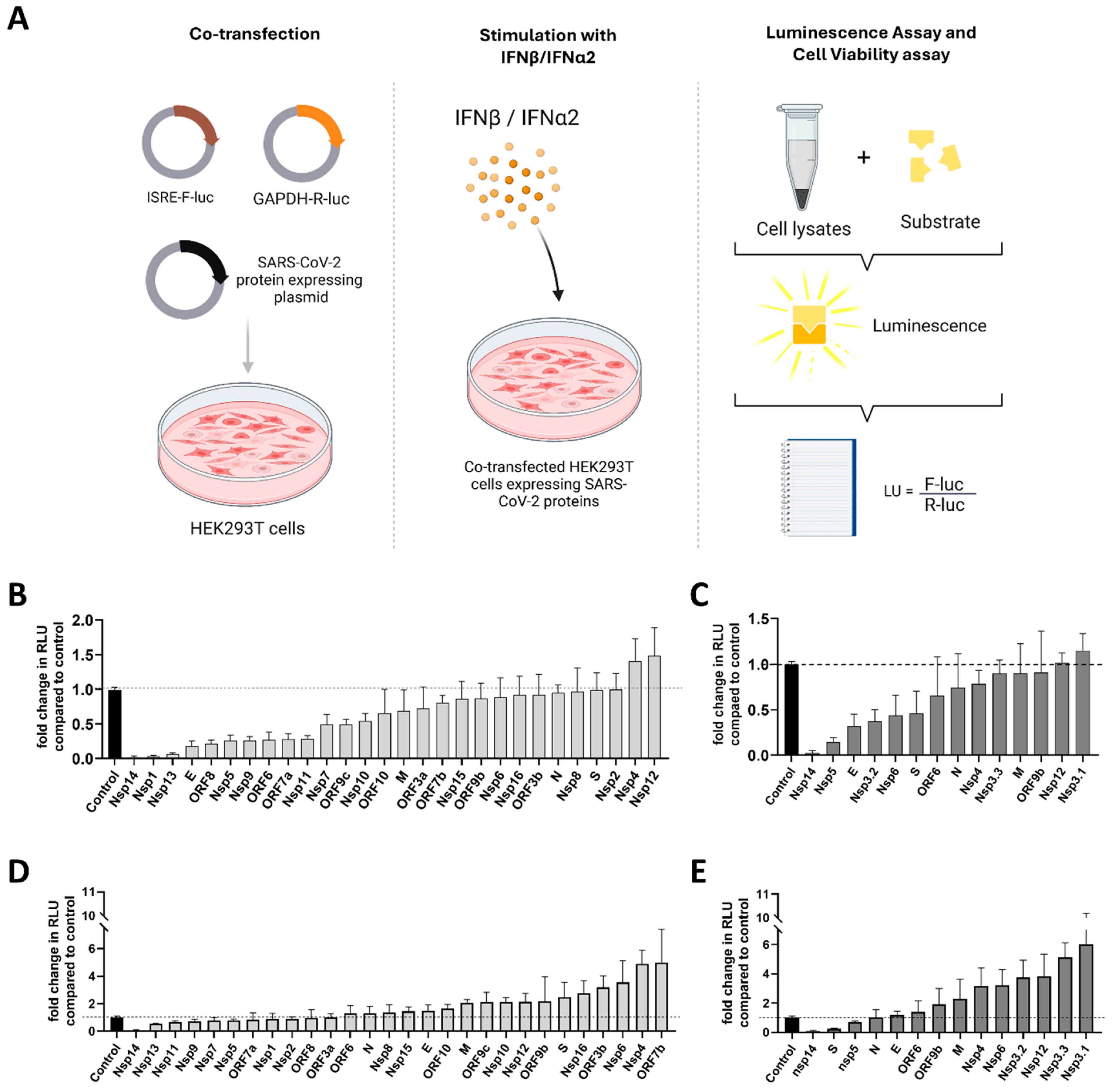
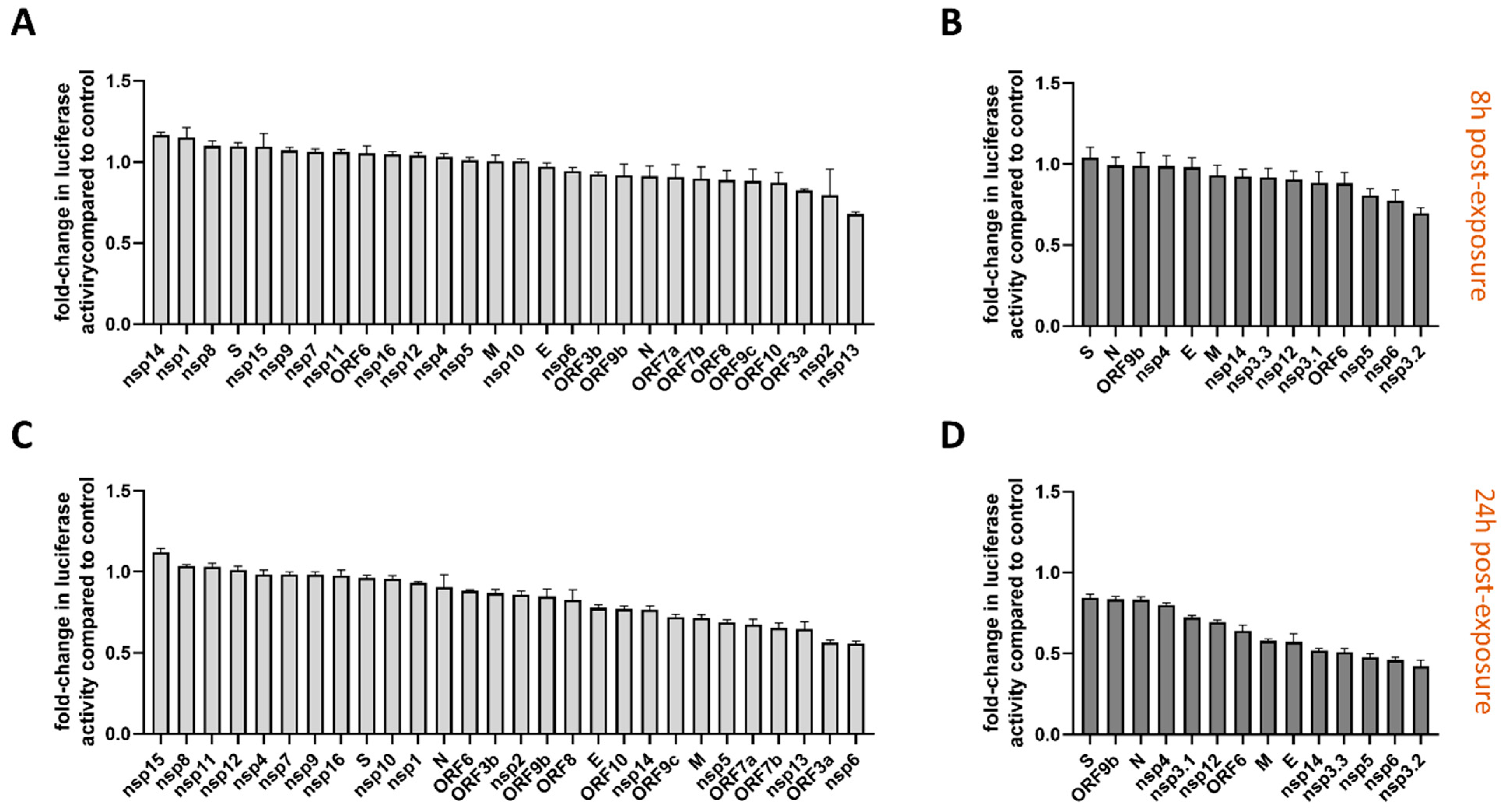
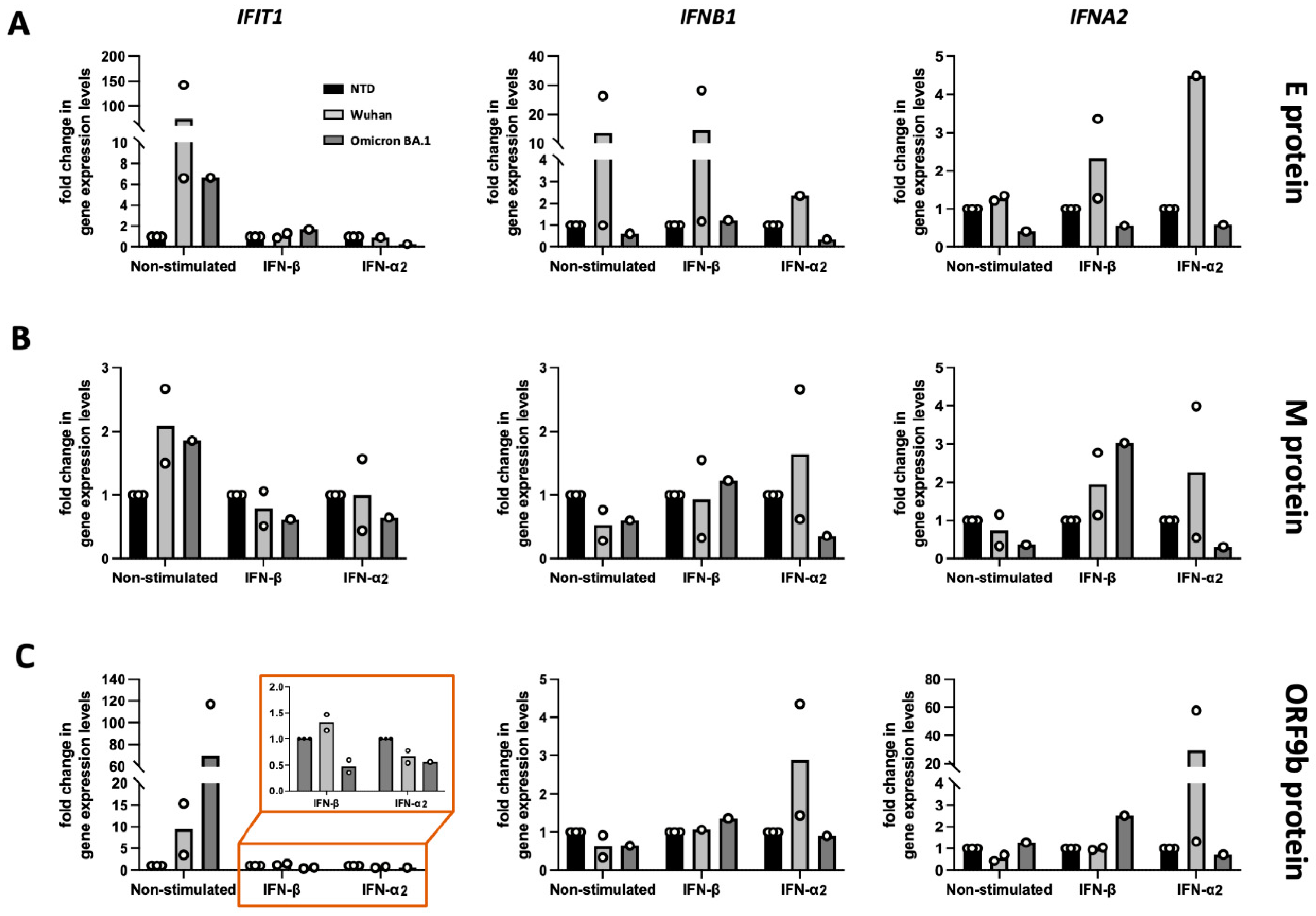
3.2. Functional Screening of SARS-CoV-2 Wuhan and Omicron Strain Proteins for Impact on Innate Immune Sensing
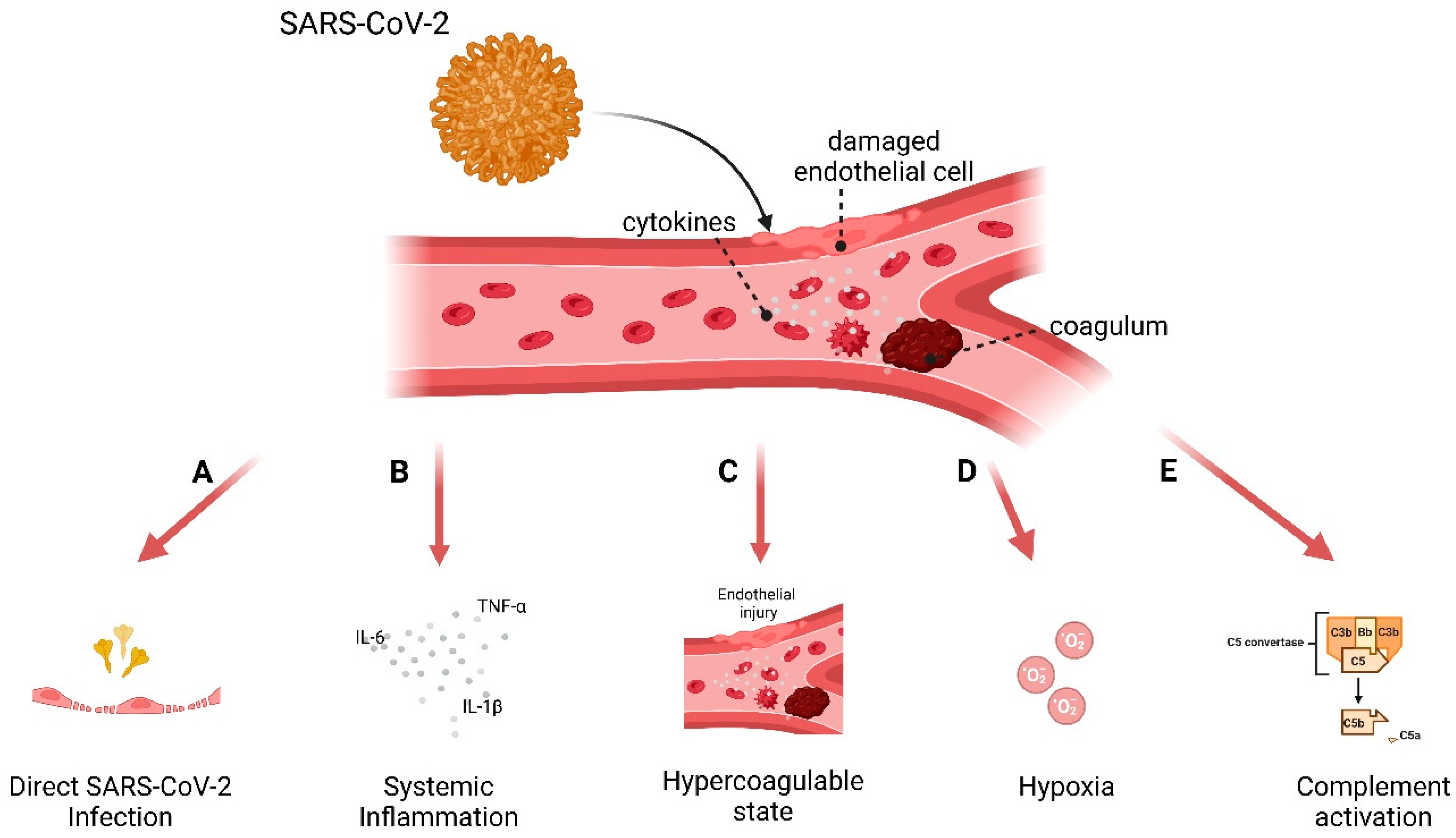
3.3. Model to Study Vascular Impact: Immune Response in Endothelial Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kirtipal, N.; Bharadwaj, S.; Kang, S.G. From SARS to SARS-CoV-2, Insights on Structure, Pathogenicity and Immunity Aspects of Pandemic Human Coronaviruses. Infect. Genet. Evol. 2020, 85, 104502. [Google Scholar] [CrossRef] [PubMed]
- Barnes, C.O.; Jette, C.A.; Abernathy, M.E.; Dam, K.-M.A.; Esswein, S.R.; Gristick, H.B.; Malyutin, A.G.; Sharaf, N.G.; Huey-Tubman, K.E.; Lee, Y.E.; et al. SARS-CoV-2 Neutralizing Antibody Structures Inform Therapeutic Strategies. Nature 2020, 588, 682–687. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Su, B.; Guo, X.; Sun, W.; Deng, Y.; Bao, L.; Zhu, Q.; Zhang, X.; Zheng, Y.; Geng, C.; et al. Potent Neutralizing Antibodies against SARS-CoV-2 Identified by High-Throughput Single-Cell Sequencing of Convalescent Patients’ B Cells. Cell 2020, 182, 73–84.e16. [Google Scholar] [CrossRef]
- Klasse, P.; Moore, J.P. Antibodies to SARS-CoV-2 and Their Potential for Therapeutic Passive Immunization. eLife 2020, 9, e57877. [Google Scholar] [CrossRef]
- Del Sole, F.; Farcomeni, A.; Loffredo, L.; Carnevale, R.; Menichelli, D.; Vicario, T.; Pignatelli, P.; Pastori, D. Features of Severe COVID-19: A Systematic Review and Meta-analysis. Eur. J. Clin. Investig. 2020, 50, e13378. [Google Scholar] [CrossRef] [PubMed]
- Rossouw, T.M.; Anderson, R.; Manga, P.; Feldman, C. Emerging Role of Platelet-Endothelium Interactions in the Pathogenesis of Severe SARS-CoV-2 Infection-Associated Myocardial Injury. Front. Immunol. 2022, 13, 776861. [Google Scholar] [CrossRef]
- Sievers, B.L.; Cheng, M.T.K.; Csiba, K.; Meng, B.; Gupta, R.K. SARS-CoV-2 and Innate Immunity: The Good, the Bad, and the “Goldilocks”. Cell Mol. Immunol. 2023, 21, 171–183. [Google Scholar] [CrossRef]
- Onomoto, K.; Onoguchi, K.; Yoneyama, M. Regulation of RIG-I-like Receptor-Mediated Signaling: Interaction between Host and Viral Factors. Cell Mol. Immunol. 2021, 18, 539–555. [Google Scholar] [CrossRef]
- Alfaro, E.; Díaz-García, E.; García-Tovar, S.; Galera, R.; Casitas, R.; Torres-Vargas, M.; López-Fernández, C.; Añón, J.M.; García-Río, F.; Cubillos-Zapata, C. Endothelial Dysfunction and Persistent Inflammation in Severe Post-COVID-19 Patients: Implications for Gas Exchange. BMC Med. 2024, 22, 242. [Google Scholar] [CrossRef]
- Ackermann, M.; Kamp, J.C.; Werlein, C.; Walsh, C.L.; Stark, H.; Prade, V.; Surabattula, R.; Wagner, W.L.; Disney, C.; Bodey, A.J.; et al. The Fatal Trajectory of Pulmonary COVID-19 Is Driven by Lobular Ischemia and Fibrotic Remodelling. EBioMedicine 2022, 85, 104296. [Google Scholar] [CrossRef]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial Cell Infection and Endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef] [PubMed]
- Becker, R.C.; Tantry, U.S.; Khan, M.; Gurbel, P.A. The COVID-19 Thrombus: Distinguishing Pathological, Mechanistic, and Phenotypic Features and Management. J. Thromb. Thrombolysis 2024, 58, 15–49. [Google Scholar] [CrossRef]
- Bai, B.; Yang, Y.; Wang, Q.; Li, M.; Tian, C.; Liu, Y.; Aung, L.H.H.; Li, P.; Yu, T.; Chu, X. NLRP3 Inflammasome in Endothelial Dysfunction. Cell Death Dis. 2020, 11, 776. [Google Scholar] [CrossRef] [PubMed]
- Birnhuber, A.; Fließer, E.; Gorkiewicz, G.; Zacharias, M.; Seeliger, B.; David, S.; Welte, T.; Schmidt, J.; Olschewski, H.; Wygrecka, M.; et al. Between Inflammation and Thrombosis: Endothelial Cells in COVID-19. Eur. Respir. J. 2021, 58, 2100377. [Google Scholar] [CrossRef]
- Guney, C.; Akar, F. Epithelial and Endothelial Expressions of ACE2: SARS-CoV-2 Entry Routes. J. Pharm. Pharm. Sci. 2021, 24, 84–93. [Google Scholar] [CrossRef]
- Lei, Y.; Zhang, J.; Schiavon, C.R.; He, M.; Chen, L.; Shen, H.; Zhang, Y.; Yin, Q.; Cho, Y.; Andrade, L.; et al. SARS-CoV-2 Spike Protein Impairs Endothelial Function via Downregulation of ACE 2. Circ. Res. 2021, 128, 1323–1326. [Google Scholar] [CrossRef] [PubMed]
- Perico, L.; Morigi, M.; Pezzotta, A.; Locatelli, M.; Imberti, B.; Corna, D.; Cerullo, D.; Benigni, A.; Remuzzi, G. SARS-CoV-2 Spike Protein Induces Lung Endothelial Cell Dysfunction and Thrombo-Inflammation Depending on the C3a/C3a Receptor Signalling. Sci. Rep. 2023, 13, 11392. [Google Scholar] [CrossRef]
- Kang, S.; Tanaka, T.; Inoue, H.; Ono, C.; Hashimoto, S.; Kioi, Y.; Matsumoto, H.; Matsuura, H.; Matsubara, T.; Shimizu, K.; et al. IL-6 Trans-Signaling Induces Plasminogen Activator Inhibitor-1 from Vascular Endothelial Cells in Cytokine Release Syndrome. Proc. Natl. Acad. Sci. USA 2020, 117, 22351–22356. [Google Scholar] [CrossRef]
- Kang, S.; Kishimoto, T. Interplay between Interleukin-6 Signaling and the Vascular Endothelium in Cytokine Storms. Exp. Mol. Med. 2021, 53, 1116–1123. [Google Scholar] [CrossRef]
- Puhlmann, M.; Weinreich, D.M.; Farma, J.M.; Carroll, N.M.; Turner, E.M.; Alexander, H.R. Interleukin-1β Induced Vascular Permeability Is Dependent on Induction of Endothelial Tissue Factor (TF) Activity. J. Transl. Med. 2005, 3, 37. [Google Scholar] [CrossRef]
- Kandhaya-Pillai, R.; Yang, X.; Tchkonia, T.; Martin, G.M.; Kirkland, J.L.; Oshima, J. TNF-α/IFN-γ Synergy Amplifies Senescence-Associated Inflammation and SARS-CoV-2 Receptor Expression via Hyper-Activated JAK/STAT1. Aging Cell 2022, 21, e13646. [Google Scholar] [CrossRef] [PubMed]
- Valencia, I.; Lumpuy-Castillo, J.; Magalhaes, G.; Sánchez-Ferrer, C.F.; Lorenzo, Ó.; Peiró, C. Mechanisms of Endothelial Activation, Hypercoagulation and Thrombosis in COVID-19: A Link with Diabetes Mellitus. Cardiovasc. Diabetol. 2024, 23, 75. [Google Scholar] [CrossRef] [PubMed]
