
Relationship Between Gut Microbiota and the Clinical Course of COVID-19 Disease


 ,
,  , ,
, ,  ,
,  , , ,
, , ,  ,
,  , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Clinical Classification
2.3. Sample Preparation and 16S rRNA Gene Sequencing
2.4. Bioinformatic Analyses
3. Results
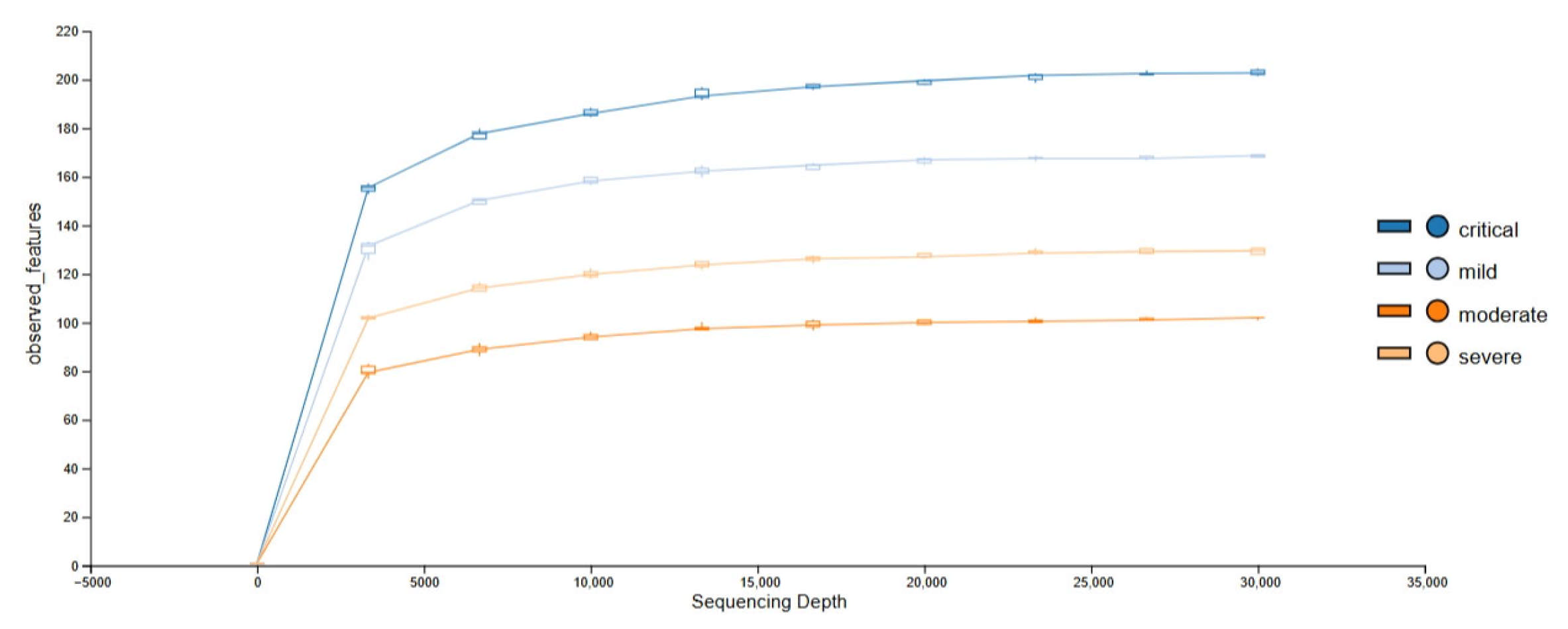
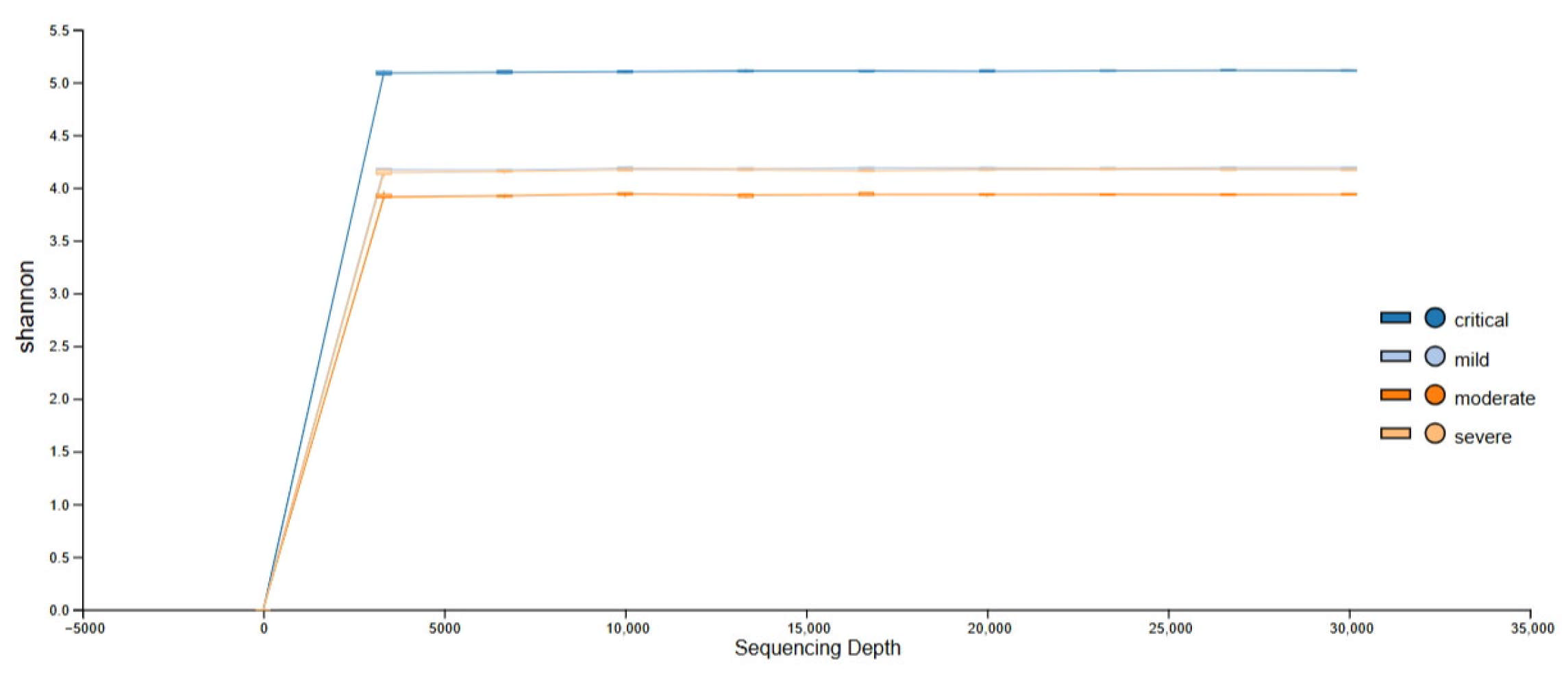
