
Genomic and Epidemiological Investigations Reveal Chromosomal Integration of the Acipenserid Herpesvirus 3 Genome in Lake Sturgeon Acipenser fulvescens

 ,
,  , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Field Study of Lake Sturgeon in the Hudson Bay Drainage Basin
2.2. Other Sample Collections
2.3. Viruses and General Virology
2.4. Viral Nucleic Acid
2.4.1. DNA Synthesis and Plasmid Purification
2.4.2. Nucleic Acid Extraction
2.5. Next-Generation Sequencing, Assembly and Annotation
2.6. Genome Nucleotide Sequence Accession Number
2.7. Evaluation of Alloherpesvirus Core Proteins
2.8. PCR Tests
2.8.1. qPCR Test (Q1mcp)
2.8.2. Genotyping PCR (GCmcp)
2.9. Optimization and Analytical Validation of Q1mcp
2.9.1. qPCR Assay Development and Optimization
2.9.2. Analytical Validation
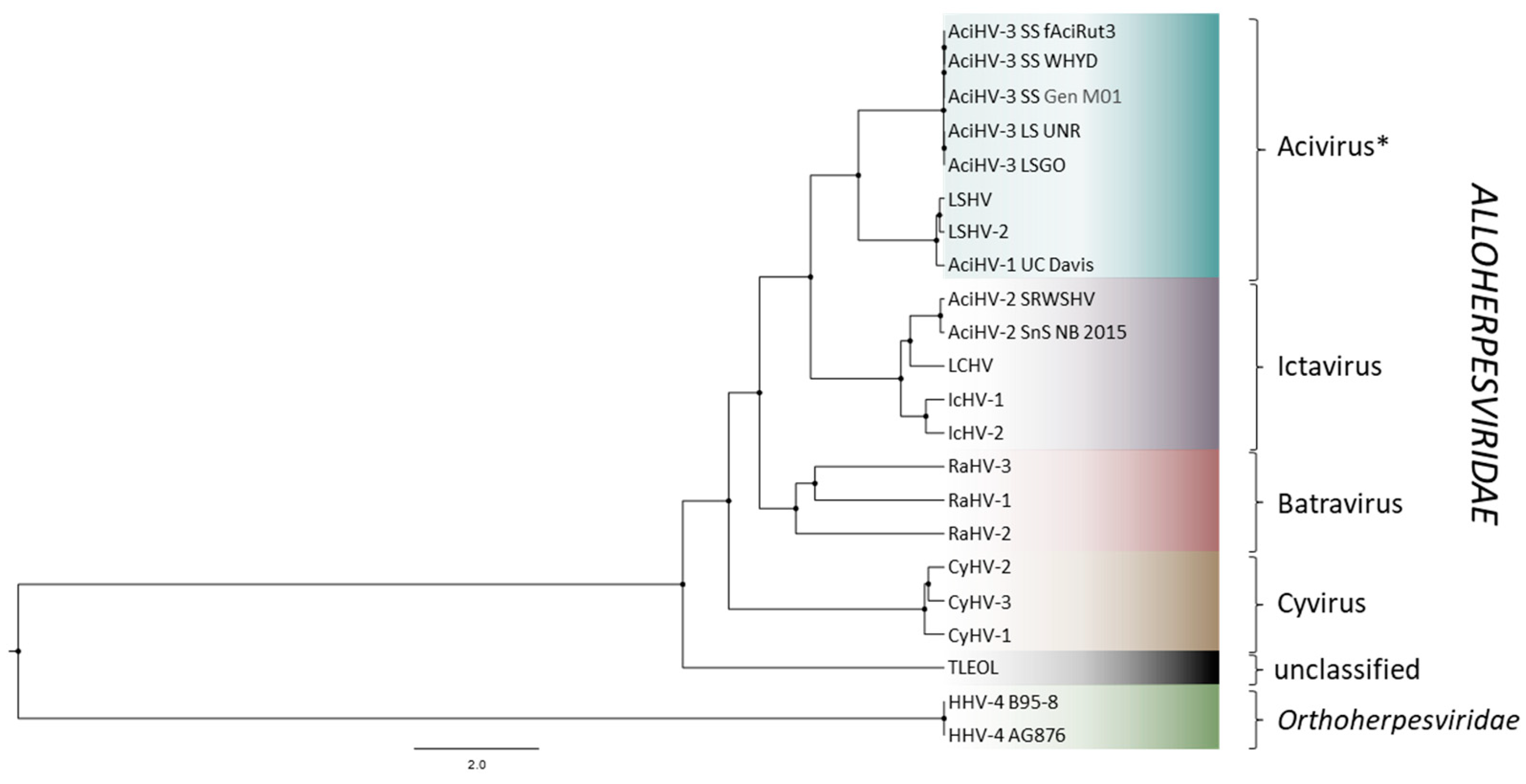
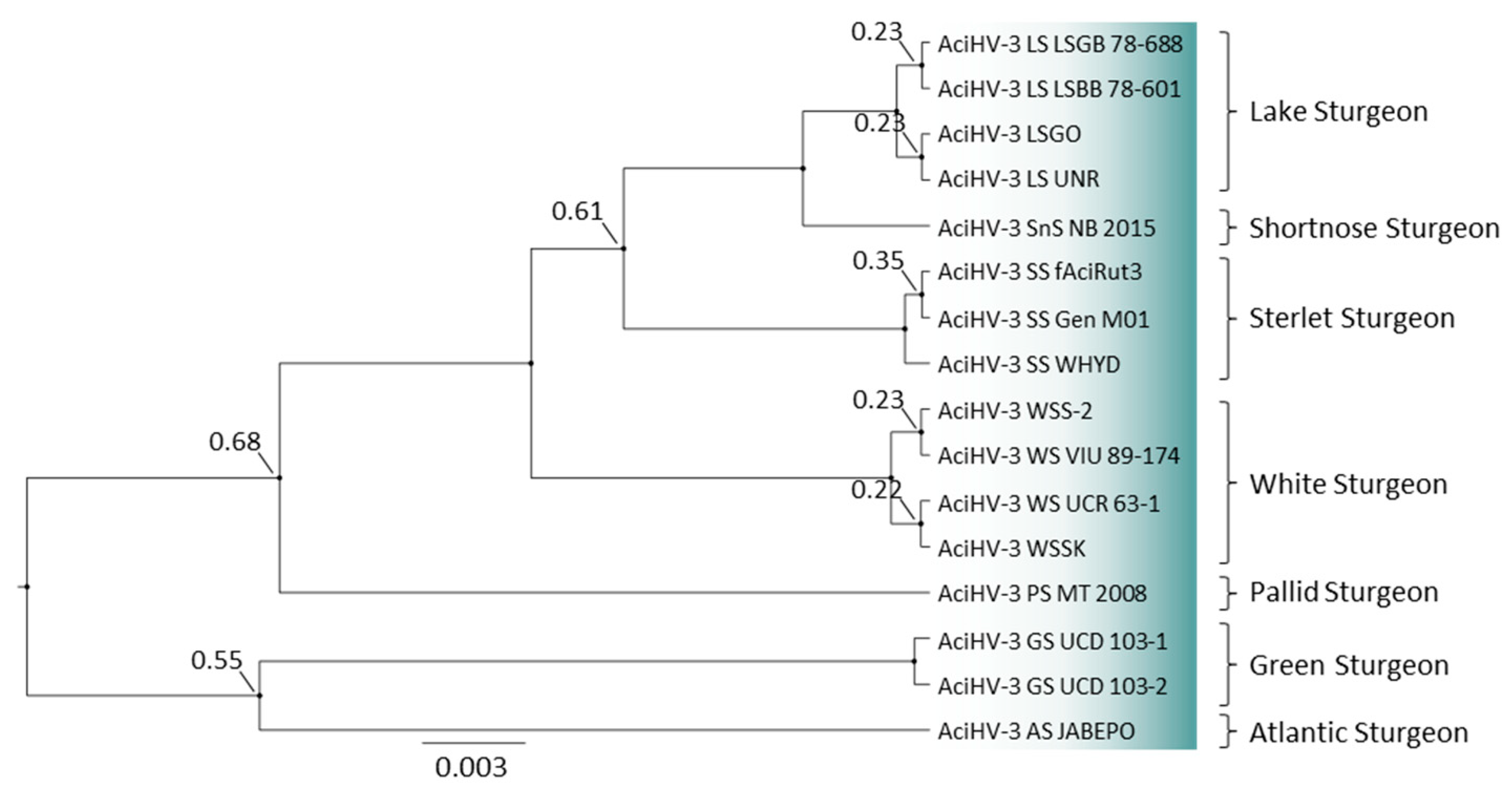
2.10. Phylogenetic Analyses
3. Results
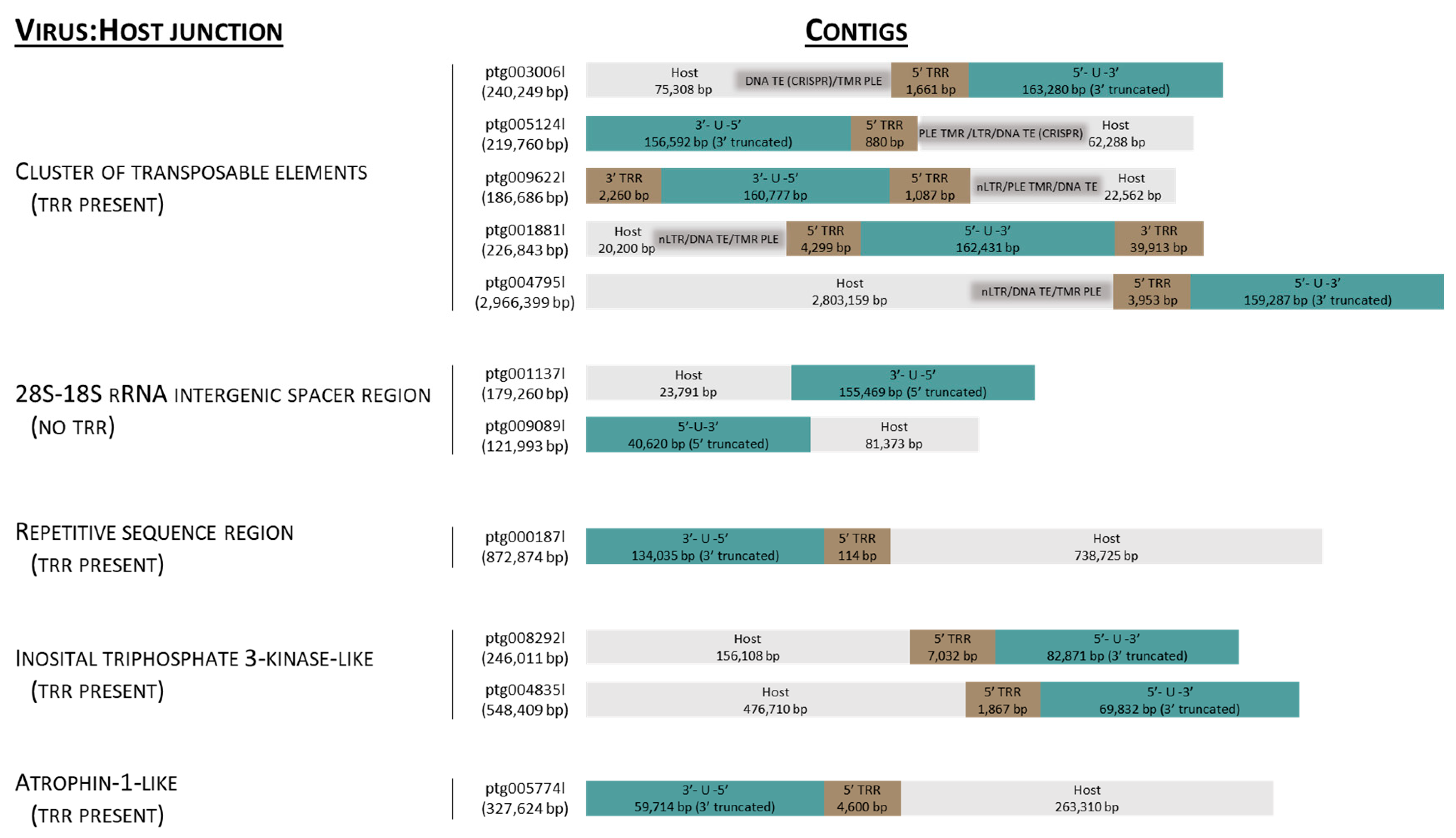
3.1. Identification of Contigs Containing AciHV-3 Sequence
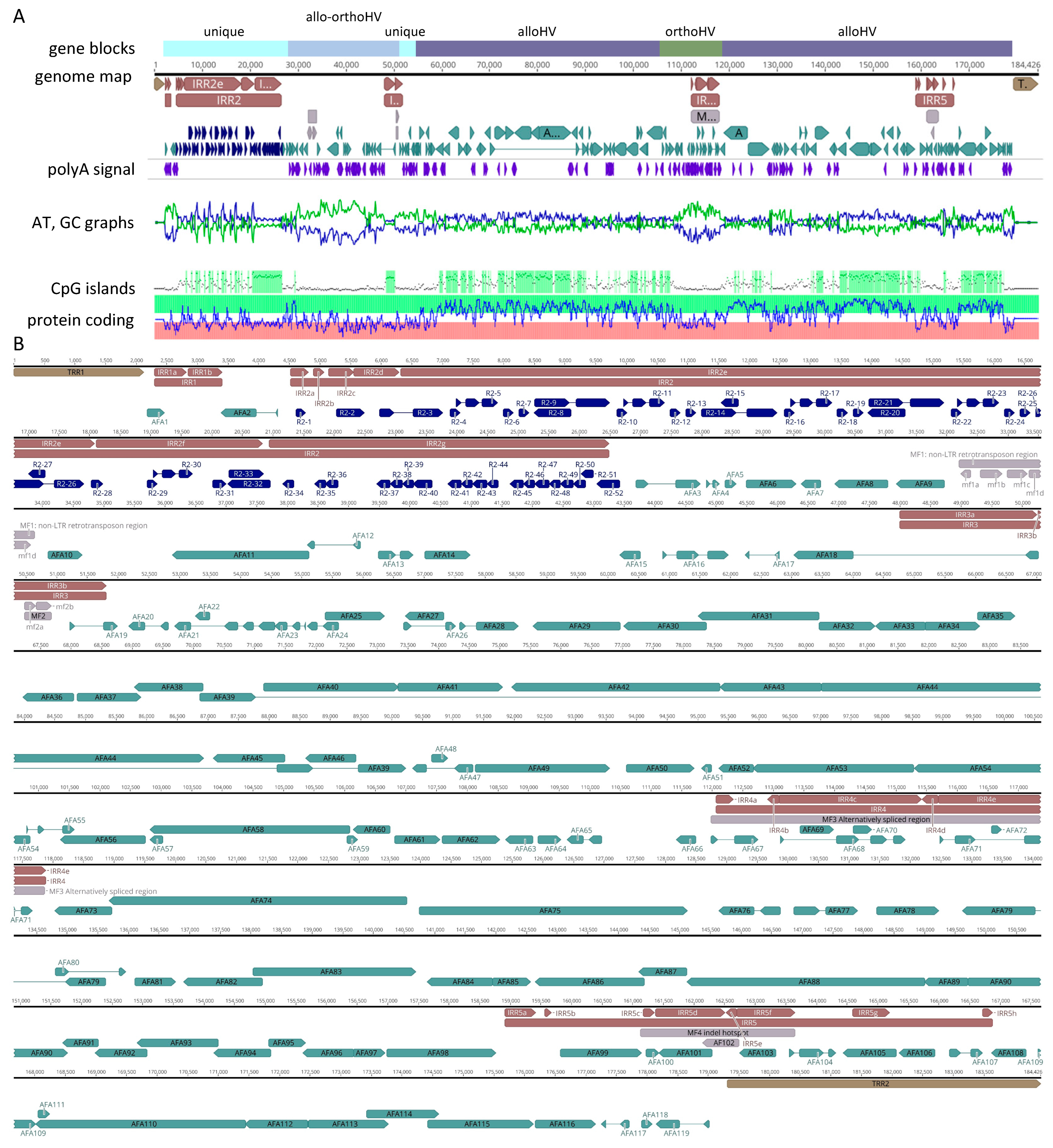
3.2. Genome Structure and Composition
3.3. Gene Arrangement
3.4. AciHV-3 Genome Is Contiguous with Host DNA
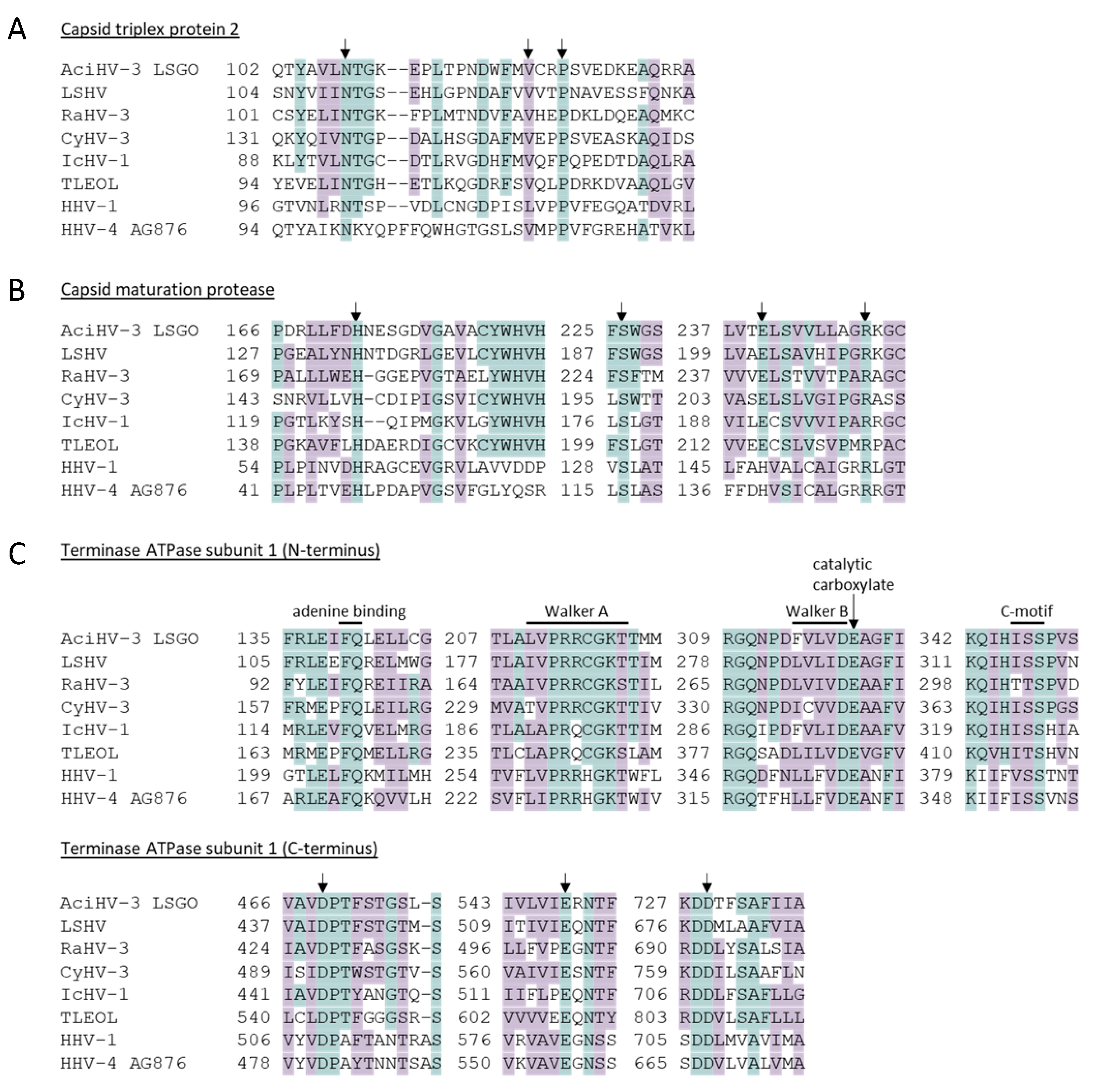
3.5. Genes Encoding Core Herpesvirus Proteins Are Intact
3.6. AciHV-3 LSGO Is Distinct from Teratorn-like Herpesviruses
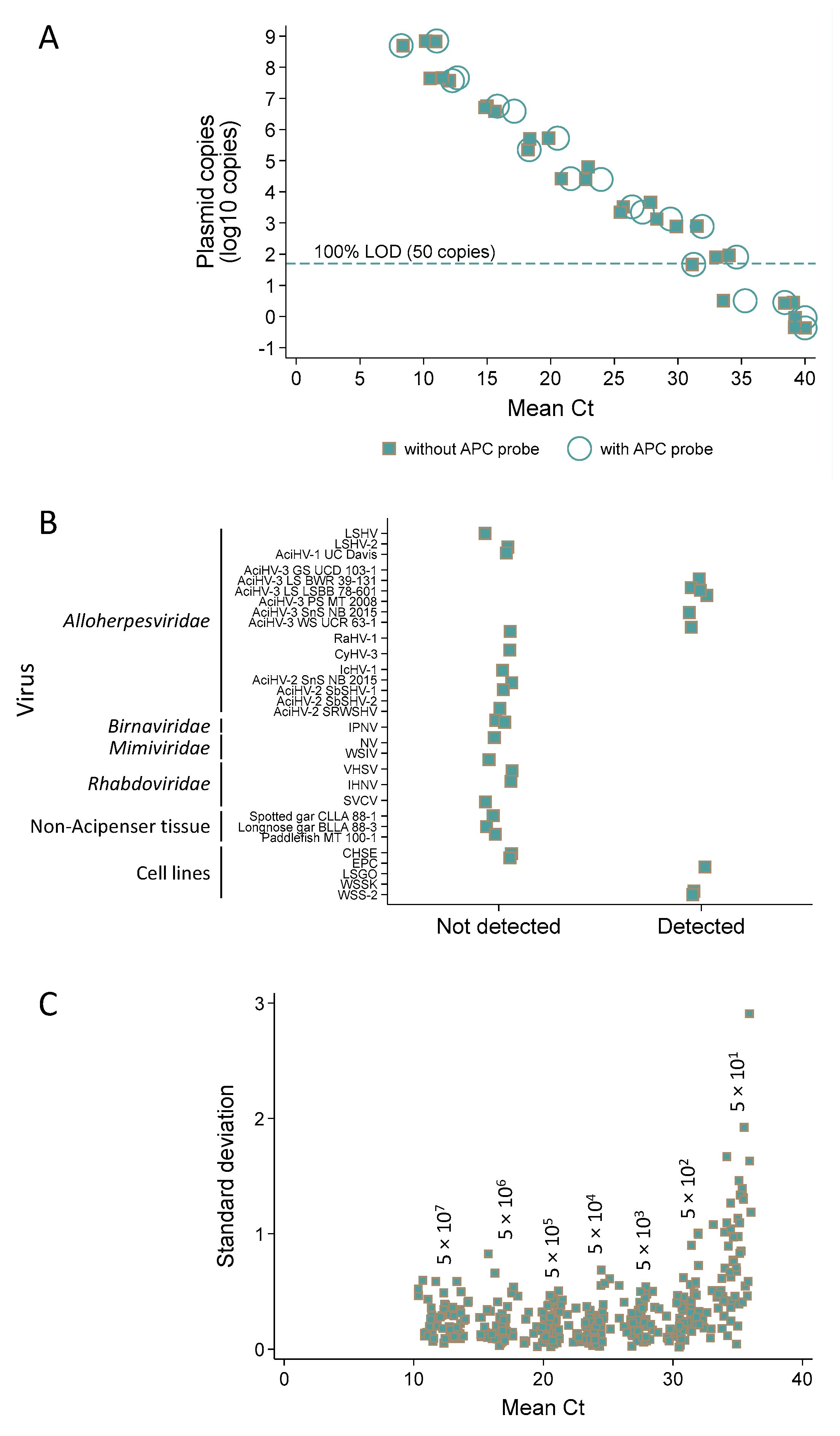
3.7. Q1mcp, a Sensitive Test for Specific Detection of AciHV-3 DNA
3.8. AciHV-3 DNA Is Detected in High Titers in Somatic and Germ Line Tissues
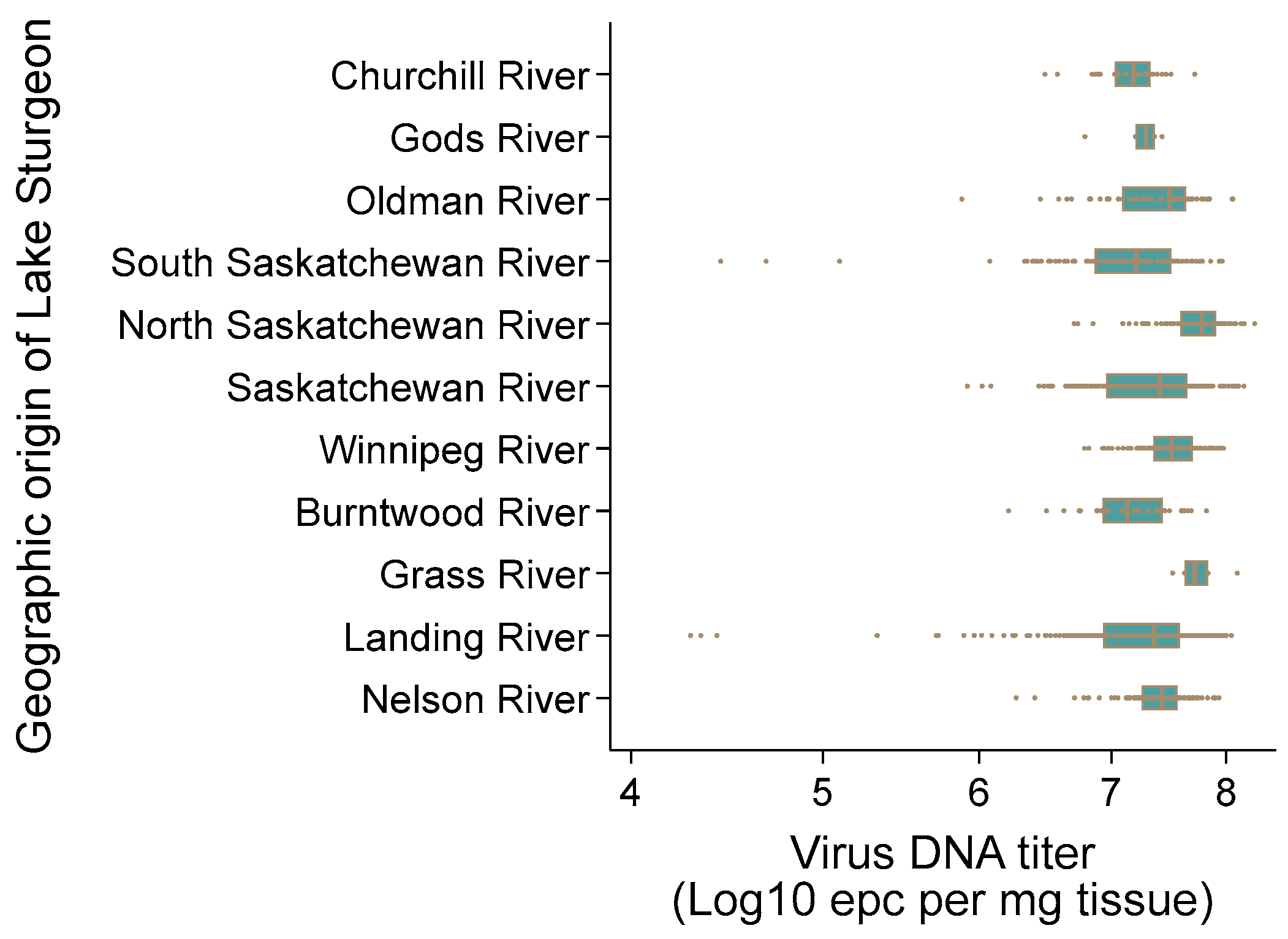
3.9. AciHV-3 DNA Is Present in Lake Sturgeon Populations from Two Drainage Basins in Canada
3.10. A New Genotype of AciHV-3 Is Detected in Green Sturgeon Acipenser Medirostris