- Won, T.; Wood, M.K.; Hughes, D.M.; Talor, M.V.; Ma, Z.; Schneider, J.; Skinner, J.T.; Asady, B.; Goerlich, E.; Halushka, M.K.; et al. Endothelial Thrombomodulin Downregulation Caused by Hypoxia Contributes to Severe Infiltration and Coagulopathy in COVID-19 Patient Lungs. EBioMedicine 2022, 75, 103812. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Ji, W.; Yang, H.; Chen, S.; Zhang, W.; Duan, G. Endothelial Activation and Dysfunction in COVID-19: From Basic Mechanisms to Potential Therapeutic Approaches. Signal Transduct. Target. Ther. 2020, 5, 293. [Google Scholar] [CrossRef]
- Lim, E.H.T.; van Amstel, R.B.E.; de Boer, V.V.; van Vught, L.A.; de Bruin, S.; Brouwer, M.C.; Vlaar, A.P.J.; van de Beek, D. Complement Activation in COVID-19 and Targeted Therapeutic Options: A Scoping Review. Blood Rev. 2023, 57, 100995. [Google Scholar] [CrossRef]
- Conway, E.M.; Pryzdial, E.L.G. Is the COVID-19 Thrombotic Catastrophe Complement-connected? J. Thromb. Haemost. 2020, 18, 2812–2822. [Google Scholar] [CrossRef]
- Baudin, B.; Bruneel, A.; Bosselut, N.; Vaubourdolle, M. A Protocol for Isolation and Culture of Human Umbilical Vein Endothelial Cells. Nat. Protoc. 2007, 2, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 Protein Interaction Map Reveals Targets for Drug Repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef]
- Bouhaddou, M.; Memon, D.; Meyer, B.; White, K.M.; Rezelj, V.V.; Correa Marrero, M.; Polacco, B.J.; Melnyk, J.E.; Ulferts, S.; Kaake, R.M.; et al. The Global Phosphorylation Landscape of SARS-CoV-2 Infection. Cell 2020, 182, 685–712.e19. [Google Scholar] [CrossRef]
- Hirschenberger, M.; Hayn, M.; Laliberté, A.; Koepke, L.; Kirchhoff, F.; Sparrer, K.M.J. Luciferase Reporter Assays to Monitor Interferon Signaling Modulation by SARS-CoV-2 Proteins. STAR Protoc. 2021, 2, 100781. [Google Scholar] [CrossRef]
- Vermeire, J.; Naessens, E.; Vanderstraeten, H.; Landi, A.; Iannucci, V.; van Nuffel, A.; Taghon, T.; Pizzato, M.; Verhasselt, B. Quantification of Reverse Transcriptase Activity by Real-Time PCR as a Fast and Accurate Method for Titration of HIV, Lenti- and Retroviral Vectors. PLoS ONE 2012, 7, e50859. [Google Scholar] [CrossRef] [PubMed]
- Laha, S.; Chakraborty, J.; Das, S.; Manna, S.K.; Biswas, S.; Chatterjee, R. Characterizations of SARS-CoV-2 Mutational Profile, Spike Protein Stability and Viral Transmission. Infect. Genet. Evol. 2020, 85, 104445. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Allen, J.D.; Wrapp, D.; McLellan, J.S.; Crispin, M. Site-Specific Glycan Analysis of the SARS-CoV-2 Spike. Science 2020, 369, 330–333. [Google Scholar] [CrossRef]
- Gong, Y.; Qin, S.; Dai, L.; Tian, Z. The Glycosylation in SARS-CoV-2 and Its Receptor ACE2. Signal Transduct. Target. Ther. 2021, 6, 396. [Google Scholar] [CrossRef] [PubMed]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; Peacock, S.J.; et al. SARS-CoV-2 Variants, Spike Mutations and Immune Escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef]
- Montezano, A.C.; Camargo, L.L.; Mary, S.; Neves, K.B.; Rios, F.J.; Stein, R.; Lopes, R.A.; Beattie, W.; Thomson, J.; Herder, V.; et al. SARS-CoV-2 Spike Protein Induces Endothelial Inflammation via ACE2 Independently of Viral Replication. Sci. Rep. 2023, 13, 14086. [Google Scholar] [CrossRef]
- Urata, R.; Ikeda, K.; Yamazaki, E.; Ueno, D.; Katayama, A.; Shin-Ya, M.; Ohgitani, E.; Mazda, O.; Matoba, S. Senescent Endothelial Cells Are Predisposed to SARS-CoV-2 Infection and Subsequent Endothelial Dysfunction. Sci. Rep. 2022, 12, 11855. [Google Scholar] [CrossRef]
- Li, F.; Li, J.; Wang, P.H.; Yang, N.; Huang, J.; Ou, J.; Xu, T.; Zhao, X.; Liu, T.; Huang, X.; et al. SARS-CoV-2 Spike Promotes Inflammation and Apoptosis through Autophagy by ROS-Suppressed PI3K/AKT/MTOR Signaling. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166260. [Google Scholar] [CrossRef]
- Scheim, D.E.; Vottero, P.; Santin, A.D.; Hirsh, A.G. Sialylated Glycan Bindings from SARS-CoV-2 Spike Protein to Blood and Endothelial Cells Govern the Severe Morbidities of COVID-19. Int. J. Mol. Sci. 2023, 24, 17039. [Google Scholar] [CrossRef]
- Savellini, G.G.; Anichini, G.; Gandolfo, C.; Cusi, M.G. SARS-CoV-2 n Protein Targets TRIM25-Mediated RIG-I Activation to Suppress Innate Immunity. Viruses 2021, 13, 1439. [Google Scholar] [CrossRef]
- Oh, S.J.; Shin, O.S. Sars-Cov-2 Nucleocapsid Protein Targets Rig-i-like Receptor Pathways to Inhibit the Induction of Interferon Response. Cells 2021, 10, 530. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Chen, J.; Yu, X.; Lan, H.Y. Signaling Mechanisms of SARS-CoV-2 Nucleocapsid Protein in Viral Infection, Cell Death and Inflammation. Int. J. Biol. Sci. 2022, 18, 4704–4713. [Google Scholar] [CrossRef]
- Zhao, Y.; Sui, L.; Wu, P.; Wang, W.; Wang, Z.; Yu, Y.; Hou, Z.; Tan, G.; Liu, Q.; Wang, G. A Dual-Role of SARS-CoV-2 Nucleocapsid Protein in Regulating Innate Immune Response. Signal Transduct. Target. Ther. 2021, 6, 331. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Cao, Z.; Xie, X.; Zhang, X.; Chen, J.Y.C.; Wang, H.; Menachery, V.D.; Rajsbaum, R.; Shi, P.Y. Evasion of Type I Interferon by SARS-CoV-2. Cell Rep. 2020, 33, 108234. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Yin, Y.; Pan, P.; Huang, Y.; Chen, S.; Chen, J.; Wang, J.; Xu, G.; Tao, X.; Xiao, X.; et al. The Interaction between SARS-CoV-2 Nucleocapsid Protein and UBC9 Inhibits MAVS Ubiquitination by Enhancing Its SUMOylation. Viruses 2023, 15, 2304. [Google Scholar] [CrossRef]
- Guo, X.; Yang, S.; Cai, Z.; Zhu, S.; Wang, H.; Liu, Q.; Zhang, Z.; Feng, J.; Chen, X.; Li, Y.; et al. SARS-CoV-2 Specific Adaptations in N Protein Inhibit NF-ΚB Activation and Alter Pathogenesis. J. Cell Biol. 2025, 224, e202404131. [Google Scholar] [CrossRef]
- Carlson, C.R.; Asfaha, J.B.; Ghent, C.M.; Howard, C.J.; Hartooni, N.; Safari, M.; Frankel, A.D.; Morgan, D.O. Phosphoregulation of Phase Separation by the SARS-CoV-2 N Protein Suggests a Biophysical Basis for Its Dual Functions. Mol. Cell 2020, 80, 1092–1103.e4. [Google Scholar] [CrossRef]
- Fu, Y.Z.; Wang, S.Y.; Zheng, Z.Q.; Yi, H.; Li, W.W.; Xu, Z.S.; Wang, Y.Y. SARS-CoV-2 Membrane Glycoprotein M Antagonizes the MAVS-Mediated Innate Antiviral Response. Cell Mol. Immunol. 2021, 18, 613–620. [Google Scholar] [CrossRef]