3.1. Obtained Sequencing Depth
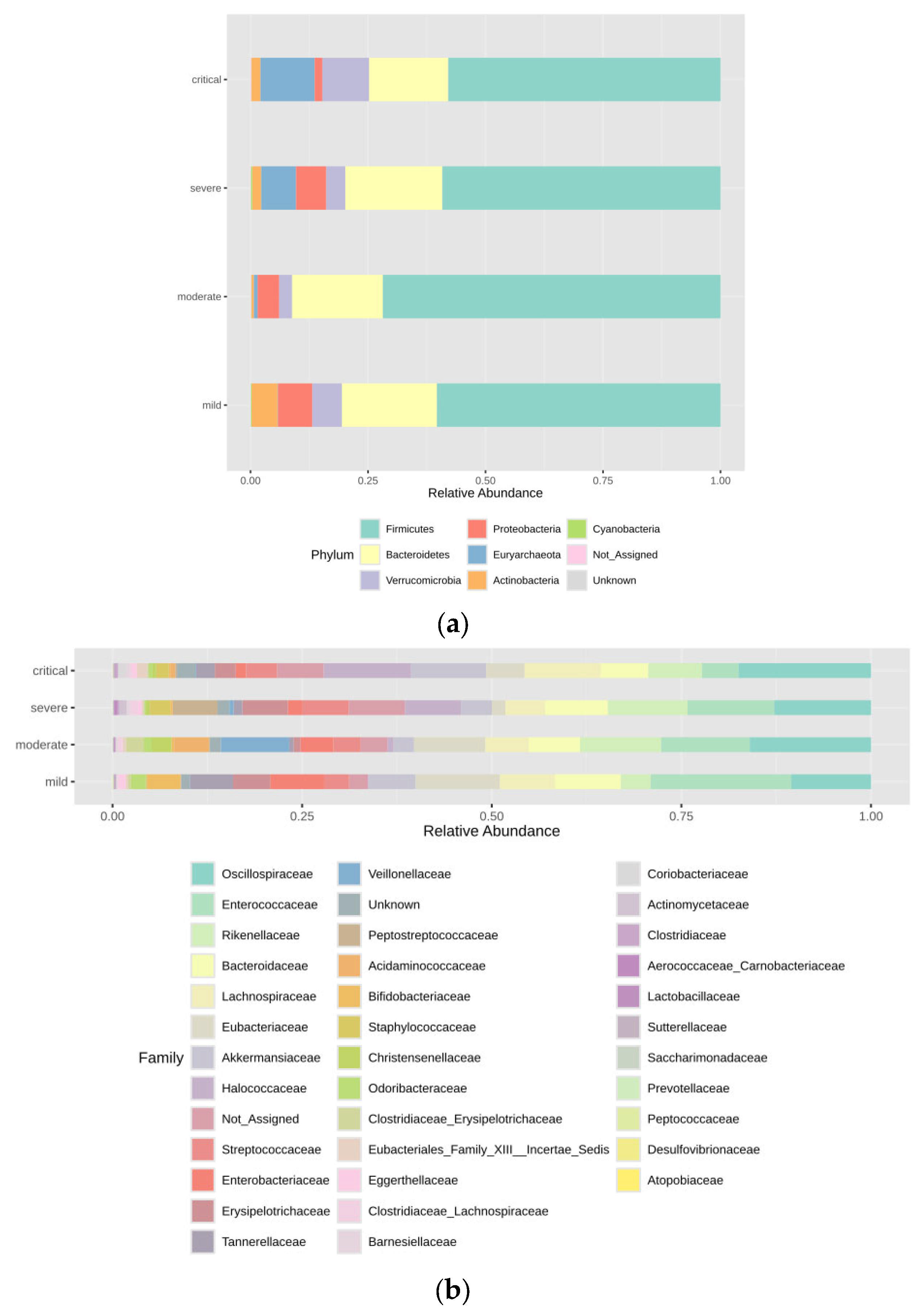
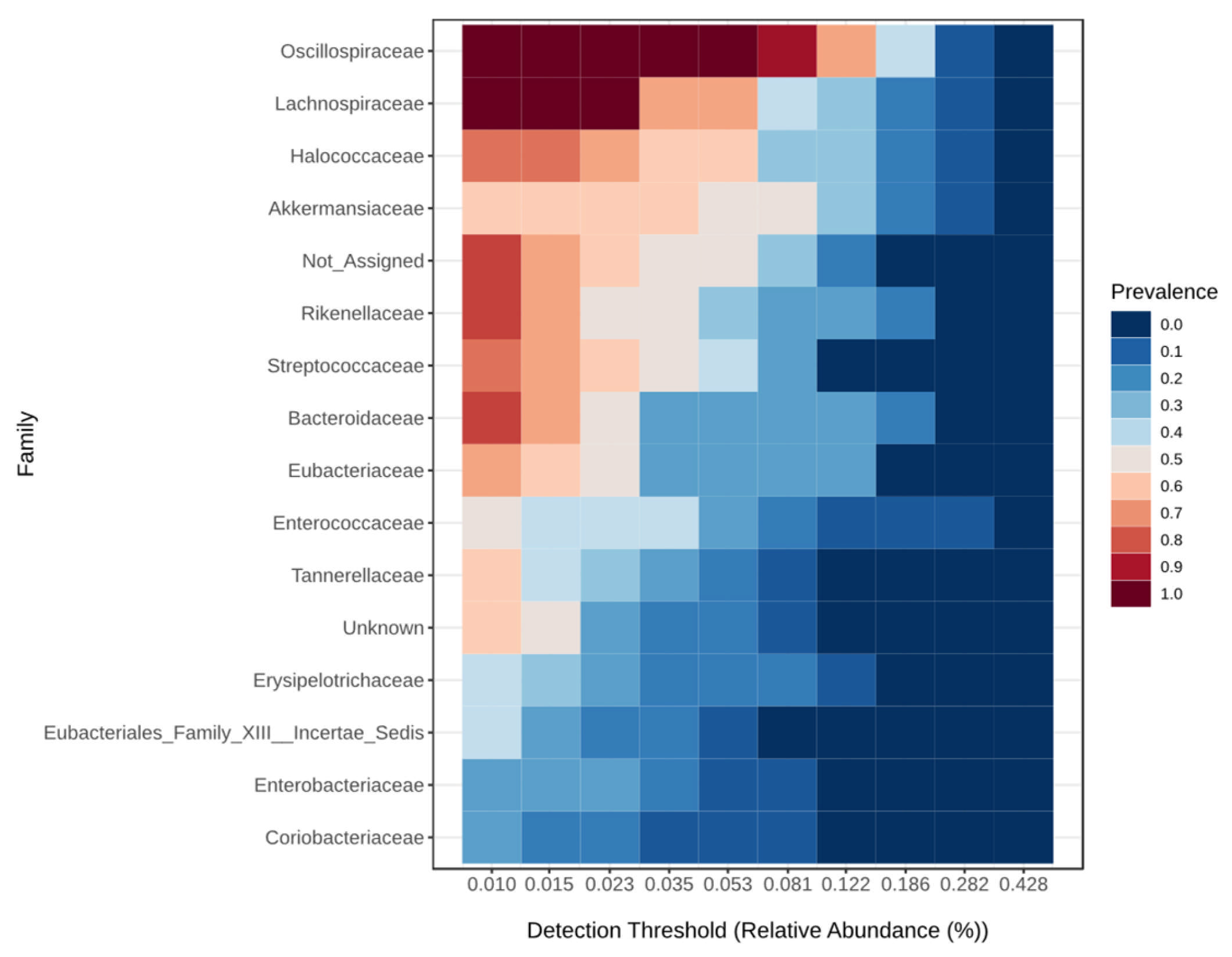
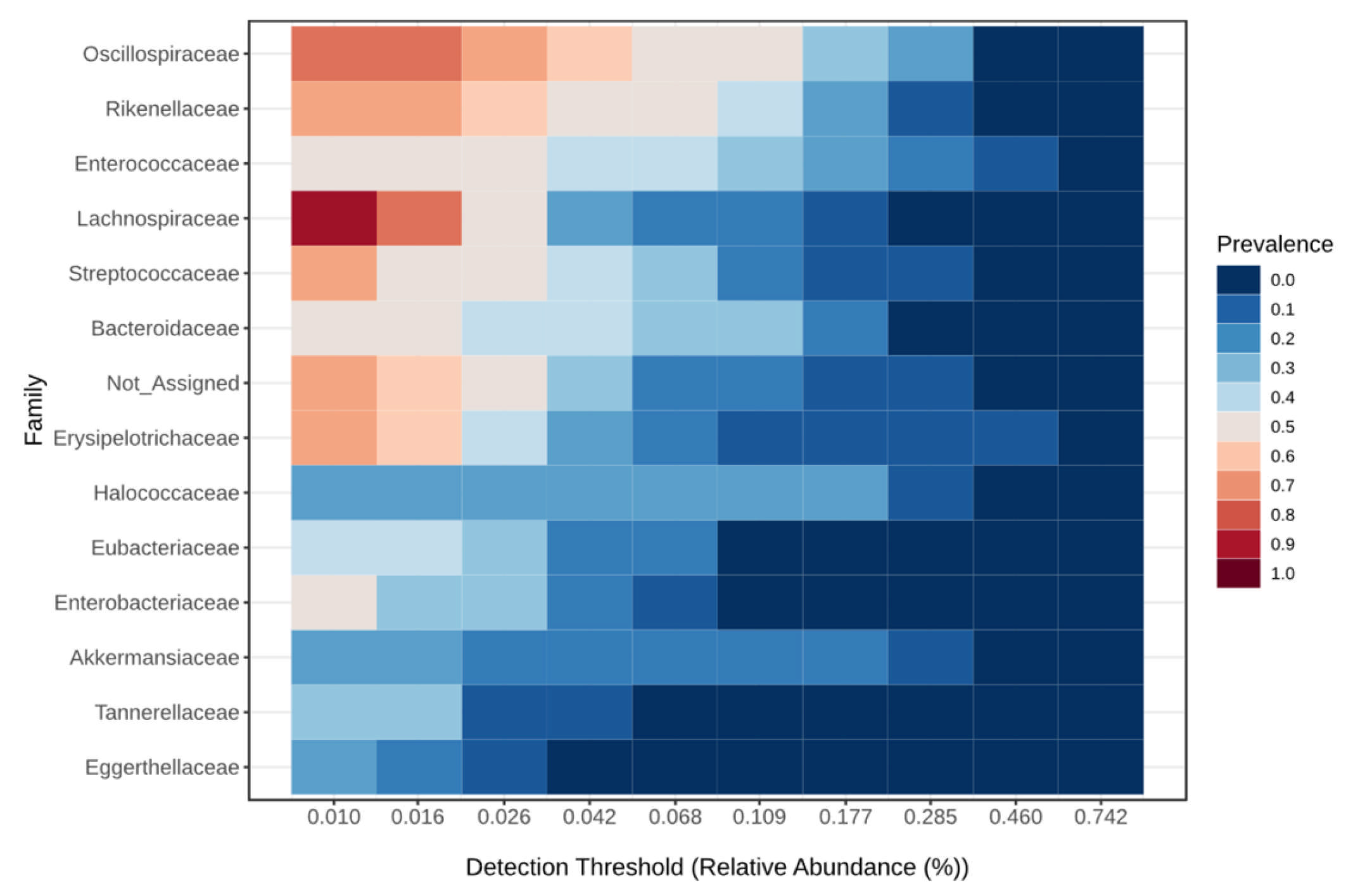
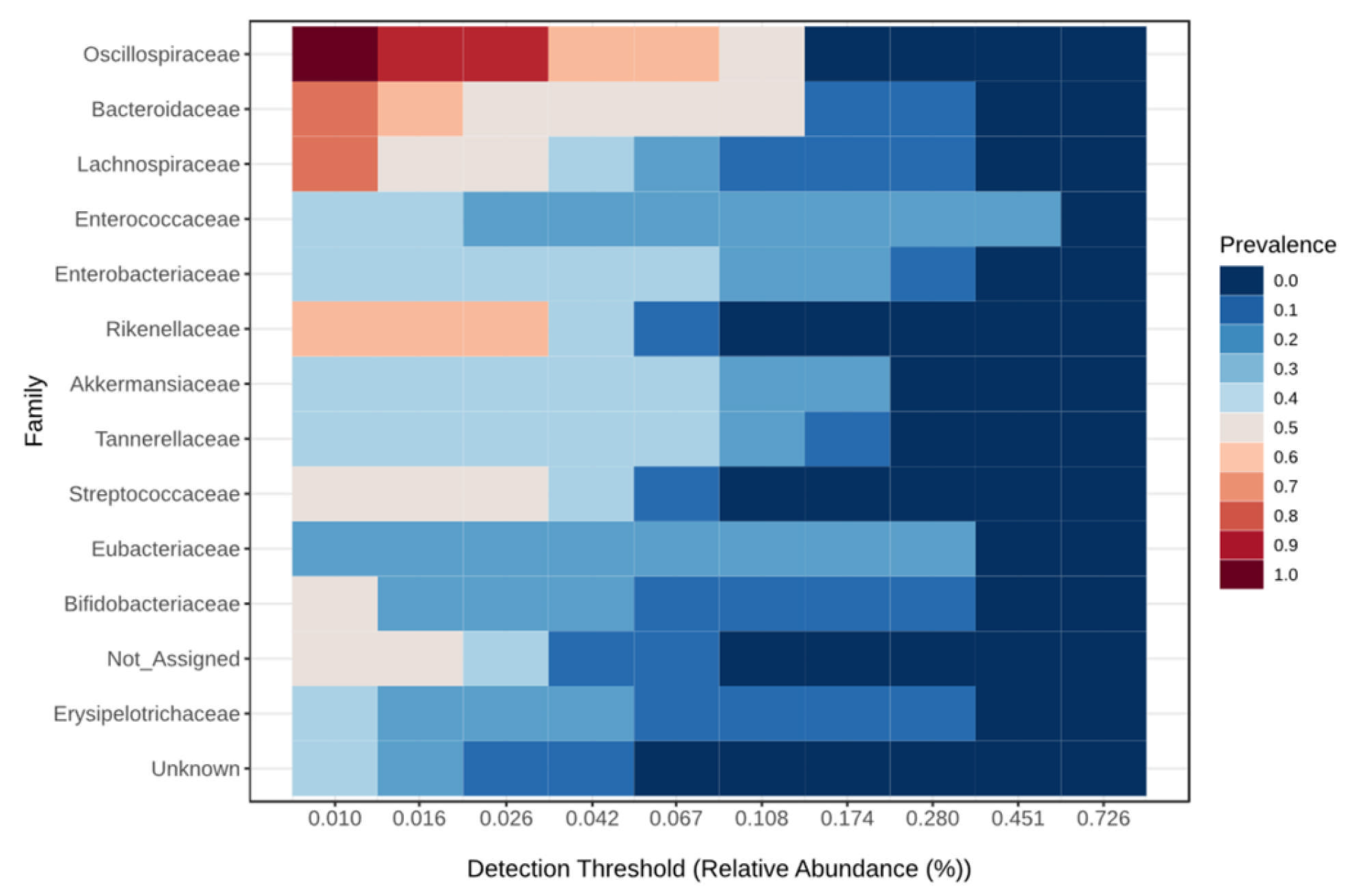
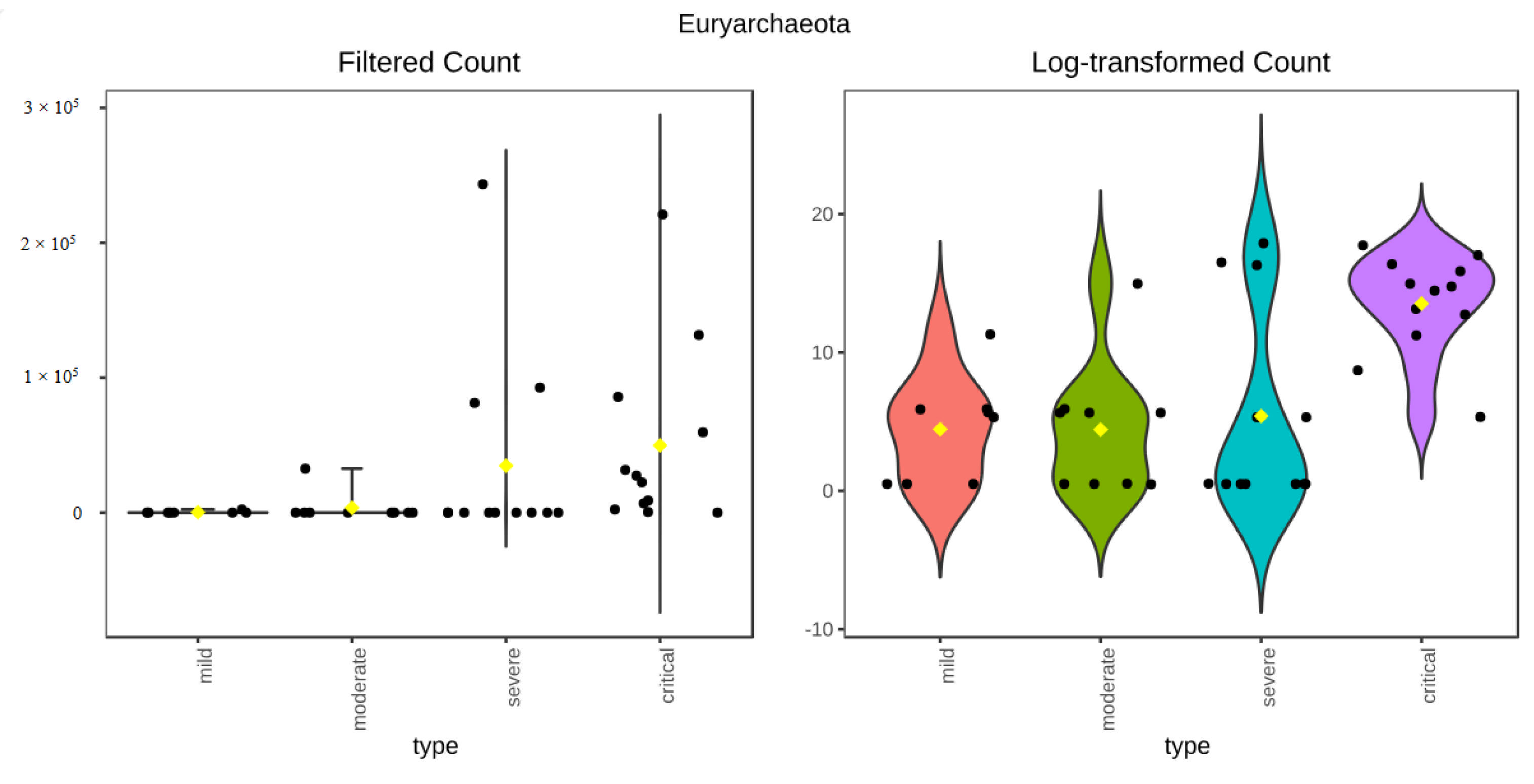
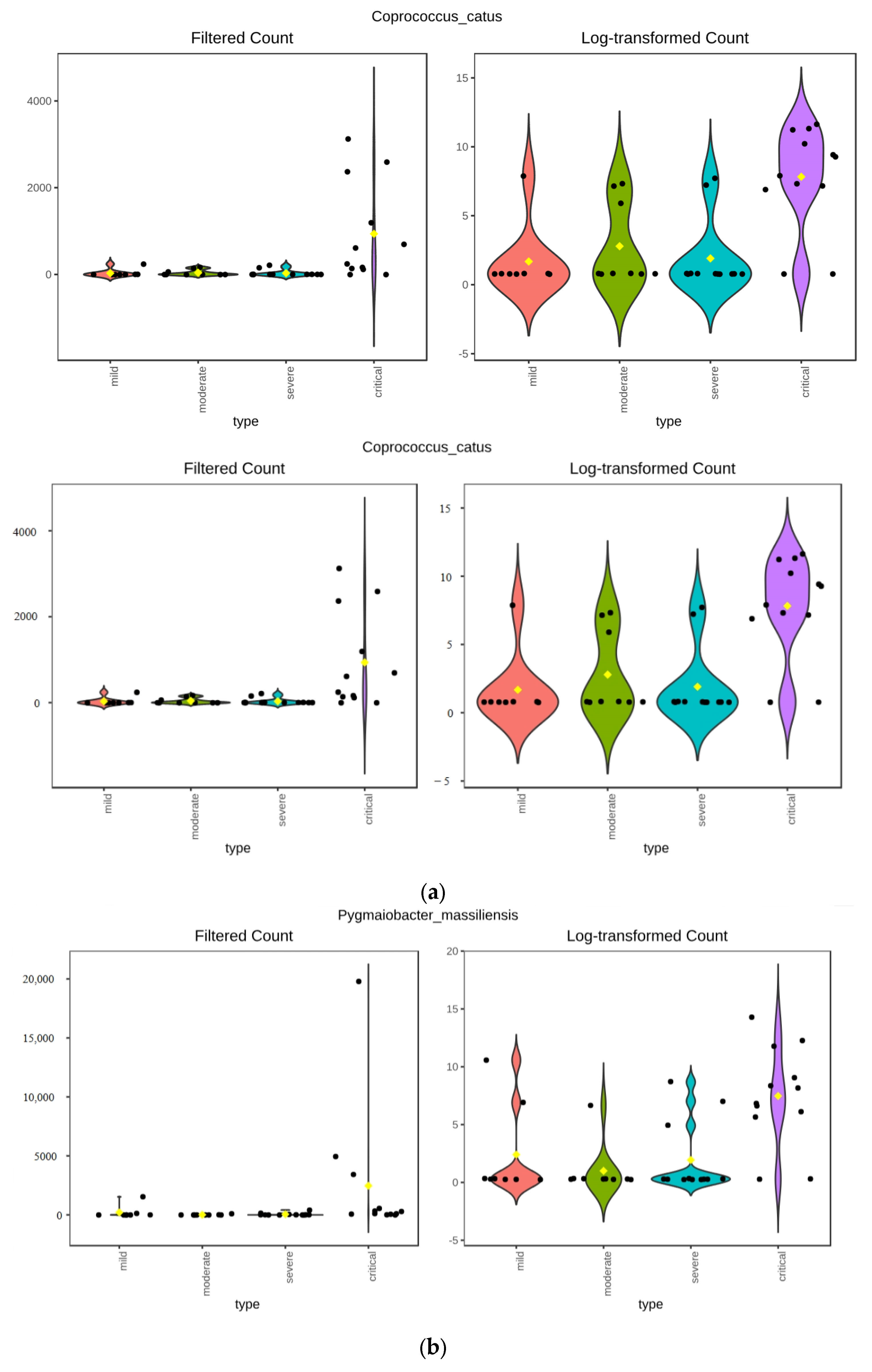
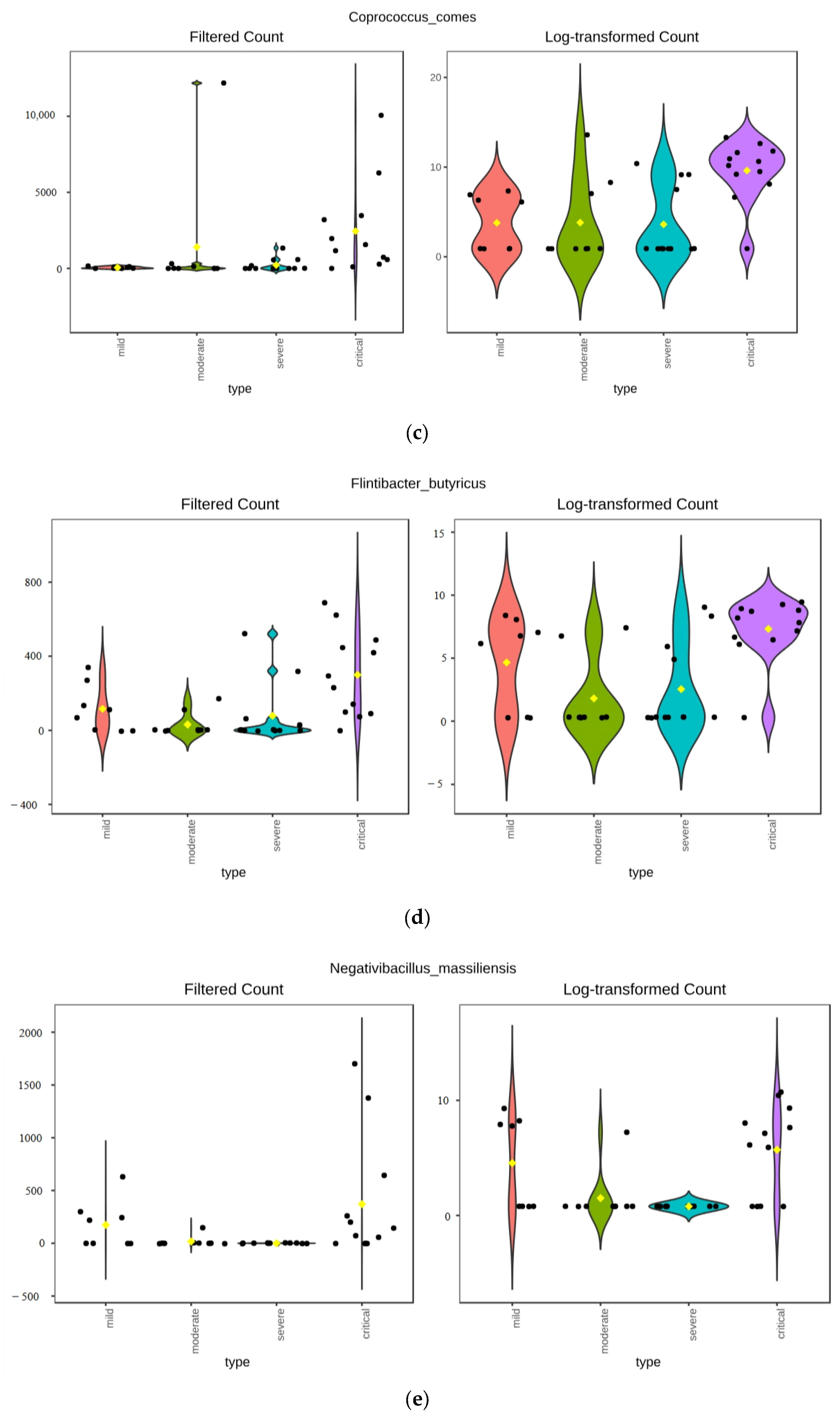
3.2. Taxonomic Abundance