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- ICTV 2024 (International Committee on Taxonomy of Viruses). 2023 Taxonomy Release. Available online: https://ictv.global/taxonomy (accessed on 20 September 2024).
- McGeoch, D.J.; Rixon, F.J.; Davison, A.J. Topics in herpesvirus genomics and evolution. Virus Res. 2006, 117, 90–104. [Google Scholar] [PubMed]
- McGeoch, D.J.; Davison, A.J.; Dolan, A.; Gatherer, D.; Sevilla-Reyes, E.E. Molecular evolution of the Herpesvirales. In Origin and Evolution of Viruses, 2nd ed.; Domingo, E., Parrish, C.R., Holland, J.J., Eds.; Academic Press: London, UK, 2008; pp. 447–475. [Google Scholar]
- Roizman, B.; Pellett, P.E. The family of Herpesviridae: A brief introduction. In Fields Virology; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2001; pp. 2381–2397. [Google Scholar]
- Roizman, B.; Knipe, D.M.; Whitley, R.J. Herpes simplex viruses. In Fields Virology, 5th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2007; pp. 2501–2601. [Google Scholar]
- McVoy, M.A.; Adler, S.P. Human cytomegalovirus DNA replicates after early circularization by concatemer formation, and inversion occurs within the concatemer. J. Virol. 1994, 68, 1040–1051. [Google Scholar] [PubMed]
- Morissette, G.; Flamand, L. Herpesviruses and chromosomal integration. J. Virol. 2010, 84, 12100–12109. [Google Scholar] [PubMed]
- Delecluse, H.J.; Bartnizke, S.; Hammerschmidt, W.; Bullerdiek, J.; Bornkamm, G.W. Episomal and integrated copies of Epstein-Barr virus coexist in Burkitt lymphoma cell lines. J. Virol. 1993, 67, 1292–1299. [Google Scholar]