- Lopandić, Z.; Protić-Rosić, I.; Todorović, A.; Glamočlija, S.; Gnjatović, M.; Ćujic, D.; Gavrović-Jankulović, M. Igm and Igg Immunoreactivity of Sars-Cov-2 Recombinant m Protein. Int. J. Mol. Sci. 2021, 22, 4951. [Google Scholar] [CrossRef]
- Liu, J.; Wu, S.; Zhang, Y.; Wang, C.; Liu, S.; Wan, J.; Yang, L. SARS-CoV-2 Viral Genes Nsp6, Nsp8, and M Compromise Cellular ATP Levels to Impair Survival and Function of Human Pluripotent Stem Cell-Derived Cardiomyocytes. Stem Cell Res. Ther. 2023, 14, 249. [Google Scholar] [CrossRef]
- Cao, Y.; Yang, R.; Lee, I.; Zhang, W.; Sun, J.; Wang, W.; Meng, X. Characterization of the SARS-CoV-2 E Protein: Sequence, Structure, Viroporin, and Inhibitors. Protein Sci. 2021, 30, 1114–1130. [Google Scholar] [CrossRef] [PubMed]
- Xia, B.; Shen, X.; He, Y.; Pan, X.; Liu, F.L.; Wang, Y.; Yang, F.; Fang, S.; Wu, Y.; Duan, Z.; et al. SARS-CoV-2 Envelope Protein Causes Acute Respiratory Distress Syndrome (ARDS)-like Pathological Damages and Constitutes an Antiviral Target. Cell Res. 2021, 31, 847–860. [Google Scholar] [CrossRef]
- Planès, R.; Bert, J.B.; Tairi, S.; Benmohamed, L.; Bahraoui, E. SARS-CoV-2 Envelope (E) Protein Binds and Activates TLR2 Pathway: A Novel Molecular Target for COVID-19 Interventions. Viruses 2022, 14, 999. [Google Scholar] [CrossRef] [PubMed]
- Geanes, E.S.; McLennan, R.; Pierce, S.H.; Menden, H.L.; Paul, O.; Sampath, V.; Bradley, T. SARS-CoV-2 Envelope Protein Regulates Innate Immune Tolerance. iScience 2024, 27, 109975. [Google Scholar] [CrossRef]
- Zheng, M.; Karki, R.; Williams, E.P.; Yang, D.; Fitzpatrick, E.; Vogel, P.; Jonsson, C.B.; Kanneganti, T.D. TLR2 Senses the SARS-CoV-2 Envelope Protein to Produce Inflammatory Cytokines. Nat. Immunol. 2021, 22, 829–838. [Google Scholar] [CrossRef]
- Lu, H.; Liu, Z.; Deng, X.; Chen, S.; Zhou, R.; Zhao, R.; Parandaman, R.; Thind, A.; Henley, J.; Tian, L.; et al. Potent NKT Cell Ligands Overcome SARS-CoV-2 Immune Evasion to Mitigate Viral Pathogenesis in Mouse Models. PLoS Pathog. 2023, 19, e1011240. [Google Scholar] [CrossRef]
- Bhat, S.; Rishi, P.; Chadha, V.D. Understanding the Epigenetic Mechanisms in SARS CoV-2 Infection and Potential Therapeutic Approaches. Virus Res. 2022, 318, 198853. [Google Scholar] [CrossRef] [PubMed]
- Schubert, K.; Karousis, E.D.; Jomaa, A.; Scaiola, A.; Echeverria, B.; Gurzeler, L.A.; Leibundgut, M.; Thiel, V.; Mühlemann, O.; Ban, N. SARS-CoV-2 Nsp1 Binds the Ribosomal MRNA Channel to Inhibit Translation. Nat. Struct. Mol. Biol. 2020, 27, 959–966. [Google Scholar] [CrossRef]
- Frolov, I.; Agback, T.; Palchevska, O.; Dominguez, F.; Lomzov, A.; Agback, P.; Frolova, E.I. All Domains of SARS-CoV-2 Nsp1 Determine Translational Shutoff and Cytotoxicity of the Protein. J. Virol. 2023, 97, e0186522. [Google Scholar] [CrossRef]
- Zhang, K.; Miorin, L.; Makio, T.; Dehghan, I.; Gao, S.; Xie, Y.; Zhong, H.; Esparza, M.; Kehrer, T.; Kumar, A.; et al. Nsp1 Protein of SARS-CoV-2 Disrupts the MRNA Export Machinery to Inhibit Host Gene Expression. Sci. Adv. 2021, 7, eabe7386. [Google Scholar] [CrossRef]
- Fisher, T.; Gluck, A.; Narayanan, K.; Kuroda, M.; Nachshon, A.; Hsu, J.C.; Halfmann, P.J.; Yahalom-Ronen, Y.; Tamir, H.; Finkel, Y.; et al. Parsing the Role of NSP1 in SARS-CoV-2 Infection. Cell Rep. 2022, 39, 110954. [Google Scholar] [CrossRef]
- Lokugamage, K.G.; Narayanan, K.; Huang, C.; Makino, S. Severe Acute Respiratory Syndrome Coronavirus Protein Nsp1 Is a Novel Eukaryotic Translation Inhibitor That Represses Multiple Steps of Translation Initiation. J. Virol. 2012, 86, 13598–13608. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, C.; Swanson, S.E.; Negatu, S.G.; Dittmar, M.; Miller, J.; Ramage, H.R.; Cherry, S.; Jurado, K.A. SARS-CoV-2 Viral Proteins NSP1 and NSP13 Inhibit Interferon Activation through Distinct Mechanisms. PLoS ONE 2021, 16, e0253089. [Google Scholar] [CrossRef] [PubMed]
- Lui, W.Y.; Ong, C.P.; Cheung, P.H.H.; Ye, Z.W.; Chan, C.P.; To, K.K.W.; Yuen, K.S.; Jin, D.Y. Nsp1 Facilitates SARS-CoV-2 Replication through Calcineurin-NFAT Signaling. mBio 2024, 15, e0039224. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Leong, M.W.; Rustagi, A.; Beck, A.; Zeng, L.; Holmes, S.; Qi, L.S.; Blish, C.A. SARS-CoV-2 Escapes Direct NK Cell Killing through Nsp1-Mediated Downregulation of Ligands for NKG2D. Cell Rep. 2022, 41, 111892. [Google Scholar] [CrossRef]
- Xu, Z.; Choi, J.-H.; Dai, D.L.; Luo, J.; Ladak, R.J.; Li, Q.; Wang, Y.; Zhang, C.; Wiebe, S.; Liu, A.C.H.; et al. SARS-CoV-2 Impairs Interferon Production via NSP2-Induced Repression of MRNA Translation. Proc. Natl. Acad. Sci. USA 2022, 119, e2204539119. [Google Scholar] [CrossRef]
- Zou, L.; Moch, C.; Graille, M.; Chapat, C. The SARS-CoV-2 Protein NSP2 Impairs the Silencing Capacity of the Human 4EHP-GIGYF2 Complex. iScience 2022, 25, 104646. [Google Scholar] [CrossRef]
- Lacasse, É.; Gudimard, L.; Dubuc, I.; Gravel, A.; Allaeys, I.; Boilard, É.; Flamand, L. SARS-CoV-2 Nsp2 Contributes to Inflammation by Activating NF-ΚB. Viruses 2023, 15, 334. [Google Scholar] [CrossRef]
- Khan, M.T.; Zeb, M.T.; Ahsan, H.; Ahmed, A.; Ali, A.; Akhtar, K.; Malik, S.I.; Cui, Z.; Ali, S.; Khan, A.S.; et al. SARS-CoV-2 Nucleocapsid and Nsp3 Binding: An in Silico Study. Arch. Microbiol. 2021, 203, 59–66. [Google Scholar] [CrossRef]
- Fu, Z.; Huang, B.; Tang, J.; Liu, S.; Liu, M.; Ye, Y.; Liu, Z.; Xiong, Y.; Zhu, W.; Cao, D.; et al. The Complex Structure of GRL0617 and SARS-CoV-2 PLpro Reveals a Hot Spot for Antiviral Drug Discovery. Nat. Commun. 2021, 12, 488. [Google Scholar] [CrossRef]
- Clemente, V.; D’Arcy, P.; Bazzaro, M. Deubiquitinating Enzymes in Coronaviruses and Possible Therapeutic Opportunities for COVID-19. Int. J. Mol. Sci. 2020, 21, 3492. [Google Scholar] [CrossRef] [PubMed]
- Russo, L.C.; Tomasin, R.; Matos, I.A.; Manucci, A.C.; Sowa, S.T.; Dale, K.; Caldecott, K.W.; Lehtiö, L.; Schechtman, D.; Meotti, F.C.; et al. The SARS-CoV-2 Nsp3 Macrodomain Reverses PARP9/DTX3L-Dependent ADP-Ribosylation Induced by Interferon Signaling. J. Biol. Chem. 2021, 297, 101041. [Google Scholar] [CrossRef] [PubMed]