3.3. Differential Abundance
3.3.1. ANCOM Results
3.3.2. Kruskal–Wallis Results
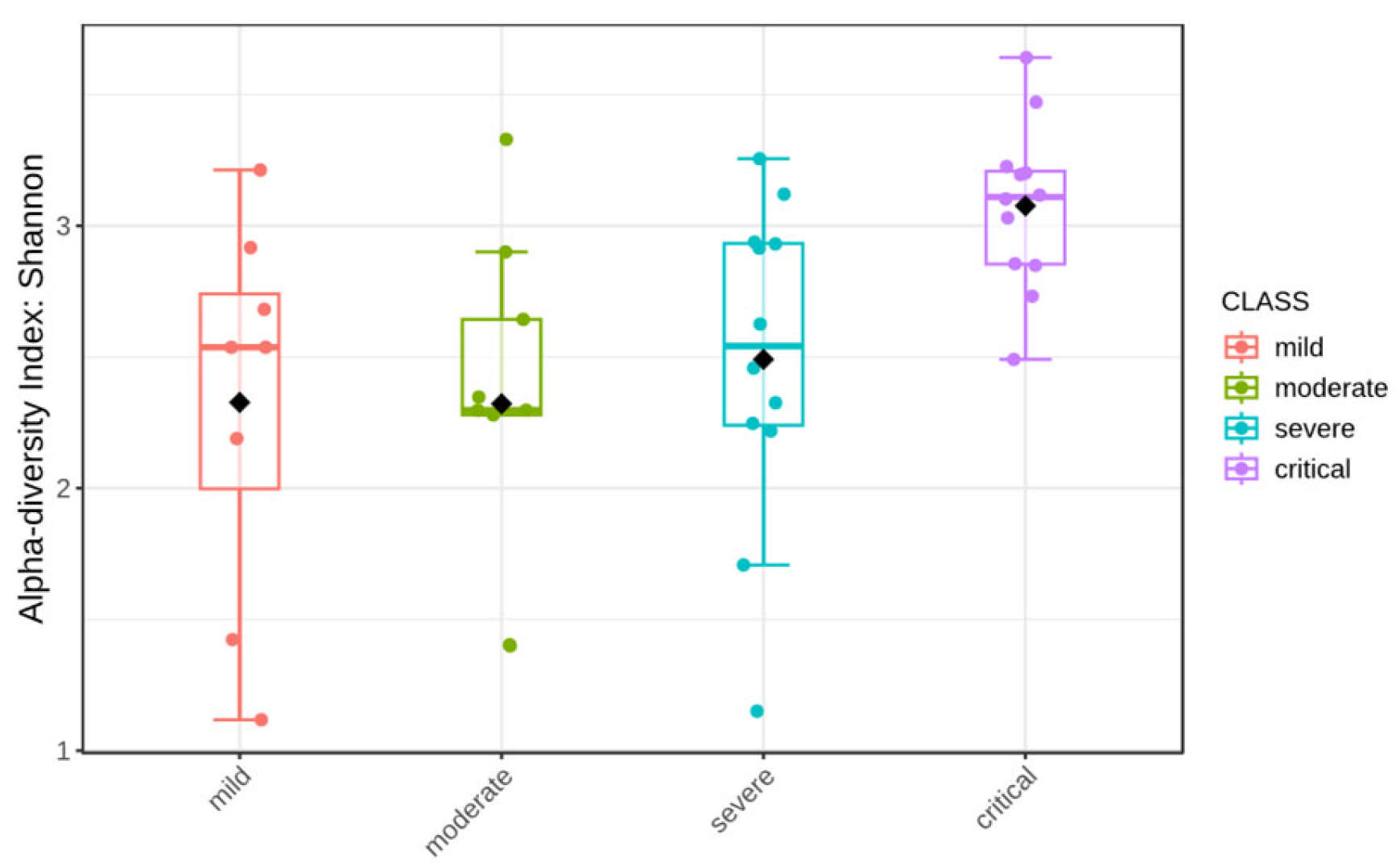
3.4. Alpha Diversity
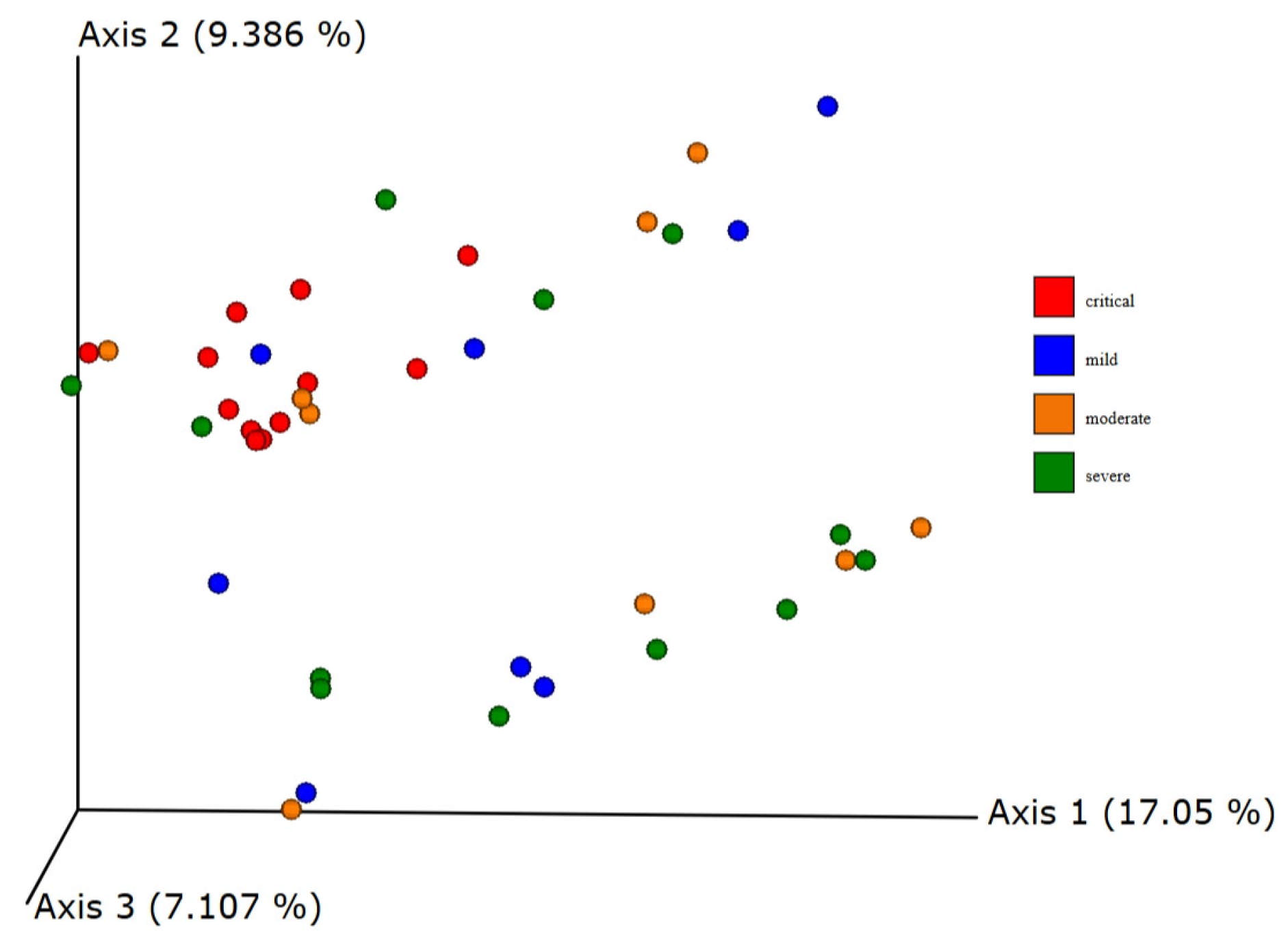
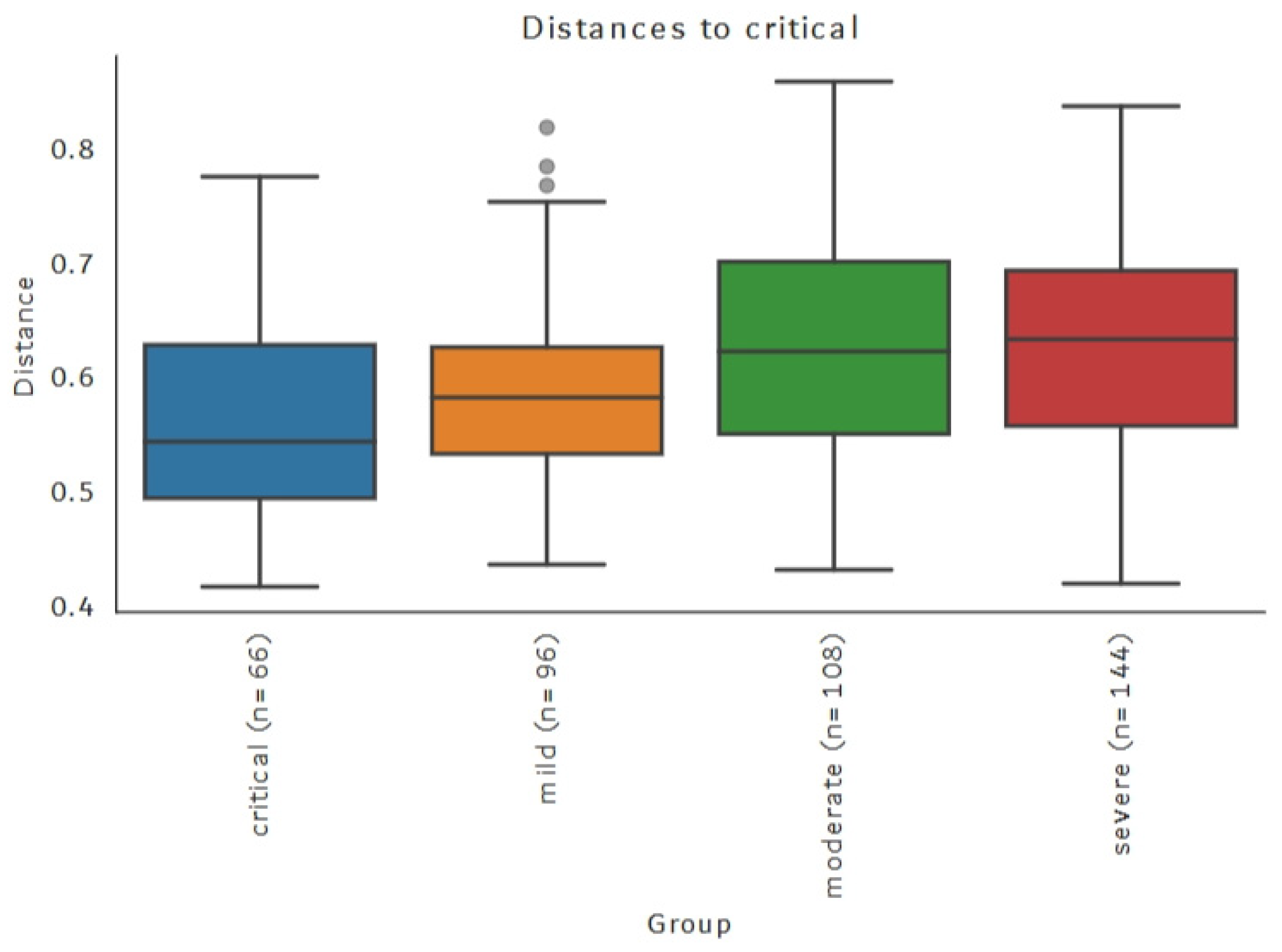
3.5. Beta Diversity
3.6. Differential Abundance Between Groups Based on Antibiotic Use