- Delecluse, H.J.; Hammerschmidt, W. Status of Marek’s disease virus in established lymphoma cell lines: Herpesvirus integration is common. J. Virol. 1993, 67, 82–92. [Google Scholar]
- Arbuckle, J.H.; Medveczky, M.M.; Luka, J.; Hadley, S.H.; Luegmayr, A.; Ablashi, D.; Lund, T.C.; Tolar, J.; De Meirleir, K.; Montoya, J.G.; et al. The latent human herpesvirus-6A genome specifically integrates in telomeres of human chromosomes in vivo and in vitro. Proc. Natl. Acad. Sci. USA 2010, 107, 5563–5568. [Google Scholar] [CrossRef]
- Delecluse, H.; Schüller, S.; Hammerschmidt, W. Latent Marek’s disease virus can be activated from its chromosomally integrated state in herpesvirus-transformed lymphoma cells. EMBO J. 1993, 12, 3277–3286. [Google Scholar] [CrossRef]
- Damania, B.; Kenney, S.C.; Raab-Traub, N. Epstein-Barr virus: Biology and clinical disease. Cell 2022, 185, 3652–3670. [Google Scholar]
- Frappier, L. Epstein-Barr virus: Current questions and challenges. Tumour Virus Res. 2021, 12, 200218. [Google Scholar]
- Aimola, G.; Beythien, G.; Aswad, A.; Kaufer, B.B. Current understanding of human herpesvirus 6 (HHV-6) chromosomal integration. Antivir. Res. 2020, 176, 104720. [Google Scholar]
- Birstein, V.J. Sturgeons and paddlefishes: Threatened fishes in need of conservation. Conserv. Biol. 1993, 7, 773–787. [Google Scholar]
- IUCN. The IUCN Red List of Threatened Species. Version 2024-1. Available online: https://www.iucnredlist.org (accessed on 9 September 2024).
- Fontana, F.; Congiu, L.; Mudrak, V.A.; Quattro, J.M.; Smith, T.I.; Ware, K.; Doroshov, S.I. Evidence of hexaploidy karyotype in shortnose sturgeon. Genome 2008, 51, 113–119. [Google Scholar]