- Garvanska, D.H.; Alvarado, R.E.; Mundt, F.O.; Lindqvist, R.; Duel, J.K.; Coscia, F.; Nilsson, E.; Lokugamage, K.; Johnson, B.A.; Plante, J.A.; et al. The NSP3 Protein of SARS-CoV-2 Binds Fragile X Mental Retardation Proteins to Disrupt UBAP2L Interactions. EMBO Rep. 2024, 25, 902–926. [Google Scholar] [CrossRef]
- Lavigne, M.; Helynck, O.; Rigolet, P.; Boudria-Souilah, R.; Nowakowski, M.; Baron, B.; Brülé, S.; Hoos, S.; Raynal, B.; Guittat, L.; et al. SARS-CoV-2 Nsp3 Unique Domain SUD Interacts with Guanine Quadruplexes and G4-Ligands Inhibit This Interaction. Nucleic Acids Res. 2021, 49, 7695–7712. [Google Scholar] [CrossRef] [PubMed]
- Faizan, M.I.; Chaudhuri, R.; Sagar, S.; Albogami, S.; Chaudhary, N.; Azmi, I.; Akhtar, A.; Ali, S.M.; Kumar, R.; Iqbal, J.; et al. NSP4 and ORF9b of SARS-CoV-2 Induce Pro-Inflammatory Mitochondrial DNA Release in Inner Membrane-Derived Vesicles. Cells 2022, 11, 2969. [Google Scholar] [CrossRef]
- Zimmermann, L.; Zhao, X.; Makroczyova, J.; Wachsmuth-Melm, M.; Prasad, V.; Hensel, Z.; Bartenschlager, R.; Chlanda, P. SARS-CoV-2 Nsp3 and Nsp4 Are Minimal Constituents of a Pore Spanning Replication Organelle. Nat. Commun. 2023, 14, 7894. [Google Scholar] [CrossRef]
- Li, W.; Qiao, J.; You, Q.; Zong, S.; Peng, Q.; Liu, Y.; Hu, S.; Liu, W.; Li, S.; Shu, X.; et al. SARS-CoV-2 Nsp5 Activates NF-ΚB Pathway by Upregulating SUMOylation of MAVS. Front. Immunol. 2021, 12, 750969. [Google Scholar] [CrossRef]
- Liu, Y.; Qin, C.; Rao, Y.; Ngo, C.; Feng, J.J.; Zhao, J.; Zhang, S.; Wang, T.-Y.; Carriere, E.; Savas, A.C.; et al. SARS-CoV-2 Nsp5 Demonstrates Two Distinct Mechanisms Targeting RIG-I and MAVS To Evade the Innate Immune Response. mBio 2021, 12, e0233521. [Google Scholar] [CrossRef]
- Zheng, Y.; Deng, J.; Han, L.; Zhuang, M.W.; Xu, Y.; Zhang, J.; Nan, M.L.; Xiao, Y.; Zhan, P.; Liu, X.; et al. SARS-CoV-2 NSP5 and N Protein Counteract the RIG-I Signaling Pathway by Suppressing the Formation of Stress Granules. Signal Transduct. Target. Ther. 2022, 7, 22. [Google Scholar] [CrossRef]
- Shemesh, M.; Aktepe, T.E.; Deerain, J.M.; McAuley, J.L.; Audsley, M.D.; David, C.T.; Purcell, D.F.J.; Urin, V.; Hartmann, R.; Moseley, G.W.; et al. SARS-CoV-2 Suppresses IFNβ Production Mediated by NSP1, 5, 6, 15, ORF6 and ORF7b but Does Not Suppress the Effects of Added Interferon. PLoS Pathog. 2021, 17, e1009800. [Google Scholar] [CrossRef]
- Chen, J.; Li, Z.; Guo, J.; Xu, S.; Zhou, J.; Chen, Q.; Tong, X.; Wang, D.; Peng, G.; Fang, L.; et al. SARS-CoV-2 Nsp5 Exhibits Stronger Catalytic Activity and Interferon Antagonism than Its SARS-CoV Ortholog. J. Virol. 2022, 96, e0003722. [Google Scholar] [CrossRef]
- Lu, J.L.; Zhou, X.L. SARS-CoV-2 Main Protease Nsp5 Cleaves and Inactivates Human TRNA Methyltransferase TRMT1. J. Mol. Cell Biol. 2023, 15, mjad024. [Google Scholar] [CrossRef] [PubMed]
- Ju, X.; Wang, Z.; Wang, P.; Ren, W.; Yu, Y.; Yu, Y.; Yuan, B.; Song, J.; Zhang, X.; Zhang, Y.; et al. SARS-CoV-2 Main Protease Cleaves MAGED2 to Antagonize Host Antiviral Defense. mBio 2023, 14, e0137323. [Google Scholar] [CrossRef] [PubMed]
- Naik, N.G.; Lee, S.-C.; Veronese, B.H.S.; Ma, Z.; Toth, Z. Interaction of HDAC2 with SARS-CoV-2 NSP5 and IRF3 Is Not Required for NSP5-Mediated Inhibition of Type I Interferon Signaling Pathway. Microbiol. Spectr. 2022, 10, e0232222. [Google Scholar] [CrossRef]
- Taefehshokr, N.; Lac, A.; Vrieze, A.M.; Dickson, B.H.; Guo, P.N.; Jung, C.; Blythe, E.N.; Fink, C.; Aktar, A.; Dikeakos, J.D.; et al. SARS-CoV-2 NSP5 Antagonizes MHC II Expression by Subverting Histone Deacetylase 2. J. Cell Sci. 2024, 137, jcs262172. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yu, Q.; Huang, R.; Chen, H.; Ren, H.; Ma, L.; He, Y.; Li, W. SARS-CoV-2 SUD2 and Nsp5 Conspire to Boost Apoptosis of Respiratory Epithelial Cells via an Augmented Interaction with the G-Quadruplex of BclII. mBio 2023, 14, e0335922. [Google Scholar] [CrossRef]
- Zhang, C.; Jiang, Q.; Liu, Z.; Li, N.; Hao, Z.; Song, G.; Li, D.; Chen, M.; Lin, L.; Liu, Y.; et al. SARS-CoV-2 NSP6 Reduces Autophagosome Size and Affects Viral Replication via Sigma-1 Receptor. J. Virol. 2024, 98, e0075424. [Google Scholar] [CrossRef]
- Jiao, P.; Fan, W.; Ma, X.; Lin, R.; Zhao, Y.; Li, Y.; Zhang, H.; Jia, X.; Bi, Y.; Feng, X.; et al. SARS-CoV-2 Nonstructural Protein 6 Triggers Endoplasmic Reticulum Stress-Induced Autophagy to Degrade STING1. Autophagy 2023, 19, 3113–3131. [Google Scholar] [CrossRef]
- Benvenuto, D.; Angeletti, S.; Giovanetti, M.; Bianchi, M.; Pascarella, S.; Cauda, R.; Ciccozzi, M.; Cassone, A. Evolutionary Analysis of SARS-CoV-2: How Mutation of Non-Structural Protein 6 (NSP6) Could Affect Viral Autophagy. J. Infect. 2020, 81, e24–e27. [Google Scholar] [CrossRef]
- Nishitsuji, H.; Iwahori, S.; Ohmori, M.; Shimotohno, K.; Murata, T. Ubiquitination of SARS-CoV-2 NSP6 and ORF7a Facilitates NF-ΚB Activation. mBio 2022, 13, e0097122. [Google Scholar] [CrossRef]
- Bills, C.J.; Xia, H.; Chen, J.Y.C.; Yeung, J.; Kalveram, B.K.; Walker, D.; Xie, X.; Shi, P.Y. Mutations in SARS-CoV-2 Variant Nsp6 Enhance Type-I Interferon Antagonism. Emerg. Microbes Infect. 2023, 12, 2209208. [Google Scholar] [CrossRef]
- Sun, X.; Liu, Y.; Huang, Z.; Xu, W.; Hu, W.; Yi, L.; Liu, Z.; Chan, H.; Zeng, J.; Liu, X.; et al. SARS-CoV-2 Non-Structural Protein 6 Triggers NLRP3-Dependent Pyroptosis by Targeting ATP6AP1. Cell Death Differ. 2022, 29, 1240–1254. [Google Scholar] [CrossRef]
- Zhu, J.; Wang, G.; Huang, X.; Lee, H.; Lee, J.G.; Yang, P.; van de Leemput, J.; Huang, W.; Kane, M.A.; Yang, P.; et al. SARS-CoV-2 Nsp6 Damages Drosophila Heart and Mouse Cardiomyocytes through MGA/MAX Complex-Mediated Increased Glycolysis. Commun. Biol. 2022, 5, 1039. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Zheng, Y.; Zheng, S.N.; Nan, M.L.; Han, L.; Zhang, J.; Jin, Y.; Pan, J.A.; Gao, C.; Wang, P.H. SARS-CoV-2 NSP7 Inhibits Type I and III IFN Production by Targeting the RIG-I/MDA5, TRIF, and STING Signaling Pathways. J. Med. Virol. 2023, 95, e28561. [Google Scholar] [CrossRef] [PubMed]