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yang, H.; Rao, Z. Structural biology of SARS-CoV-2 and implications for therapeutic development. Nat. Rev. Microbiol. 2021, 19, 685–700. [Google Scholar] [CrossRef] [PubMed]
- Schirmer, M.; Smeekens, S.P.; Vlamakis, H.; Jaeger, M.; Oosting, M.; Franzosa, E.A.; Ter Horst, R.; Jansen, T.; Jacobs, L.; Bonder, M.J.; et al. Linking the Human Gut Microbiome to Inflammatory Cytokine Production Capacity. Cell 2016, 167, 1125–1136.e8. [Google Scholar] [CrossRef] [PubMed]
- Yeoh, Y.K.; Zuo, T.; Lui, G.C.-Y.; Zhang, F.; Liu, Q.; Li, A.Y.; Chung, A.C.; Cheung, C.P.; Tso, E.Y.; Fung, K.S.; et al. Gut microbiota composition reflects disease severity and dysfunctional immune responses in patients with COVID-19. Gut 2021, 70, 698–706. [Google Scholar] [CrossRef] [PubMed]
- Shruti Ahlawat, A.; Sharma, K.K. Immunological co-ordination between gut and lungs in SARS-CoV-2 infection. Virus Res. 2020, 286, 198103. [Google Scholar] [CrossRef]
- World Health Organisation. Coronavirus Disease. Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019 (accessed on 1 October 2024).