- Krieger, J.; Fuerst, P.A. Evidence for a slowed rate of molecular evolution in the order Acipenseriformes. Mol. Biol. Evol. 2002, 19, 891–897. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Fujimoto, T.; Adachi, S.; Yamaha, E.; Arai, K. Genome Size Variation Estimated by Flow Cytometry in Acipenser Mikadoi, Huso Dauricus in Relation to Other Species of Acipenseriformes. J. Appl. Ichthyol. 2011, 27, 484–491. [Google Scholar]
- Havelka, M.; Bytyutskyy, D.; Symonová, R.; Ráb, P.; Flajšhans, M. The second highest chromosome count among vertebrates is observed in cultured sturgeon and is associated with genome plasticity. Genet. Sel. Evol. 2016, 48, 12. [Google Scholar] [PubMed]
- Lebeda, I.; Ráb, P.; Majtánová, Z.; Flajšhans, M. Artificial whole genome duplication in paleopolyploid sturgeons yields highest documented chromosome number in vertebrates. Sci. Rep. 2020, 10, 19705. [Google Scholar]
- Havelka, M.; Kaspar, V.; Hulak, M.; Flajshans, M. Sturgeon genetics and cytogenetics: A review related to ploidy levels and interspecific hybridization. Folia Zool. 2011, 60, 93–103. [Google Scholar]
- Bruch, R.M.; Haxton, T.J.; Koenigs, R.; Welsh, A.; Kerr, S.J. Status of Lake Sturgeon (Acipenser fulvescens Rafinesque 1817) in North America. J. Appl. Ichthyol. 2016, 32, 162–190. [Google Scholar]
- COSEWIC. COSEWIC Assessment and Status Report on the Lake Sturgeon Acipenser fulvescens, Western Hudson Bay populations, Saskatchewan-Nelson River populations, Southern Hudson Bay- James Bay populations and Great Lakes-Upper St. Lawrence populations in Canada. Committee on the Status of Endangered Wildlife in Canada. Ottawa. 2023. Available online: https://wildlife-species.canada.ca/species-risk-registry/virtual_sara/files/cosewic/sr_Lake%20Sturgeon_2017_e.pdf (accessed on 14 January 2024).