- Ghelichkhani, F.; Gonzalez, F.A.; Kapitonova, M.A.; Rozovsky, S. Selenoprotein S Interacts with the Replication and Transcription Complex of SARS-CoV-2 by Binding Nsp7. J. Mol. Biol. 2023, 435, 168008. [Google Scholar] [CrossRef] [PubMed]
- Miah, S.M.S.; Lelias, S.; Gutierrez, A.H.; McAllister, M.; Boyle, C.M.; Moise, L.; De Groot, A.S. A SARS-CoV-2 NSP7 Homolog of a Treg Epitope Suppresses CD4+ and CD8+ T Cell Memory Responses. Front. Immunol. 2023, 14, 1290688. [Google Scholar] [CrossRef]
- Guo, J.; Li, W.L.; Huang, M.; Qiao, J.; Wan, P.; Yao, Y.; Ye, L.; Ding, Y.; Wang, J.; Peng, Q.; et al. SARS-CoV-2 Nsp7 Plays a Role in Cognitive Dysfunction by Impairing Synaptic Plasticity. Front. Neurosci. 2024, 18, 1490099. [Google Scholar] [CrossRef]
- Yang, Z.; Zhang, X.; Wang, F.; Wang, P.; Kuang, E.; Li, X. Suppression of MDA5-Mediated Antiviral Immune Responses by NSP8 of SARS-CoV-2 2020. BioRxiv 2020. [Google Scholar] [CrossRef]
- Zhang, X.; Yang, Z.; Pan, T.; Sun, Q.; Chen, Q.; Wang, P.H.; Li, X.; Kuang, E. SARS-CoV-2 Nsp8 Suppresses MDA5 Antiviral Immune Responses by Impairing TRIM4-Mediated K63-Linked Polyubiquitination. PLoS Pathog. 2023, 19, e1011792. [Google Scholar] [CrossRef]
- Zong, S.; Wu, Y.; Li, W.; You, Q.; Peng, Q.; Wang, C.; Wan, P.; Bai, T.; Ma, Y.; Sun, B.; et al. SARS-CoV-2 Nsp8 Induces Mitophagy by Damaging Mitochondria. Virol. Sin. 2023, 38, 520–530. [Google Scholar] [CrossRef]
- Makiyama, K.; Hazawa, M.; Kobayashi, A.; Lim, K.; Voon, D.C.; Wong, R.W. NSP9 of SARS-CoV-2 Attenuates Nuclear Transport by Hampering Nucleoporin 62 Dynamics and Functions in Host Cells. Biochem. Biophys. Res. Commun. 2022, 586, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xin, B.; Liu, Y.; Jiang, W.; Han, W.; Deng, J.; Wang, P.; Hong, X.; Yan, D. SARS-COV-2 Protein NSP9 Promotes Cytokine Production by Targeting TBK1. Front. Immunol. 2023, 14, 1211816. [Google Scholar] [CrossRef] [PubMed]
- Lundrigan, E.; Toudic, C.; Pennock, E.; Pezacki, J.P. SARS-CoV-2 Protein Nsp9 Is Involved in Viral Evasion through Interactions with Innate Immune Pathways. ACS Omega 2024, 9, 26428–26438. [Google Scholar] [CrossRef] [PubMed]
- Benoni, R.; Krafcikova, P.; Baranowski, M.R.; Kowalska, J.; Boura, E.; Cahová, H. Substrate Specificity of Sars-Cov-2 Nsp10-Nsp16 Methyltransferase. Viruses 2021, 13, 1722. [Google Scholar] [CrossRef]
- Wang, H.; Rizvi, S.R.; Dong, D.; Lou, J.; Wang, Q.; Sopipong, W.; Su, Y.; Najar, F.; Agarwal, P.K.; Kozielski, F.; et al. Emerging Variants of SARS-CoV-2 NSP10 Highlight Strong Functional Conservation of Its Binding to Two Non-Structural Proteins, NSP14 and NSP16. eLife 2023, 12, e87884. [Google Scholar] [CrossRef]
- Yang, L.; Zeng, X.T.; Luo, R.H.; Ren, S.X.; Liang, L.L.; Huang, Q.X.; Tang, Y.; Fan, H.; Ren, H.Y.; Zhang, W.J.; et al. SARS-CoV-2 NSP12 Utilizes Various Host Splicing Factors for Replication and Splicing Regulation. J. Med. Virol. 2024, 96, e29396. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhou, Z.; Xiao, X.; Tian, Z.; Dong, X.; Wang, C.; Li, L.; Ren, L.; Lei, X.; Xiang, Z.; et al. SARS-CoV-2 Nsp12 Attenuates Type I Interferon Production by Inhibiting IRF3 Nuclear Translocation. Cell Mol. Immunol. 2021, 18, 945–953. [Google Scholar] [CrossRef]
- Feng, K.; Zhang, H.J.; Min, Y.Q.; Zhou, M.; Deng, F.; Wang, H.L.; Li, P.Q.; Ning, Y.J. SARS-CoV-2 NSP13 Interacts with Host IRF3, Blocking Antiviral Immune Responses. J. Med. Virol. 2023, 95, e28881. [Google Scholar] [CrossRef]
- Fung, S.Y.; Siu, K.L.; Lin, H.; Chan, C.P.; Yeung, M.L.; Jin, D.Y. SARS-CoV-2 NSP13 Helicase Suppresses Interferon Signaling by Perturbing JAK1 Phosphorylation of STAT1. Cell Biosci. 2022, 12, 36. [Google Scholar] [CrossRef]
- Yuen, C.K.; Lam, J.Y.; Wong, W.M.; Mak, L.F.; Wang, X.; Chu, H.; Cai, J.P.; Jin, D.Y.; To, K.K.W.; Chan, J.F.W.; et al. SARS-CoV-2 Nsp13, Nsp14, Nsp15 and Orf6 Function as Potent Interferon Antagonists. Emerg. Microbes Infect. 2020, 9, 1418–1428. [Google Scholar] [CrossRef]
- Li, T.W.; Kenney, A.D.; Park, J.G.; Fiches, G.N.; Liu, H.; Zhou, D.; Biswas, A.; Zhao, W.; Que, J.; Santoso, N.; et al. SARS-CoV-2 Nsp14 Protein Associates with IMPDH2 and Activates NF-ΚB Signaling. Front. Immunol. 2022, 13, 1007089. [Google Scholar] [CrossRef]
- Zaffagni, M.; Harris, J.M.; Patop, I.L.; Reddy Pamudurti, N.; Nguyen, S.; Kadener, S. SARS-CoV-2 Nsp14 Mediates the Effects of Viral Infection on the Host Cell Transcriptome. eLife 2022, 11, e71945. [Google Scholar] [CrossRef] [PubMed]
- Tofaute, M.J.; Weller, B.; Graß, C.; Halder, H.; Dohai, B.; Falter-Braun, P.; Krappmann, D. SARS-CoV-2 NSP14 MTase Activity Is Critical for Inducing Canonical NF-ΚB Activation. Biosci. Rep. 2024, 44, BSR20231418. [Google Scholar] [CrossRef]
- Moeller, N.H.; Passow, K.T.; Harki, D.A.; Aihara, H. SARS-CoV-2 Nsp14 Exoribonuclease Removes the Natural Antiviral 3′-Deoxy-3′,4′-Didehydro-Cytidine Nucleotide from RNA. Viruses 2022, 14, 1790. [Google Scholar] [CrossRef]
- Walter, M.; Chen, I.P.; Vallejo-Gracia, A.; Kim, I.-J.; Bielska, O.; Lam, V.L.; Hayashi, J.M.; Cruz, A.; Shah, S.; Soveg, F.W.; et al. SIRT5 Is a Proviral Factor That Interacts with SARS-CoV-2 Nsp14 Protein. PLoS Pathog. 2022, 18, e1010811. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Ji, L.; Chen, X.; He, Y.; Sun, Y.; Ji, L.; Zhang, T.; Shen, Q.; Wang, X.; Wang, Y.; et al. SARS-CoV-2 Nsp15 Suppresses Type I Interferon Production by Inhibiting IRF3 Phosphorylation and Nuclear Translocation. iScience 2023, 26, 107705. [Google Scholar] [CrossRef] [PubMed]
- Otter, C.J.; Bracci, N.; Parenti, N.A.; Ye, C.; Asthana, A.; Blomqvist, E.K.; Tan, L.H.; Pfannenstiel, J.J.; Jackson, N.; Fehr, A.R.; et al. SARS-CoV-2 Nsp15 Endoribonuclease Antagonizes DsRNA-Induced Antiviral Signaling. Proc. Natl. Acad. Sci. USA 2024, 121, e2320194121. [Google Scholar] [CrossRef]
- Wang, X.; Zhu, B. SARS-CoV-2 Nsp15 Preferentially Degrades AU-Rich DsRNA via Its DsRNA Nickase Activity. Nucleic Acids Res. 2024, 52, 5257–5272. [Google Scholar] [CrossRef]