- Ahmadi Badi, S.; Tarashi, S.; Fateh, A.; Rohani, P.; Masotti, A.; Siadat, S.D. From the Role of Microbiota in Gut-Lung Axis to SARS-CoV-2 Pathogenesis. Mediat. Inflamm. 2021, 2021, 6611222. [Google Scholar] [CrossRef]
- Brogna, C.; Brogna, B.; Bisaccia, D.R.; Lauritano, F.; Marino, G.; Montano, L.; Cristoni, S.; Prisco, M.; Piscopo, M. Could SARS-CoV-2 Have Bacteriophage Behavior or Induce the Activity of Other Bacteriophages? Vaccines 2022, 10, 708. [Google Scholar] [CrossRef]
- Brogna, C.; Costanzo, V.; Brogna, B.; Bisaccia, D.R.; Brogna, G.; Giuliano, M.; Montano, L.; Viduto, V.; Cristoni, S.; Fabrowski, M.; et al. Analysis of Bacteriophage Behavior of a Human RNA Virus, SARS-CoV-2, through the Integrated Approach of Immunofluorescence Microscopy, Proteomics and D-Amino Acid Quantification. Int. J. Mol. Sci. 2023, 24, 3929. [Google Scholar] [CrossRef]
- Kau, A.L.; Ahern, P.P.; Griffin, N.W.; Goodman, A.L.; Gordon, J.I. Human nutrition, the gut microbiome and the immune system. Nature 2011, 474, 327–336. [Google Scholar] [CrossRef]
- Luchen, C.C.; Chibuye, M.; Spijker, R.; Simuyandi, M.; Chisenga, C.; Bosomprah, S.; Chilengi, R.; Schultsz, C.; Mende, D.R.; Harris, V.C. Impact of antibiotics on gut microbiome composition and resistome in the first years of life in low- to middle-income countries: A systematic review. PLoS Med. 2023, 20, e1004235. [Google Scholar] [CrossRef]
- Ramirez, J.; Guarner, F.; Bustos Fernandez, L.; Maruy, A.; Sdepanian, V.L.; Cohen, H. Antibiotics as Major Disruptors of Gut Microbiota. Front. Cell. Infect. Microbiol. 2020, 10, 572912. [Google Scholar] [CrossRef]
- McDonald, D.; Ackermann, G.; Khailova, L.; Baird, C.; Heyland, D.; Kozar, R.; Lemieux, M.; Derenski, K.; King, J.; Vis-Kampen, C.; et al. Extreme Dysbiosis of the Microbiome in Critical Illness. mSphere 2016, 1, 1–6. [Google Scholar]
- Beam, A.; Clinger, E.; Hao, L. Effect of Diet and Dietary Components on the Composition of the Gut Microbiota. Nutrients 2021, 13, 2795. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Living Guidance for Clinical Management of COVID-19. Available online: https://iris.who.int/bitstream/handle/10665/349321/WHO-2019-nCoV-clinical-2021.2-eng.pdf (accessed on 1 October 2024).
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059. [Google Scholar]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Gregory Caporaso, J. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef]
- Molano, L.A.G.; Vega-Abellaneda, S.; Manichanh, C. GSR-DB: A manually curated and optimized taxonomical database for 16S rRNA amplicon analysis. mSystems 2024, 9, e0095023. [Google Scholar] [CrossRef]
- Mandal, S.; Van Treuren, W.; White, R.A.; Eggesbø, M.; Knight, R.; Peddada, S.D. Analysis of composition of microbiomes: A novel method for studying microbial composition. Microb. Ecol. Health Dis. 2015, 26, 27663. [Google Scholar]
- Zhou, J.; Yang, X.; Yang, Y.; Wei, Y.; Lu, D.; Xie, Y.; Liang, H.; Cui, P.; Ye, L.; Huang, J. Human microbiota dysbiosis after SARS-CoV-2 infection have the potential to predict disease prognosis. BMC Infect. Dis. 2023, 23, 841. [Google Scholar] [CrossRef]
- Herrera, G.; Paredes-Sabja, D.; Patarroyo, M.A.; Ramírez, J.D.; Muñoz, M. Updating changes in human gut microbial communities associated with Clostridioides difficile infection. Gut Microbes 2021, 13, 1966277. [Google Scholar]
- Demonfort Nkamga, V.; Henrissat, B.; Drancourt, M. Archaea: Essential inhabitants of the human digestive microbiota. Hum. Microbiome J. 2017, 3, 1–8. [Google Scholar]
- Gaci, N.; Borrel, G.; Tottey, W.; O’Toole, P.W.; Brugère, J.F. Archaea and the human gut: New beginning of an old story. World J. Gastroenterol. 2014, 20, 16062. [Google Scholar]
- Duller, S.; Moissl-Eichinger, C. Archaea in the Human Microbiome and Potential Effects on Human Infectious Disease. Emerg. Infect. Dis. 2024, 30, 1505. [Google Scholar]
- Kim, J.Y.; Whon, T.W.; Lim, M.Y.; Kim, Y.B.; Kim, N.; Kwon, M.S.; Kim, J.; Lee, S.H.; Choi, H.J.; Nam, I.H.; et al. The human gut archaeome: Identification of diverse haloarchaea in Korean subjects. Microbiome 2020, 8, 114. [Google Scholar]
- Kouzuma, A.; Kato, S.; Watanabe, K. Microbial interspecies interactions: Recent findings in syntrophic consortia. Front. Microbiol. 2015, 6, 477. [Google Scholar]
- DeGruttola, A.K.; Low, D.; Mizoguchi, A.; Mizoguchi, E. Current Understanding of Dysbiosis in Disease in Human and Animal Models. Inflamm. Bowel Dis. 2016, 22, 1137. [Google Scholar]
- Zhang, P.; Kong, L.; Huang, H.; Pan, Y.; Zhang, D.; Jiang, J.; Shen, Y.; Xi, C.; Lai, J.; Ng, C.H.; et al. Gut Microbiota—A Potential Contributor in the Pathogenesis of Bipolar Disorder. Front. Neurosci. 2022, 16, 830748. [Google Scholar]
- Tao, W.; Zhang, G.; Wang, X.; Guo, M.; Zeng, W.; Xu, Z. Analysis of the intestinal microbiota in COVID-19 patients and its correlation with the inflammatory factor IL-18. Med. Microecol. 2020, 5, 100023. [Google Scholar]
- Wang, M.; Zhang, Y.; Li, C.; Chang, W.; Zhang, L. The relationship between gut microbiota and COVID-19 progression: New insights into immunopathogenesis and treatment. Front. Immunol. 2023, 14, 1180336. [Google Scholar] [CrossRef]