- Clouthier, S.; Tomczyk, M.; Schroeder, T.; Klassen, C.; Dufresne, A.; Emmenegger, E.; Nalpathamkalam, T.; Wang, Z.; Thiruvahindrapuram, B. A new sturgeon herpesvirus from juvenile Lake Sturgeon Acipenser fulvescens displaying epithelial skin lesions. Pathogens 2023, 12, 1115. [Google Scholar] [CrossRef]
- Watson, L.; Yun, S.; Groff, J.; Hedrick, R. Characteristics and pathogenicity of a novel herpesvirus isolated from adult and subadult white sturgeon Acipenser transmontanus. Dis. Aquat. Org. 1995, 22, 199–210. [Google Scholar]
- Soto, E.; Richey, C.; Stevens, B.; Yun, S.; Kenelty, K.; Reichley, S.; Griffin, M.; Kurobe, T.; Camus, A. Co-infection of Acipenserid herpesvirus 2 (AciHV-2) and Streptococcus iniae in cultured white sturgeon Acipenser transmontanus. Dis. Aquat. Org. 2017, 124, 11–20. [Google Scholar]
- LaPatra, S.E.; Groff, J.M.; Keith, I.; E Hogans, W.; Groman, D. Case report: Concurrent herpesviral and presumptive iridoviral infection associated with disease in cultured shortnose sturgeon, Acipenser brevirostrum (L.), from the Atlantic coast of Canada. J. Fish Dis. 2013, 37, 141–147. [Google Scholar] [PubMed]
- Doszpoly, A.; Shchelkunov, I. Partial genome analysis of Siberian Sturgeon alloherpesvirus suggests its close relation to AciHV-2–Short communication. Acta Vet. Hung. 2010, 58, 269–274. [Google Scholar] [PubMed]
- Doszpoly, A.; Somogyi, V.; LaPatra, S.E.; Benkő, M. Partial genome characterization of acipenserid herpesvirus 2: Taxonomical proposal for the demarcation of three subfamilies in Alloherpesviridae. Arch. Virol. 2011, 156, 2291–2296. [Google Scholar]
- Doszpoly, A.; Kalabekov, I.M.; Breyta, R.; Shchelkunov, I.S. Isolation and characterization of an atypical Siberian Sturgeon herpesvirus strain in Russia: Novel North American Acipenserid herpesvirus 2 strain in Europe? J. Fish Dis. 2017, 40, 1363–1372. [Google Scholar]
- Hedrick, R.P.; Groff, J.M.; McDowell, T.S. Isolation of an epitheliotropic herpesvirus from white sturgeon (Acipenser transmontanus). Dis. Aquat. Org. 1991, 11, 49–56. [Google Scholar]
- Kurobe, T.; Kelley, G.O.; Waltzek, T.B.; Hedrick, R.P. Revised phylogenetic relationships among herpesviruses isolated from sturgeons. J. Aquat. Anim. Heal. 2008, 20, 96–102. [Google Scholar]
- Kelley, G.O.; Waltzek, T.B.; McDowell, T.S.; Yun, S.C.; LaPatra, S.E.; Hedrick, R.P. Genetic relationships among herpes-like viruses isolated from sturgeon. J. Aquat. Anim. Heal. 2005, 17, 297–303. [Google Scholar]
- Walker, L.; Subramaniam, K.; Viadanna, P.; Vann, J.; Marcquenski, S.; Godard, D.; Kieran, E.; Frasca, S.; Popov, V.; Kerr, K.; et al. Characterization of an alloherpesvirus from wild lake sturgeon Acipenser fulvescens in Wisconsin (USA). Dis. Aquat. Org. 2022, 14, 83–96. [Google Scholar]
- Johnston, A.E.; Shavalier, M.A.; Scribner, K.T.; Soto, E.; Griffin, M.J.; Waldbieser, G.C.; Richardson, B.M.; Winters, A.D.; Yun, S.; Baker, E.A.; et al. First isolation of a herpesvirus (Family Alloherpesviridae) from Great Lakes Lake Sturgeon (Acipenser fulvescens). Animals 2022, 12, 3230. [Google Scholar] [CrossRef]
- Hanson, L.; Doszpoly, A.; van Beurden, S.; Viadanna, P.d.O.; Waltzek, T. Alloherpesviruses of fish. In Aquaculture Virology; Kibenge, F.S.B., Godoy, M.G., Eds.; Academic Press: London, UK, 2016; pp. 153–172. [Google Scholar]
- Georgiadis, M.P.; Hedrick, R.P.; Johnson, W.O.; Yun, S.; Gardner, I.A. Risk factors for outbreaks of disease attributable to white sturgeon iridovirus and white sturgeon herpesvirus-2 at a commercial sturgeon farm. Am. J. Vet. Res. 2000, 61, 1232–1240. [Google Scholar] [PubMed]
- Shchelkunov, I.; Shchelkunova, T.; Shchelkunov, A.; Kolbassova, Y.; Didenko, L.; Bykovsky, A. First detection of a viral agent causing disease in farmed sturgeon in Russia. Dis. Aquat. Org. 2009, 86, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Hedrick, R.P.; McDowell, T.S.; Rosemark, R.; Aronstein, D.; Lannan, C.N. Two cell lines from White Sturgeon. Trans. Am. Fish. Soc. 1991, 120, 528–534. [Google Scholar] [CrossRef]
- Quijano Cardé, E.M.; Anenson, K.; Waldbieser, G.; Brown, C.T.; Griffin, M.; Henderson, E.; Yun, S.; Soto, E. Acipenserid herpesvirus 2 genome and partial validation of a qPCR for its detection in white sturgeon Acipenser transmontanus. Dis. Aquat. Organ. 2024, 157, 45–59. [Google Scholar]
- Doszpoly, A.; Kovács, E.R.; Bovo, G.; LaPatra, S.E.; Harrach, B.; Benkő, M. Molecular confirmation of a new herpesvirus from catfish (Ameiurus melas) by testing the performance of a novel PCR method, designed to target the DNA polymerase gene of alloherpesviruses. Arch. Virol. 2008, 153, 2123–2127. [Google Scholar]
- Clouthier, S.; VanWalleghem, E.; Copeland, S.; Klassen, C.; Hobbs, G.; Nielsen, O.; Anderson, E. A new species of nucleocytoplasmic large DNA virus (NCLDV) associated with mortalities in Manitoba Lake Sturgeon Acipenser fulvescens. Dis. Aquat. Org. 2013, 102, 195–209. [Google Scholar]
- ESRI. ArcGIS Desktop: Release 10.6.1; Environmental Systems Research Institute: Redlands, CA, USA, 2016. [Google Scholar]
- NRC. Government of Canada, Natural Resources Canada—CANVec Series. 2019. Available online: https://open.canada.ca/data/en/dataset/9d96e8c9-22fe-4ad2-b5e8-94a6991b744b (accessed on 24 May 2019).