- Vithani, N.; Ward, M.D.; Zimmerman, M.I.; Novak, B.; Borowsky, J.H.; Singh, S.; Bowman, G.R. SARS-CoV-2 Nsp16 Activation Mechanism and a Cryptic Pocket with Pan-Coronavirus Antiviral Potential. Biophys. J. 2021, 120, 2880–2889. [Google Scholar] [CrossRef]
- Russ, A.; Wittmann, S.; Tsukamoto, Y.; Herrmann, A.; Deutschmann, J.; Lagisquet, J.; Ensser, A.; Kato, H.; Gramberg, T. Nsp16 Shields SARS-CoV-2 from Efficient MDA5 Sensing and IFIT1-mediated Restriction. EMBO Rep. 2022, 23, e55648. [Google Scholar] [CrossRef]
- Xu, H.; Akinyemi, I.A.; Chitre, S.A.; Loeb, J.C.; Lednicky, J.A.; McIntosh, M.T.; Bhaduri-McIntosh, S. SARS-CoV-2 Viroporin Encoded by ORF3a Triggers the NLRP3 Inflammatory Pathway. Virology 2022, 568, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Nie, Y.; Mou, L.; Long, Q.; Deng, D.; Hu, R.; Cheng, J.; Wu, J. SARS-CoV-2 ORF3a Positively Regulates NF-ΚB Activity by Enhancing IKKβ-NEMO Interaction. Virus Res. 2023, 328, 199086. [Google Scholar] [CrossRef] [PubMed]
- Arshad, N.; Laurent-Rolle, M.; Ahmed, W.S.; Hsu, J.C.C.; Mitchell, S.M.; Pawlak, J.; Sengupta, D.; Biswas, K.H.; Cresswell, P. SARS-CoV-2 Accessory Proteins ORF7a and ORF3a Use Distinct Mechanisms to down-Regulate MHC-I Surface Expression. Proc. Natl. Acad. Sci. USA 2023, 120, e2208525120. [Google Scholar] [CrossRef]
- Ren, Y.; Shu, T.; Wu, D.; Mu, J.; Wang, C.; Huang, M.; Han, Y.; Zhang, X.Y.; Zhou, W.; Qiu, Y.; et al. The ORF3a Protein of SARS-CoV-2 Induces Apoptosis in Cells. Cell Mol. Immunol. 2020, 17, 881–883. [Google Scholar] [CrossRef] [PubMed]
- Stewart, H.; Palmulli, R.; Johansen, K.H.; McGovern, N.; Shehata, O.M.; Carnell, G.W.; Jackson, H.K.; Lee, J.S.; Brown, J.C.; Burgoyne, T.; et al. Tetherin Antagonism by SARS-CoV -2 ORF3a and Spike Protein Enhances Virus Release. EMBO Rep. 2023, 24, e57224. [Google Scholar] [CrossRef]
- Chen, D.; Zheng, Q.; Sun, L.; Ji, M.; Li, Y.; Deng, H.; Zhang, H. ORF3a of SARS-CoV-2 Promotes Lysosomal Exocytosis-Mediated Viral Egress. Dev. Cell 2021, 56, 3250–3263.e5. [Google Scholar] [CrossRef]
- Miao, G.; Zhao, H.; Li, Y.; Ji, M.; Chen, Y.; Shi, Y.; Bi, Y.; Wang, P.; Zhang, H. ORF3a of the COVID-19 Virus SARS-CoV-2 Blocks HOPS Complex-Mediated Assembly of the SNARE Complex Required for Autolysosome Formation. Dev. Cell 2021, 56, 427–442.e5. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, H.; Pei, R.; Mao, B.; Zhao, Z.; Li, H.; Lin, Y.; Lu, K. The SARS-CoV-2 Protein ORF3a Inhibits Fusion of Autophagosomes with Lysosomes. Cell Discov. 2021, 7, 31. [Google Scholar] [CrossRef]
- Walia, K.; Sharma, A.; Paul, S.; Chouhan, P.; Kumar, G.; Ringe, R.; Sharma, M.; Tuli, A. SARS-CoV-2 Virulence Factor ORF3a Blocks Lysosome Function by Modulating TBC1D5-Dependent Rab7 GTPase Cycle. Nat. Commun. 2024, 15, 2053. [Google Scholar] [CrossRef]
- Suleman, M.; Said, A.; Khan, H.; Rehman, S.U.; Alshammari, A.; Crovella, S.; Yassine, H.M. Mutational Analysis of SARS-CoV-2 ORF6-KPNA2 Binding Interface and Identification of Potent Small Molecule Inhibitors to Recuse the Host Immune System. Front. Immunol. 2024, 14, 1266776. [Google Scholar] [CrossRef]
- Addetia, A.; Lieberman, N.A.P.; Phung, Q.; Hsiang, T.-Y.; Xie, H.; Roychoudhury, P.; Shrestha, L.; Loprieno, M.A.; Huang, M.-L.; Gale, M.; et al. SARS-CoV-2 ORF6 Disrupts Bidirectional Nucleocytoplasmic Transport through Interactions with Rae1 and Nup98. mBio 2021, 12, e00065-21. [Google Scholar] [CrossRef] [PubMed]
- Miorin, L.; Kehrer, T.; Teresa Sanchez-Aparicio, M.; Zhang, K.; Cohen, P.; Patel, R.S.; Cupic, A.; Makio, T.; Mei, M.; Moreno, E.; et al. SARS-CoV-2 Orf6 Hijacks Nup98 to Block STAT Nuclear Import and Antagonize Interferon Signaling. Proc. Natl. Acad. Sci. USA 2020, 117, 28344–28354. [Google Scholar] [CrossRef]
- Kehrer, T.; Cupic, A.; Ye, C.; Yildiz, S.; Bouhaddou, M.; Crossland, N.A.; Barrall, E.A.; Cohen, P.; Tseng, A.; Çağatay, T.; et al. Impact of SARS-CoV-2 ORF6 and Its Variant Polymorphisms on Host Responses and Viral Pathogenesis. Cell Host Microbe 2023, 31, 1668–1684.e12. [Google Scholar] [CrossRef] [PubMed]
- Hall, R.; Guedan, A.; Yap, M.W.; Young, G.R.; Harvey, R.; Stoye, J.P.; Bishop, K.N. SARS-CoV-2 ORF6 Disrupts Innate Immune Signalling by Inhibiting Cellular MRNA Export. PLoS Pathog. 2022, 18, e1010349. [Google Scholar] [CrossRef]
- Miyamoto, Y.; Itoh, Y.; Suzuki, T.; Tanaka, T.; Sakai, Y.; Koido, M.; Hata, C.; Wang, C.X.; Otani, M.; Moriishi, K.; et al. SARS-CoV-2 ORF6 Disrupts Nucleocytoplasmic Trafficking to Advance Viral Replication. Commun. Biol. 2022, 5, 483. [Google Scholar] [CrossRef] [PubMed]
- Khatun, O.; Sharma, M.; Narayan, R.; Tripathi, S. SARS-CoV-2 ORF6 Protein Targets TRIM25 for Proteasomal Degradation to Diminish K63-Linked RIG-I Ubiquitination and Type-I Interferon Induction. Cell. Mol. Life Sci. 2023, 80, 364. [Google Scholar] [CrossRef]
- López-Ayllón, B.D.; de Lucas-Rius, A.; Mendoza-García, L.; García-García, T.; Fernández-Rodríguez, R.; Suárez-Cárdenas, J.M.; Santos, F.M.; Corrales, F.; Redondo, N.; Pedrucci, F.; et al. SARS-CoV-2 Accessory Proteins Involvement in Inflammatory and Profibrotic Processes through IL11 Signaling. Front. Immunol. 2023, 14, 1220306. [Google Scholar] [CrossRef]
- Cao, Z.; Xia, H.; Rajsbaum, R.; Xia, X.; Wang, H.; Shi, P.Y. Ubiquitination of SARS-CoV-2 ORF7a Promotes Antagonism of Interferon Response. Cell Mol. Immunol. 2021, 18, 746–748. [Google Scholar] [CrossRef]
- Liu, Z.; Fu, Y.; Huang, Y.; Zeng, F.; Rao, J.; Xiao, X.; Sun, X.; Jin, H.; Li, J.; Yang, J.; et al. Ubiquitination of SARS-CoV-2 ORF7a Prevents Cell Death Induced by Recruiting BclXL to Activate ER Stress. Microbiol. Spectr. 2022, 10, e0150922. [Google Scholar] [CrossRef]
- Hou, P.; Wang, X.; Wang, H.; Wang, T.; Yu, Z.; Xu, C.; Zhao, Y.; Wang, W.; Zhao, Y.; Chu, F.; et al. The ORF7a Protein of SARS-CoV-2 Initiates Autophagy and Limits Autophagosome-Lysosome Fusion via Degradation of SNAP29 to Promote Virus Replication. Autophagy 2023, 19, 551–569. [Google Scholar] [CrossRef]
- Timilsina, U.; Umthong, S.; Ivey, E.B.; Waxman, B.; Stavrou, S. SARS-CoV-2 ORF7a Potently Inhibits the Antiviral Effect of the Host Factor SERINC5. Nat. Commun. 2022, 13, 2935. [Google Scholar] [CrossRef]