- Li, S.; Yang, S.; Zhou, Y.; Disoma, C.; Dong, Z.; Du, A.; Zhang, Y.; Chen, Y.; Huang, W.; Chen, J.; et al. Microbiome Profiling Using Shotgun Metagenomic Sequencing Identified Unique Microorganisms in COVID-19 Patients with Altered Gut Microbiota. Front. Microbiol. 2021, 12, 712081. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Cheng, X.; Jiang, G.; Tang, H.; Ming, S.; Tang, L.; Lu, J.; Guo, C.; Shan, H.; Huang, X. Author Correction: Altered oral and gut microbiota and its association with SARS-CoV-2 viral load in COVID-19 patients during hospitalization. npj Biofilms Microbiomes 2021, 7, 90. [Google Scholar] [CrossRef] [PubMed]
- Notting, F.; Pirovano, W.; Sybesma, W.; Kort, R. The butyrate-producing and spore-forming bacterial genus Coprococcus as a potential biomarker for neurological disorders. Gut Microbiome 2023, 4, e16. [Google Scholar] [CrossRef] [PubMed]
- Tomova, A.; Bukovsky, I.; Rembert, E.; Yonas, W.; Alwarith, J.; Barnard, N.D.; Kahleova, H. The Effects of Vegetarian and Vegan Diets on Gut Microbiota. Front. Nutr. 2019, 6, 47. [Google Scholar] [CrossRef]
- Guo, H.; Chen, Y.; Dong, W.; Lu, S.; Du, Y.; Duan, L. Fecal Coprococcus, hidden behind abdominal symptoms in patients with small intestinal bacterial overgrowth. J. Transl. Med. 2024, 22, 496. [Google Scholar] [CrossRef]
- Cao, J.; Wang, C.; Zhang, Y.; Lei, G.; Xu, K.; Zhao, N.; Lu, J.; Meng, F.; Yu, L.; Yan, J.; et al. Integrated gut virome and bacteriome dynamics in COVID-19 patients. Gut Microbes 2021, 13, 1887722. [Google Scholar] [CrossRef]
- Galperine, T.; Choi, Y.; Pagani, J.L.; Kritikos, A.; Papadimitriou-Olivgeris, M.; Méan, M.; Scherz, V.; Opota, O.; Greub, G.; Guery, B.; et al. Temporal changes in fecal microbiota of patients infected with COVID-19: A longitudinal cohort. BMC Infect. Dis. 2023, 23, 537. [Google Scholar] [CrossRef]
- Righi, E.; Vecchia, I.D.; Auerbach, N.; Morra, M.; Górska, A.; Sciammarella, C.; Lambertenghi, L.; Gentilotti, E.; Mirandola, M.; Tacconelli, E.; et al. Gut Microbiome Disruption Following SARS-CoV-2: A Review. Microorganisms 2024, 12, 131. [Google Scholar] [CrossRef]
- Lin, R.; Xiao, M.; Cao, S.; Sun, Y.; Zhao, L.; Mao, X.; Chen, P.; Tong, X.; Ou, Z.; Zhu, H.; et al. Distinct gut microbiota and health outcomes in asymptomatic infection, viral nucleic acid test re-positive, and convalescent COVID-19 cases. mLife 2022, 1, 183–197. [Google Scholar] [CrossRef]
- Shen, Y.; Yu, F.; Zhang, D.; Zou, Q.; Xie, M.; Chen, X.; Yuan, L.; Lou, B.; Xie, G.; Wang, R.; et al. Dynamic Alterations in the Respiratory Tract Microbiota of Patients with COVID-19 and its Association with Microbiota in the Gut. Adv. Sci. 2022, 9, 2200956. [Google Scholar] [CrossRef]
- Mazzarelli, A.; Giancola, M.L.; Fontana, A.; Piselli, P.; Binda, E.; Trivieri, N.; Mencarelli, G.; Marchioni, L.; Vulcano, A.; De Giuli, C.; et al. Gut microbiota composition in COVID-19 hospitalized patients with mild or severe symptoms. Front. Microbiol. 2022, 13, 1049215. [Google Scholar] [CrossRef] [PubMed]
- Fabbrini, M.; D’amico, F.; van der Gun, B.T.F.; Barone, M.; Conti, G.; Roggiani, S.; Wold, K.I.; Vincenti-Gonzalez, M.F.; de Boer, G.C.; Veloo, A.C.M.; et al. The gut microbiota as an early predictor of COVID-19 severity. mSphere 2024, 19, e0018124. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.; Chen, Y.; Wu, Z.; Chen, Y.; Gao, H.; Lv, L.; Guo, F.; Zhang, X.; Luo, R.; Huang, C.; et al. Alterations of the Gut Microbiota in Patients with Coronavirus Disease 2019 or H1N1 Influenza. Clin. Infect. Dis. 2020, 71, 2669–2678. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Taxonomic Level | Taxa | Kruskal–Wallis Test | ANCOM | |
|---|---|---|---|---|
| p-Value | FDR | W Value | ||
| Phylum | Euryarchaeota | 0.0023509 | 0.021158 | 16 |
| Class | Halobacteria | 0.0023509 | 0.039965 | 30 |
| Order | Halobacteriales | 0.0023509 | 0.02586 | 58 |
| Family | Halococcaceae | 0.0023509 | 0.086984 | 68 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jonjić, A.; Dolanc, I.; Slivšek, G.; Bočkor, L.; Tarle, M.; Mustapić, S.; Kmet, M.; Orehovec, B.; Kučan Brlić, P.; Cokarić Brdovčak, M.; et al. Relationship Between Gut Microbiota and the Clinical Course of COVID-19 Disease. Viruses 2025, 17, 520. https://doi.org/10.3390/v17040520
Jonjić A, Dolanc I, Slivšek G, Bočkor L, Tarle M, Mustapić S, Kmet M, Orehovec B, Kučan Brlić P, Cokarić Brdovčak M, et al. Relationship Between Gut Microbiota and the Clinical Course of COVID-19 Disease. Viruses. 2025; 17(4):520. https://doi.org/10.3390/v17040520
Chicago/Turabian StyleJonjić, Antonija, Ivan Dolanc, Goran Slivšek, Luka Bočkor, Marko Tarle, Sanda Mustapić, Marta Kmet, Biserka Orehovec, Paola Kučan Brlić, Maja Cokarić Brdovčak, and et al. 2025. "Relationship Between Gut Microbiota and the Clinical Course of COVID-19 Disease" Viruses 17, no. 4: 520. https://doi.org/10.3390/v17040520
APA StyleJonjić, A., Dolanc, I., Slivšek, G., Bočkor, L., Tarle, M., Mustapić, S., Kmet, M., Orehovec, B., Kučan Brlić, P., Cokarić Brdovčak, M., Obad, A., Walenta, M., Dražić, I., Bilić-Zulle, L., Lukšić, I., Bulić, N., Goessler, W., Jonjić, S., Čoklo, M., & Žučko, J. (2025). Relationship Between Gut Microbiota and the Clinical Course of COVID-19 Disease. Viruses, 17(4), 520. https://doi.org/10.3390/v17040520