- Clouthier, S.; Caskenette, A.; Van Walleghem, E.; Schroeder, T.; Macdonald, D.; Anderson, E.D. Molceular phylogeny of sturgeon mimiviruses and Bayesian hierarchical modeling of their effect on wild Lake Sturgeon (Acipenser fulvescens) in central Canada. Infect. Genet. Evol. 2020, 84, 104491. [Google Scholar]
- Fijan, N.; Sulimanović, D.; Bearzotti, M.; Muzinić, D.; Zwillenberg, L.; Chilmonczyk, S.; Vautherot, J.; de Kinkelin, P. Some properties of the Epithelioma papulosum cyprini (EPC) cell line from carp cyprinus carpio. Ann. Inst. Pasteur Virol. 1983, 134, 207–220. [Google Scholar] [CrossRef]
- Winton, J.; Batts, W.; DeKinkelin, P.; LeBerre, M.; Bremont, M.; Fijan, N. Current lineages of the Epithelioma papulosum cyprinid (EPC) cell line are contaminated with fathead minnow, Pimephales promelas cells. J. Fish Dis. 2010, 33, 701–704. [Google Scholar]
- Lannan, C.N.; Winton, J.R.; Fryer, J.L. Fish cell lines: Establishment and characterization of nine cell lines from salmonids. Vitro 1984, 20, 671–676. [Google Scholar]
- Neukirch, M.; Böttcher, K.; Bunnajjirakul, S. Isolation of a virus from koi with altered gills. Bull. Eur. Assoc. Fish Pathol. 1999, 19, 221–224. [Google Scholar]
- Snow, M.; McKay, P.; Matejusova, I. Development of a widely applicable positive control strategy to support detection of infectious salmon anaemia virus (ISAV) using Taqman real-time PCR. J. Fish Dis. 2009, 32, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Clouthier, S.C.; McClure, C.; Schroeder, T.; Desai, M.; Hawley, L.; Khatkar, S.; Lindsay, M.; Lowe, G.; Richard, J.; Anderson, E.D. Diagnostic validation of three test methods for detection of cyprinid herpesvirus 3 (CyHV-3). Dis. Aquat. Org. 2017, 123, 101–122. [Google Scholar]
- Clouthier, S.; Schroeder, T.; Bueren, E.; Anderson, E.; Emmenegger, E. Analytical validation of two RT-qPCR tests and detection of spring viremia of carp virus (SVCV) in persistently infected koi Cyprinus carpio. Dis. Aquat. Org. 2021, 143, 169–188. [Google Scholar]
- Borodovsky, M.; Lomsadze, A. Gene identification in prokaryotic genomes, phages, metagenomes, and EST sequences with GeneMarkS suite. Curr. Protoc. Microbiol. 2014, 32, 1E.7.1–1E.7.17. [Google Scholar]
- Tcherepanov, V.; Ehlers, A.; Upton, C. Genome Annotation Transfer Utility (GATU): Rapid annotation of viral genomes using a closely related reference genome. BMC Genom. 2006, 7, 150. [Google Scholar]
- Smit, A.F.A.; Hubley, R.; Green, P. RepeatMasker. 2024. Available online: http://repeatmasker.org (accessed on 5 October 2022).
- Storer, J.; Hubley, R.; Rosen, J.; Wheeler, T.J.; Smit, A.F. The Dfam community resource of transposable element families, sequence models and genome annotations. Mob. DNA 2021, 12, 2. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar]
- Scalzitti, N.; Kress, A.; Orhand, R.; Weber, T.; Moulinier, L.; Jeannin-Girardon, A.; Collet, P.; Poch, O.; Thompson, J.D. Spliceator: Multi-species splice site prediction using convolutional neural networks. BMC Bioinform. 2021, 22, 561. [Google Scholar]
- Hallgren, J.; Tsirigos, K.D.; Pedersen, M.D.; Armenteros, J.J.A.; Marcatili, P.; Nielsen, H.; Krogh, A.; Winther, O. DeepTMHMM predicts alpha and beta transmembrane proteins using deep neural networks. bioRxiv 2022. [Google Scholar] [CrossRef]
- Gough, J.; Karplus, K.; Hughey, R.; Chothia, C. Assignment of homology to genome sequences using a library of hidden markov models that represent all proteins of known structure. J. Mol. Biol. 2001, 313, 903–919. [Google Scholar] [CrossRef]
- Pandurangan, A.P.; Stahlhacke, J.; E Oates, M.; Smithers, B.; Gough, J. The SUPERFAMILY 2.0 database: A significant proteome update and a new webserver. Nucleic Acids Res. 2018, 47, D490–D494. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef]
- Remmert, M.; Biegert, A.; Hauser, A.; Söding, J. HHblits: Lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat. Methods 2011, 9, 173–175. [Google Scholar] [CrossRef] [PubMed]
- Marchler-Bauer, A.; Zheng, C.; Chitsaz, F.; Derbyshire, M.K.; Geer, L.Y.; Geer, R.C.; Gonzales, N.R.; Gwadz, M.; Hurwitz, D.I.; Lanczycki, C.J.; et al. CDD: Conserved domains and protein three-dimensional structure. Nucleic Acids Res. 2013, 41, D348–D352. [Google Scholar] [CrossRef]
- Wang, J.; Chitsaz, F.; Derbyshire, M.K.; Gonzales, N.R.; Gwadz, M.; Lu, S.; Marchler, G.H.; Song, J.S.; Thanki, N.; Yamashita, R.A.; et al. The conserved domain database in 2023. Nucleic Acids Res. 2023, 51, D384–D388. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Okoye, M.E.; Sexton, G.L.; Huang, E.; McCaffery, J.M.; Desai, P. Functional analysis of the triplex proteins (VP19C and VP23) of herpes simplex virus type 1. J. Virol. 2006, 80, 929–940. [Google Scholar] [CrossRef]
- Yuan, S.; Wang, J.; Zhu, D.; Wang, N.; Gao, Q.; Chen, W.; Tang, H.; Wang, J.; Zhang, X.; Liu, H.; et al. Cryo-EM structure of a herpesvirus capsid at 3.1 Å. Science 2018, 360, eaao7283. [Google Scholar] [PubMed]
- Cheng, H.; Shen, N.; Pei, J.; Grishin, N.V. Double-stranded DNA bacteriophage prohead protease is homologous to herpesvirus protease. Prot. Sci. 2004, 13, 2260–2269. [Google Scholar]
- Rao, V.B.; Feiss, M. The bacteriophage DNA packaging motor. Annu. Rev. Genet. 2008, 42, 647–681. [Google Scholar] [PubMed]
- Rao, V.B.; Feiss, M. Mechanisms of DNA packaging by large double-stranded DNA viruses. Annu. Rev. Virol. 2015, 2, 351–378. [Google Scholar] [CrossRef]
- Sigamani, S.S.; Zhao, H.; Kamau, Y.N.; Baines, J.D.; Tang, L. The structure of the herpes simplex virus DNA-packaging terminase pUL15 nuclease domain suggests an evolutionary lineage among eukaryotic and prokaryotic viruses. J. Virol. 2013, 87, 7140–7148. [Google Scholar]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467. [Google Scholar] [CrossRef]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES science gateway for inference of large phylogenetic trees. In Proceedings of the SC10Workshop on Gateway Computing Environments (GCE10), New Orleans, LA, USA, 14 November 2010. [Google Scholar]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. ClustalW and Clustal Z version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar]
- Schwarz, G. Estimating the dimension of a model. Ann. Stat. 1978, 6, 461–464. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar]
- Thiebaux, H.J.; Zwiers, F.W. The interpretation and estimation of effective sample size. J. Clim. Appl. Meteorol. 1984, 23, 800–811. [Google Scholar]
- Rambaut, A.; Suchard, M.A.; Xie, D.; Drummond, A.J. Tracer v1.7.1. 2018. Available online: https://github.com/beast-dev/tracer/releases/tag/v1.7.1 (accessed on 1 June 2023).
- Rambaut, A. FigTree, a Graphical Viewer of Phylogenetic Trees v1.4.4. 2018. Available online: https://github.com/rambaut/figtree/releases (accessed on 1 June 2023).