- Yang, R.; Zhao, Q.; Rao, J.; Zeng, F.; Yuan, S.; Ji, M.; Sun, X.; Li, J.; Yang, J.; Cui, J.; et al. SARS-CoV-2 Accessory Protein ORF7b Mediates Tumor Necrosis Factor-α-Induced Apoptosis in Cells. Front. Microbiol. 2021, 12, 654709. [Google Scholar] [CrossRef]
- García-García, T.; Fernández-Rodríguez, R.; Redondo, N.; de Lucas-Rius, A.; Zaldívar-López, S.; López-Ayllón, B.D.; Suárez-Cárdenas, J.M.; Jiménez-Marín, Á.; Montoya, M.; Garrido, J.J. Impairment of Antiviral Immune Response and Disruption of Cellular Functions by SARS-CoV-2 ORF7a and ORF7b. iScience 2022, 25, 105444. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Fu, Y.; You, W.; Huang, C.; Zeng, F.; Gu, X.; Sun, X.; Li, J.; Zhang, Q.; Du, W.; et al. Inhibition of the RLR Signaling Pathway by SARS-CoV-2 ORF7b Is Mediated by MAVS and Abrogated by ORF7b-Homologous Interfering Peptide. J. Virol. 2024, 98, e0157323. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, Y.; Li, Y.; Huang, F.; Luo, B.; Yuan, Y.; Xia, B.; Ma, X.; Yang, T.; Yu, F.; et al. The ORF8 Protein of SARS-CoV-2 Mediates Immune Evasion through down-Regulating MHC-Ι. Proc. Natl. Acad. Sci. USA 2021, 118, e2024202118. [Google Scholar] [CrossRef]
- Beaudoin-Bussières, G.; Arduini, A.; Bourassa, C.; Medjahed, H.; Gendron-Lepage, G.; Richard, J.; Pan, Q.; Wang, Z.; Liang, C.; Finzi, A. SARS-CoV-2 Accessory Protein ORF8 Decreases Antibody-Dependent Cellular Cytotoxicity. Viruses 2022, 14, 1237. [Google Scholar] [CrossRef] [PubMed]
- Móvio, M.I.; de Almeida, G.W.C.; Martines, I.d.G.L.; de Lima, G.B.; Sasaki, S.D.; Kihara, A.H.; Poole, E.; Nevels, M.; da Silva, M.C.C. SARS-CoV-2 ORF8 as a Modulator of Cytokine Induction: Evidence and Search for Molecular Mechanisms. Viruses 2024, 16, 161. [Google Scholar] [CrossRef] [PubMed]
- Kumar, J.; Dhyani, S.; Kumar, P.; Sharma, N.R.; Ganguly, S. SARS-CoV-2–Encoded ORF8 Protein Possesses Complement Inhibitory Properties. J. Biol. Chem. 2023, 299, 102930. [Google Scholar] [CrossRef]
- Arduini, A.; Laprise, F.; Liang, C. SARS-CoV-2 ORF8: A Rapidly Evolving Immune and Viral Modulator in COVID-19. Viruses 2023, 15, 871. [Google Scholar] [CrossRef]
- Gao, X.; Zhu, K.; Qin, B.; Olieric, V.; Wang, M.; Cui, S. Crystal Structure of SARS-CoV-2 Orf9b in Complex with Human TOM70 Suggests Unusual Virus-Host Interactions. Nat. Commun. 2021, 12, 2843. [Google Scholar] [CrossRef]
- Wu, J.; Shi, Y.; Pan, X.; Wu, S.; Hou, R.; Zhang, Y.; Zhong, T.; Tang, H.; Du, W.; Wang, L.; et al. SARS-CoV-2 ORF9b Inhibits RIG-I-MAVS Antiviral Signaling by Interrupting K63-Linked Ubiquitination of NEMO. Cell Rep. 2021, 34, 108761. [Google Scholar] [CrossRef]
- Han, L.; Zhuang, M.W.; Deng, J.; Zheng, Y.; Zhang, J.; Nan, M.L.; Zhang, X.J.; Gao, C.; Wang, P.H. SARS-CoV-2 ORF9b Antagonizes Type I and III Interferons by Targeting Multiple Components of the RIG-I/MDA-5–MAVS, TLR3–TRIF, and CGAS–STING Signaling Pathways. J. Med. Virol. 2021, 93, 5376–5389. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Liu, Y.; Li, C.; Stine, L.D.; Wang, P.H.; Turnbull, M.W.; Wu, H.; Liu, Q. Ectopic Expression of SARS-CoV-2 S and ORF-9B Proteins Alters Metabolic Profiles and Impairs Contractile Function in Cardiomyocytes. Front. Cell Dev. Biol. 2023, 11, 1110271. [Google Scholar] [CrossRef]
- Homma, D.; Limlingan, S.J.M.; Saito, T.; Ando, K. SARS-CoV-2-Derived Protein Orf9b Enhances MARK2 Activity via Interaction with the Autoinhibitory KA1 Domain. FEBS Lett. 2024, 598, 2385–2393. [Google Scholar] [CrossRef] [PubMed]
- Sarvari, J.; Jalili, S.; Mohammad, S.; Hashemi, A. SARS-COV-2 ORF9b Dysregulate Fibrinogen and Albumin Genes in a Liver Cell Line. Rep. Biochem. Mol. Biol. 2024, 13, 51. [Google Scholar]
- Li, X.; Hou, P.; Ma, W.; Wang, X.; Wang, H.; Yu, Z.; Chang, H.; Wang, T.; Jin, S.; Wang, X.; et al. SARS-CoV-2 ORF10 Suppresses the Antiviral Innate Immune Response by Degrading MAVS through Mitophagy. Cell Mol. Immunol. 2022, 19, 67–78. [Google Scholar] [CrossRef]
- Han, L.; Zheng, Y.; Deng, J.; Nan, M.L.; Xiao, Y.; Zhuang, M.W.; Zhang, J.; Wang, W.; Gao, C.; Wang, P.H. SARS-CoV-2 ORF10 Antagonizes STING-Dependent Interferon Activation and Autophagy. J. Med. Virol. 2022, 94, 5174–5188. [Google Scholar] [CrossRef]
- Korneeva, N.; Khalil, M.I.; Ghosh, I.; Fan, R.; Arnold, T.; De Benedetti, A. SARS-CoV-2 Viral Protein Nsp2 Stimulates Translation under Normal and Hypoxic Conditions. Virol. J. 2023, 20, 55. [Google Scholar] [CrossRef]
- Thorne, L.G.; Bouhaddou, M.; Reuschl, A.K.; Zuliani-Alvarez, L.; Polacco, B.; Pelin, A.; Batra, J.; Whelan, M.V.X.; Hosmillo, M.; Fossati, A.; et al. Evolution of Enhanced Innate Immune Evasion by SARS-CoV-2. Nature 2022, 602, 487–495. [Google Scholar] [CrossRef]
- Takata, M.A.; Gonçalves-Carneiro, D.; Zang, T.M.; Soll, S.J.; York, A.; Blanco-Melo, D.; Bieniasz, P.D. CG Dinucleotide Suppression Enables Antiviral Defence Targeting Non-Self RNA. Nature 2017, 550, 124–127. [Google Scholar] [CrossRef]
- Malone, B.; Urakova, N.; Snijder, E.J.; Campbell, E.A. Structures and Functions of Coronavirus Replication–Transcription Complexes and Their Relevance for SARS-CoV-2 Drug Design. Nat. Rev. Mol. Cell Biol. 2022, 23, 21–39. [Google Scholar] [CrossRef]
- Hagelauer, E.; Lotke, R.; Kmiec, D.; Hu, D.; Hohner, M.; Stopper, S.; Nchioua, R.; Kirchhoff, F.; Sauter, D.; Schindler, M. Tetherin Restricts SARS-CoV-2 despite the Presence of Multiple Viral Antagonists. Viruses 2023, 15, 2364. [Google Scholar] [CrossRef] [PubMed]
- Bills, C.; Xie, X.; Shi, P.Y. The Multiple Roles of Nsp6 in the Molecular Pathogenesis of SARS-CoV-2. Antiviral Res. 2023, 213, 105590. [Google Scholar] [CrossRef] [PubMed]
- Krachmarova, E.; Petkov, P.; Lilkova, E.; Ilieva, N.; Rangelov, M.; Todorova, N.; Malinova, K.; Hristova, R.; Nacheva, G.; Gospodinov, A.; et al. Insights into the SARS-CoV-2 ORF6 Mechanism of Action. Int. J. Mol. Sci. 2023, 24, 11589. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Yang, L.; Han, Z.; Zhou, X.; Zhang, Z.; Sun, T.; Zheng, F.; Yang, J.; Guan, F.; Xie, J.; et al. SARS-CoV-2 Nucleocapsid Protein Enhances the Level of Mitochondrial Reactive Oxygen Species. J. Med. Virol. 2023, 95, e29270. [Google Scholar] [CrossRef]