- Yang, Z. Likelihood Ratio Tests for Detecting Positive Selection and Application to Primate Lysozyme Evolution. Mol. Biol. Evol. 1998, 15, 568–573. [Google Scholar]
- Henikoff, S.; Henikoff, J.G. Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. USA 1992, 89, 10915–10919. [Google Scholar] [CrossRef]
- Yule, G.U. A mathematical theory of evolution, based on the conclusions of Dr. JC Willis, FRS. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1925, 213, 21–87. [Google Scholar]
- Gernhard, T. New analytic results for speciation times in neutral models. Bull. Math. Biol. 2008, 70, 1082–1097. [Google Scholar]
- Hasegawa, M.; Kishino, H.; Yano, T.-A. Dating of human-ape splitting by a molecular clock of mitochondrial DNA. J. Mol. Evol. 1985, 22, 160–174. [Google Scholar] [PubMed]
- Inoue, Y.; Saga, T.; Aikawa, T.; Kumagai, M.; Shimada, A.; Kawaguchi, Y.; Naruse, K.; Morishita, S.; Koga, A.; Takeda, H. Complete fusion of a transposon and herpesvirus created the Teratorn mobile element in medaka fish. Nat. Commun. 2017, 8, 551. [Google Scholar]
- Meyne, J.; Ratliff, R.L.; Moyzis, R.K. Conservation of the human telomere sequences (TTAGGG) among vertebrates. Proc. Natl. Acad. Sci. USA 1989, 86, 7049–7053. [Google Scholar]
- Osterrieder, N.; Wallaschek, N.; Kaufer, B.B. Herpesvirus genome integration into telomeric repeats of host cell chromosomes. Annu. Rev. Virol. 2014, 1, 215–235. [Google Scholar] [CrossRef]
- Davison, A.J. The genome of salmonid herpesvirus 1. J. Virol. 1998, 72, 1974–1982. [Google Scholar] [CrossRef] [PubMed]
- Isfort, R.; Jones, D.; Kost, R.; Witter, R.; Kung, H.-J. Retrovirus insertion into herpesvirus in vitro and in vivo. Proc. Natl. Acad. Sci. USA 1992, 89, 991–995. [Google Scholar] [CrossRef]
- WOAH (World Organization for Animal Health). Manual of Diagnostic Tests for Aquatic Animals 2019; Chapter 1.1.2. Principles and Methods of Validation of Diagnostic Assays for Infectious Diseases. Available online: https://www.woah.org/fileadmin/Home/eng/Health_standards/aahm/current/1.1.02_VALIDATION.pdf (accessed on 14 April 2023).
- Arbuckle, J.H.; Pantry, S.N.; Medveczky, M.M.; Prichett, J.; Loomis, K.S.; Ablashi, D.; Medveczky, P.G. Mapping the telomere integrated genome of human herpesvirus 6A and 6B. Virology 2013, 442, 3–11. [Google Scholar] [CrossRef]
- Robinson, C.M.; Hunt, H.D.; Cheng, H.H.; E Delany, M. Chromosomal integration of an avian oncogenic herpesvirus reveals telomeric preferences and evidence for lymphoma clonality. Herpesviridae 2010, 1, 5. [Google Scholar] [CrossRef] [PubMed]
- Glass, M.C.; Smith, J.M.; Cheng, H.H.; Delany, M.E. Marek’s Disease Virus telomeric integration profiles of neoplastic host tissues reveal unbiased chromosomal selection and loss of cellular diversity during tumorigenesis. Genes 2021, 12, e1630. [Google Scholar] [CrossRef] [PubMed]
- Lestou, V.S.; De Braekeleer, M.; Strehl, S.; Ott, G.; Gadner, H.; Ambros, P.F. Non-random integration of Epstein-Barr virus in lymphoblastoid cell lines. Genes Chromosomes Cancer 1993, 8, 38–48. [Google Scholar] [CrossRef]
- Xu, M.; Zhang, W.-L.; Zhu, Q.; Yao, Y.-Y.; Feng, Q.-S.; Zhang, Z.; Peng, R.-J.; Jia, W.-H.; He, G.-P.; Feng, L.; et al. Genome-wide profiling of Epstein-Barr virus integration by targeted sequencing in Epstein-Barr virus associated malignancies. Theranostics 2019, 9, 1115–1124. [Google Scholar] [CrossRef]
- Kaufer, B.B.; Jarosinski, K.W.; Osterrieder, N. Herpesvirus telomeric repeats facilitate genomic integration into host telomeres and mobilization of viral DNA during reactivation. J. Exp. Med. 2011, 208, 605–615. [Google Scholar] [CrossRef]
- You, Y.; Vychodil, T.; Aimola, G.; Previdelli, R.L.; Göbel, T.W.; Bertzbach, L.D.; Kaufer, B.B. A cell culture system to investigate Marek’s disease virus integration into host chromosomes. Microorganisms 2021, 9, 2489. [Google Scholar] [CrossRef]
- Wood, M.L.; Neumann, R.; Roy, P.; Nair, V.; Royle, N.J. Characterization of integrated Marek’s disease virus genomes supports a model of integration by homology-directed recombination and telomere-loop-driven excision. J. Virol. 2023, 97, e0071623. [Google Scholar] [CrossRef] [PubMed]
- Wallaschek, N.; Sanyal, A.; Pirzer, F.; Gravel, A.; Mori, Y.; Flamand, L.; Kaufer, B.B. The telomeric repeats of human herpesvirus 6A (HHV-6A) are required for efficient virus integration. PLoS Pathog. 2016, 12, e1005666. [Google Scholar] [CrossRef] [PubMed]
- Evgen’ev, M.B.; Arkhipova, I.R. Penelope-like elements—A new class of retroelements: Distribution, function and possible evolutionary significance. Cytogenet. Genome Res. 2005, 110, 510–521. [Google Scholar]
- Fontana, F.; Tagliavini, J.; Congiu, L. Sturgeon genetics and cytogenetics: Recent advancements and perspectives. Genetica 2001, 111, 359–373. [Google Scholar]
- Fontana, F.; Lanfredi, M.; Congiu, L.; Leis, M.; Chicca, M.; Rossi, R. Chromosomal mapping of 18S–28S and 5S rRNA genes by two-colour fluorescent in situ hybridization in six sturgeon species. Genome 2003, 46, 473–477. [Google Scholar]
- Vega, L.R.; Mateyak, M.K.; Zakian, V.A. Getting to the end: Telomerase access in yeast and humans. Nat. Rev. Mol. Cell Biol. 2003, 4, 948–959. [Google Scholar] [CrossRef]
- Ocalewicz, K. Telomeres in fishes. Cytogenet. Genome Res. 2013, 141, 114–125. [Google Scholar] [CrossRef]
- Jacob, R.J.; Roizman, B. Anatomy of herpes simplex virus DNA. VIII. Properties of the replicating DNA. J. Virol. 1977, 23, 394–411. [Google Scholar] [CrossRef] [PubMed]
- Jacob, R.J.; Morse, L.S.; Roizman, B. Anatomy of herpes simplex virus DNA. XII. Accumulation of head-to-tail concatemers in nuclei of infected cells and their role in the generation of the four isomeric arrangements of viral DNA. J. Virol. 1979, 29, 448–457. [Google Scholar]
- Liu, X.; Kosugi, S.; Koide, R.; Kawamura, Y.; Ito, J.; Miura, H.; Matoba, N.; Matsuzaki, M.; Fujita, M.; Kamada, A.J.; et al. Endogenization and excision of human herpesvirus 6 in human genomes. PLoS Genet. 2020, 16, e1008915. [Google Scholar]
- Wood, M.L.; Veal, C.D.; Neumann, R.; Suzrez, N.M.; Nochols, J.; Parker, A.J.; Martin, D.; Romaine, S.P.R.; Codd, V.; Samani, N.J.; et al. Variation in human herpesvirus 6B telomeric integration, excision, and transmission between tissues and individuals. eLife 2021, 10, e70452. [Google Scholar] [PubMed]
- Huang, Y.; Hidalgo-Bravo, A.; Zhang, E.; Cotton, V.E.; Mendez-Bermudez, A.; Wig, G.; Medina-Calzada, Z.; Neumann, R.; Jeffreys, A.J.; Winney, B.; et al. Human telomeres that carry an integrated copy of human herpesvirus 6 are often short and unstable, facilitating release of the viral genome from the chromosome. Nucleic Acids Res. 2013, 42, 315–327. [Google Scholar] [PubMed]
- Zhang, E.; Bell, A.J.; Wilkie, G.S.; Suárez, N.M.; Batini, C.; Veal, C.D.; Armendáriz-Castillo, I.; Neumann, R.; Cotton, V.E.; Huang, Y.; et al. Inherited chromosomally integrated human herpesvirus 6 genomes are ancient, intact, and potentially able to reactivate from telomeres. J. Virol. 2017, 91, e01137-17. [Google Scholar] [PubMed]
- Peddu, V.; Dubuc, I.; Gravel, A.; Xie, H.; Huang, M.L.; Tenenbaum, D.; Jerome, K.R.; Tardif, J.C.; Dube, M.P.; Flamand, L. Inherited chromosomally integrated human herpesvirus 6 demonstrates tissue-specific RNA expression in vivo that correlates with an increased antibody immune response. J. Virol. 2019, 96, e01418-19. [Google Scholar]
- Smith, J.M.; Haigh, J. The hitch-hiking effect of a favourable gene. Genet. Res. 1974, 23, 23–35. [Google Scholar]
- Katzourakis, A.; Gifford, R.J. Endogenous viral elements in animal genomes. PLoS Genet. 2010, 6, e1001191. [Google Scholar]
- Schreier, A.D.; Gille, D.; Mahardja, B.; May, B. Neutral markers confirm the octoploid origin and reveal spontaneous autopolyploidy in white sturgeon, Acipenser transmontanus. J. Appl. Ichthyol. 2011, 27, 24–33. [Google Scholar]
- Gorshkova, G.; Gorshkov, S.; Gordin, H.; Knibb, W. Karyological studies in hybrids of Beluga Huso Huso (L.) and the Russian Sturgeon Acipenser Guldenstadti Brandt. Isr. J Aquac. Bamidgeh 1996, 48, 35–39. [Google Scholar]
- Tie, C.H.; Rowe, H.M. Epigenetic control of retrotransposons in adult tissues: Implications for immune regulation. Curr. Opin. Virol. 2017, 25, 28–33. [Google Scholar]
- Soto, A.A.; Ortiz, G.; Contreras, S.; Soto-Rifo, R.; González, P.A. Role of Epitranscriptomic and Epigenetic Modifications during the Lytic and Latent Phases of Herpesvirus Infections. Microorganisms 2022, 10, 1754. [Google Scholar] [CrossRef]
- Engdahl, E.; Dunn, N.; Niehusmann, P.; Wideman, S.; Wipfler, P.; Becker, A.J.; Ekström, T.J.; Almgren, M.; Fogdell-Hahn, A. Human herpesvirus 6B induces hypomethylation on chromosome 17p13. 3, correlating with increased gene expression and virus integration. J. Virol. 2017, 91, e02105-16. [Google Scholar] [PubMed]
- Mariani, M.; Zimmerman, C.; Rodriguez, P.; Hasenohr, E.; Aimola, G.; Gerrard, D.L.; Richman, A.; Dest, A.; Flamand, L.; Kaufer, B.; et al. Higher-order chromatin structures of chromosomally integrated HHV-6A predict integration sites. Front. Cell. Infect. Microbiol. 2021, 11, 612656. [Google Scholar]
- Yamamoto, T.; Kelly, R.K.; Nielsen, O. Epidermal hyperplasias of Northern Pike (Esox lucius) associated with herpesvirus and C-type particles. Arch. Virol. 1983, 79, 255–272. [Google Scholar]
- Margenau, T.L.; Marcquenski, S.V.; Rasmussen, P.W.; Macconnell, E. Prevalence of Blue Spot Disease (Esocid Herpesvirus-1) on Northern Pike and Muskellunge in Wisconsin. J. Aquat. Anim. Health 1995, 7, 29–33. [Google Scholar]
- Garver, K.A.; Leskisenoja, K.; Macrae, R.; Hawley, L.M.; Subramaniam, K.; Waltzek, T.B.; Richard, J.; Josefsson, C.; Valtonen, E.T. An alloherpesvirus infection of European perch Perca fluviatilis in Finland. Dis. Aquat. Org. 2018, 128, 175–185. [Google Scholar]
- Bowser, P.R.; Plumb, J.A. Growth rates of a new cell line from channel catfish ovary and channel catfish virus replication at different temperatures. Can. J. Fish. Aquat. Sci. 1980, 37, 871–873. [Google Scholar]
- Wolf, K.; Darlington, R.W.; Taylor, W.G.; Quimby, M.C.; Nagabayashi, T. Herpesvirus salmonis: Characterization of a new pathogen of Rainbow Trout. J. Virol. 1978, 27, 659–666. [Google Scholar]
- Kjartanson, S.L.; Haxton, T.; Wozney, K.; Lovejoy, N.R.; Wilson, C.C. Conservation Genetics of Lake Sturgeon (Acipenser fulvescens): Nuclear Phylogeography Drives Contemporary Patterns of Genetic Structure and Diversity. Diversity 2023, 15, 385. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Core Protein | Length of ORF (bp) | Number of Variations | |||
|---|---|---|---|---|---|
| Minimum | Maximum | Synonymous | Nonsynonymous | ||
| Conservative | Non-Conservative | ||||
| Allo37 | 2064 | 2064 | 3 | 1 | 1 |
| Allo54 | 1983 | 1983 | 1 | 0 | 1 |
| Allo56 | 4383 | 4383 | 2 | 3 | 2 |
| Allo60 | 1041 | 1041 | 4 | 0 | 1 |
| Allo64 | 1710 | 1710 | 2 | 0 | 0 |
| Capsid maturation protease | 1782 | 1782 | 3 | 2 | 0 |
| Capsid triplex protein 2 | 987 | 987 | 2 | 1 | 0 |
| DNA polymerase catalytic subunit | 4863 | 4863 | 1 | 0 | 1 |
| Helicase primase helicase | 1722 | 1722 | 5 | 1 | 0 |
| Helicase primase primase | 2202 | 2202 | 3 | 0 | 1 |
| Major capsid protein | 3885 | 3885 | 4 | 2 | 0 |
| Terminase ATPase subunit | 2274 | 2274 | 1 | 0 | 0 |
| Total | 31 | 10 | 7 | ||
| Brood Source | Sex | Year | AciHV-3 DNA Prevalence | ||
|---|---|---|---|---|---|
| (Number of Positive Sturgeon/Total Number of Sturgeon Tested (%)) | |||||
| Brood Fin | Gametes | Larvae (46–74 dpf) | |||
| Winnipeg River | Male | 2011 | 2/2 (100) | 2/2 (100) | - |
| Female | 3/3 (100) | 3/3 (100) | |||
| Landing River | Male | 2012 | 2/2 (100) | 2/2 (100) | - |
| Female | 2/2 (100) | 2/2 (100) | |||
| Winnipeg River | Male | 2012 | 4/4 (100) | 4/4 (100) | 34/36 (94) |
| Female | 8/8 (100) | 8/8 (100) | |||
| Burntwood River | Male | 2013 | 1/1 (100) | 1/1 (100) | - |
| Female | 2/2 (100) | 2/2 (100) | |||
| Winnipeg River | Male | 2013 | 4/4 (100) | 4/4 (100) | 24/24 (100) |
| Female | 5/5 (100) | 5/5 (100) | |||
| Landing River | Male | 2014 | 4/4 (100) | 4/4 (100) | - |
| Female | 2/2 (100) | 1/1 (100) | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Clouthier, S.; Rosani, U.; Khan, A.; Ding, Q.; Emmenegger, E.; Wang, Z.; Nalpathamkalam, T.; Thiruvahindrapuram, B. Genomic and Epidemiological Investigations Reveal Chromosomal Integration of the Acipenserid Herpesvirus 3 Genome in Lake Sturgeon Acipenser fulvescens. Viruses 2025, 17, 534. https://doi.org/10.3390/v17040534
Clouthier S, Rosani U, Khan A, Ding Q, Emmenegger E, Wang Z, Nalpathamkalam T, Thiruvahindrapuram B. Genomic and Epidemiological Investigations Reveal Chromosomal Integration of the Acipenserid Herpesvirus 3 Genome in Lake Sturgeon Acipenser fulvescens. Viruses. 2025; 17(4):534. https://doi.org/10.3390/v17040534
Chicago/Turabian StyleClouthier, Sharon, Umberto Rosani, Arfa Khan, Qiuwen Ding, Eveline Emmenegger, Zhuozhi Wang, Thomas Nalpathamkalam, and Bhooma Thiruvahindrapuram. 2025. "Genomic and Epidemiological Investigations Reveal Chromosomal Integration of the Acipenserid Herpesvirus 3 Genome in Lake Sturgeon Acipenser fulvescens" Viruses 17, no. 4: 534. https://doi.org/10.3390/v17040534
APA StyleClouthier, S., Rosani, U., Khan, A., Ding, Q., Emmenegger, E., Wang, Z., Nalpathamkalam, T., & Thiruvahindrapuram, B. (2025). Genomic and Epidemiological Investigations Reveal Chromosomal Integration of the Acipenserid Herpesvirus 3 Genome in Lake Sturgeon Acipenser fulvescens. Viruses, 17(4), 534. https://doi.org/10.3390/v17040534