- Kohyama, M.; Suzuki, T.; Nakai, W.; Ono, C.; Matsuoka, S.; Iwatani, K.; Liu, Y.; Sakai, Y.; Nakagawa, A.; Tomii, K.; et al. SARS-CoV-2 ORF8 Is a Viral Cytokine Regulating Immune Responses. Int. Immunol. 2022, 35, 43–52. [Google Scholar] [CrossRef]
- Fajgenbaum, D.C.; June, C.H. Cytokine Storm. N. Engl. J. Med. 2020, 383, 2255–2273. [Google Scholar] [CrossRef]
- Freda, C.T.; Yin, W.; Ghebrehiwet, B.; Rubenstein, D.A. SARS-CoV-2 Structural Proteins Exposure Alter Thrombotic and Inflammatory Responses in Human Endothelial Cells. Cell Mol. Bioeng. 2022, 15, 43–53. [Google Scholar] [CrossRef]
- Lei, X.; Dong, X.; Ma, R.; Wang, W.; Xiao, X.; Tian, Z.; Wang, C.; Wang, Y.; Li, L.; Ren, L.; et al. Activation and Evasion of Type I Interferon Responses by SARS-CoV-2. Nat. Commun. 2020, 11, 3810. [Google Scholar] [CrossRef]
- Zhou, S.; Lv, P.; Li, M.; Chen, Z.; Xin, H.; Reilly, S.; Zhang, X. SARS-CoV-2 E Protein: Pathogenesis and Potential Therapeutic Development. Biomed. Pharmacother. 2023, 159, 114242. [Google Scholar] [CrossRef]
- Bugatti, A.; Filippini, F.; Bardelli, M.; Zani, A.; Chiodelli, P.; Messali, S.; Caruso, A.; Caccuri, F. SARS-CoV-2 Infects Human ACE2-Negative Endothelial Cells through an Avβ3 Integrin-Mediated Endocytosis Even in the Presence of Vaccine-Elicited Neutralizing Antibodies. Viruses 2022, 14, 705. [Google Scholar] [CrossRef]
- Schimmel, L.; Chew, K.Y.; Stocks, C.J.; Yordanov, T.E.; Essebier, P.; Kulasinghe, A.; Monkman, J.; dos Santos Miggiolaro, A.F.R.; Cooper, C.; de Noronha, L.; et al. Endothelial Cells Are Not Productively Infected by SARS-CoV-2. Clin. Transl. Immunol. 2021, 10, e1350. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, C.G.K.; Miao, V.N.; Owings, A.H.; Navia, A.W.; Tang, Y.; Bromley, J.D.; Lotfy, P.; Sloan, M.; Laird, H.; Williams, H.B.; et al. Impaired Local Intrinsic Immunity to SARS-CoV-2 Infection in Severe COVID-19. Cell 2021, 184, 4713–4733.e22. [Google Scholar] [CrossRef]
- Abdelmoaty, M.M.; Yeapuri, P.; Machhi, J.; Olson, K.E.; Shahjin, F.; Kumar, V.; Zhou, Y.; Liang, J.; Pandey, K.; Acharya, A.; et al. Defining the Innate Immune Responses for SARS-CoV-2-Human Macrophage Interactions. Front. Immunol. 2021, 12, 741502. [Google Scholar] [CrossRef]
- Cai, C.; Pham, T.N.Q.; Adam, D.; Brochiero, E.; Cohen, É.A. Sensing of SARS-CoV-2-Infected Cells by Plasmacytoid Dendritic Cells Is Modulated via an Interplay between CD54/ICAM-1 and CD11a/LFA-1 αL Integrin. J. Virol. 2025, 99, e0123524. [Google Scholar] [CrossRef] [PubMed]
- Di Domizio, J.; Gulen, M.F.; Saidoune, F.; Thacker, V.V.; Yatim, A.; Sharma, K.; Nass, T.; Guenova, E.; Schaller, M.; Conrad, C.; et al. The CGAS–STING Pathway Drives Type I IFN Immunopathology in COVID-19. Nature 2022, 603, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Jin, T.; Weng, J. Endothelial Cells as a Key Cell Type for Innate Immunity: A Focused Review on RIG-I Signaling Pathway. Front. Immunol. 2022, 13, 951614. [Google Scholar] [CrossRef]
- Fosse, J.H.; Haraldsen, G.; Falk, K.; Edelmann, R. Endothelial Cells in Emerging Viral Infections. Front. Cardiovasc. Med. 2021, 8, 619690. [Google Scholar] [CrossRef]
- Jiang, H.; Zhang, H.; Meng, Q.; Xie, J.; Li, Y.; Chen, H.; Zheng, Y.; Wang, X.; Qi, H.; Zhang, J.; et al. SARS-CoV-2 Orf9b Suppresses Type I Interferon Responses by Targeting TOM70. Cell Mol. Immunol. 2020, 17, 998–1000. [Google Scholar] [CrossRef]
- Gao, W.; Wang, L.; Ju, X.; Zhao, S.; Li, Z.; Su, M.; Xu, J.; Wang, P.; Ding, Q.; Lv, G.; et al. The Deubiquitinase USP29 Promotes SARS-CoV-2 Virulence by Preventing Proteasome Degradation of ORF9b. mBio 2022, 13, e0130022. [Google Scholar] [CrossRef]
- Lenhard, S.; Gerlich, S.; Khan, A.; Rödl, S.; Bökenkamp, J.E.; Peker, E.; Zarges, C.; Faust, J.; Storchova, Z.; Räschle, M.; et al. The Orf9b Protein of SARS-CoV-2 Modulates Mitochondrial Protein Biogenesis. J. Cell Biol. 2023, 222, e202303002. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.Y.C.; Hoiland, R.L.; Stukas, S.; Wellington, C.L.; Sekhon, M.S. Confronting the Controversy: Interleukin-6 and the COVID-19 Cytokine Storm Syndrome. Eur. Respir. J. 2020, 56, 2003006. [Google Scholar] [CrossRef] [PubMed]
- Carty, C.L.; Heagerty, P.; Heckbert, S.R.; Jarvik, G.P.; Lange, L.A.; Cushman, M.; Tracy, R.P.; Reiner, A.P. Interaction between Fibrinogen and IL-6 Genetic Variants and Associations with Cardiovascular Disease Risk in the Cardiovascular Health Study. Ann. Hum. Genet. 2010, 74, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, A.; Koami, H.; Shinada, K.; Sakamoto, Y. Investigation of Differences in Coagulation Characteristics between Hospitalized Patients with SARS-CoV-2 Alpha, Delta, and Omicron Variant Infection Using Rotational Thromboelastometry (ROTEM): A Single-center, Retrospective, Observational Study. J. Clin. Lab. Anal. 2022, 36, e24796. [Google Scholar] [CrossRef]
- Conway, E.M.; Mackman, N.; Warren, R.Q.; Wolberg, A.S.; Mosnier, L.O.; Campbell, R.A.; Gralinski, L.E.; Rondina, M.T.; van de Veerdonk, F.L.; Hoffmeister, K.M.; et al. Understanding COVID-19-Associated Coagulopathy. Nat. Rev. Immunol. 2022, 22, 639–649. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Janevska, M.; Naessens, E.; Verhasselt, B. Impact of SARS-CoV-2 Wuhan and Omicron Variant Proteins on Type I Interferon Response. Viruses 2025, 17, 569. https://doi.org/10.3390/v17040569
Janevska M, Naessens E, Verhasselt B. Impact of SARS-CoV-2 Wuhan and Omicron Variant Proteins on Type I Interferon Response. Viruses. 2025; 17(4):569. https://doi.org/10.3390/v17040569
Chicago/Turabian StyleJanevska, Marija, Evelien Naessens, and Bruno Verhasselt. 2025. "Impact of SARS-CoV-2 Wuhan and Omicron Variant Proteins on Type I Interferon Response" Viruses 17, no. 4: 569. https://doi.org/10.3390/v17040569
APA StyleJanevska, M., Naessens, E., & Verhasselt, B. (2025). Impact of SARS-CoV-2 Wuhan and Omicron Variant Proteins on Type I Interferon Response. Viruses, 17(4), 569. https://doi.org/10.3390/v17040569