
Analysis of Factors That Regulate HIV-1 Fusion in Reverse


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
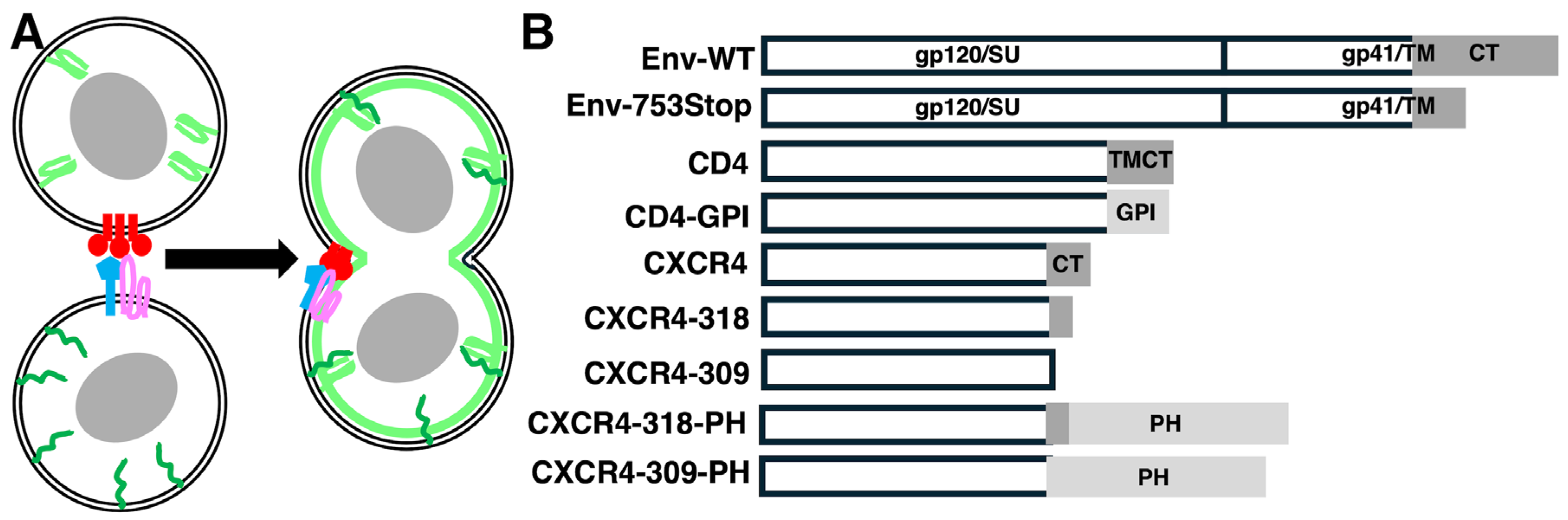
2. Materials and Methods
2.1. Recombinant DNA Constructs
2.2. Cell Culture
2.3. Microscopy
2.4. Protein Analysis
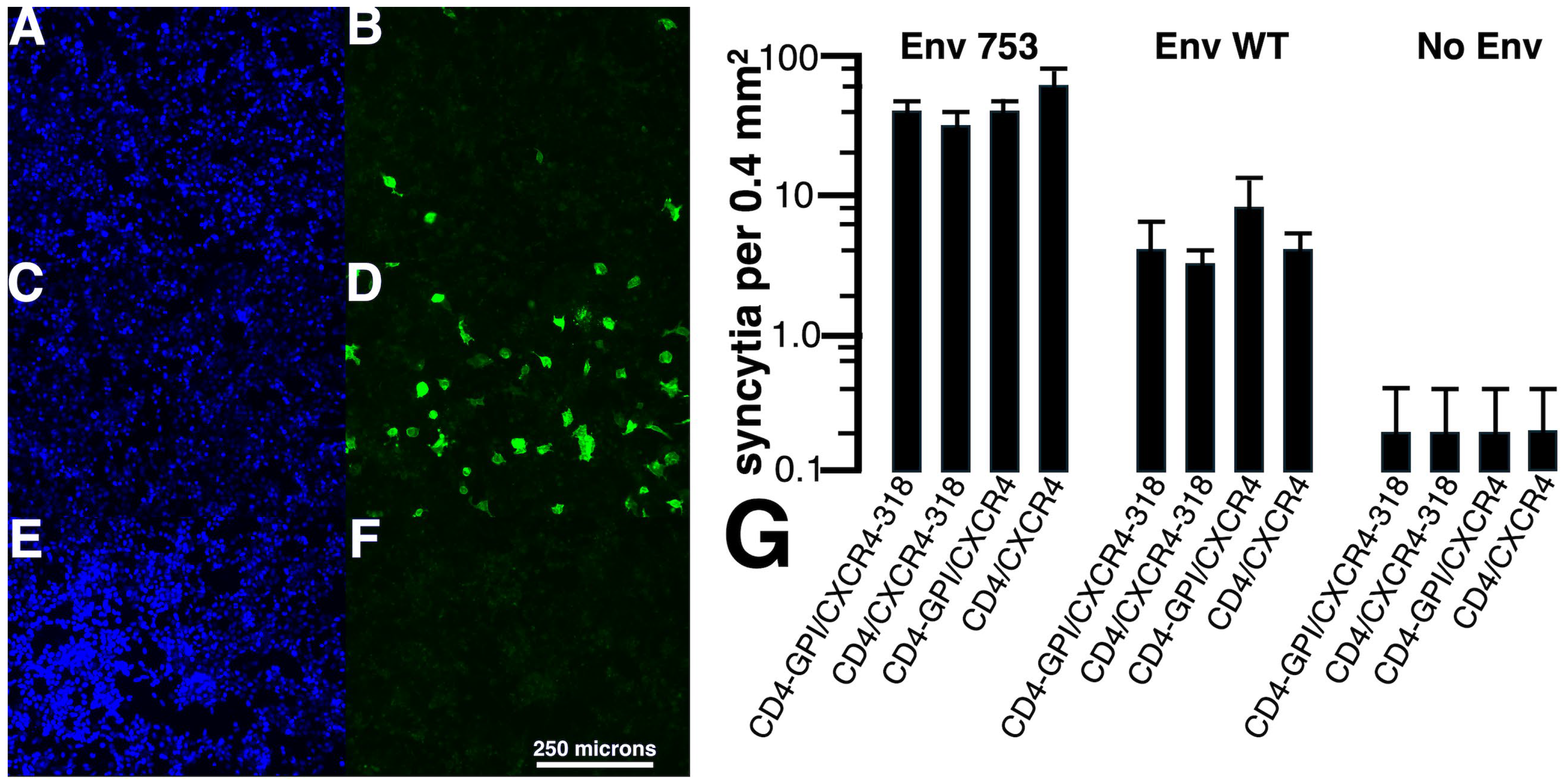
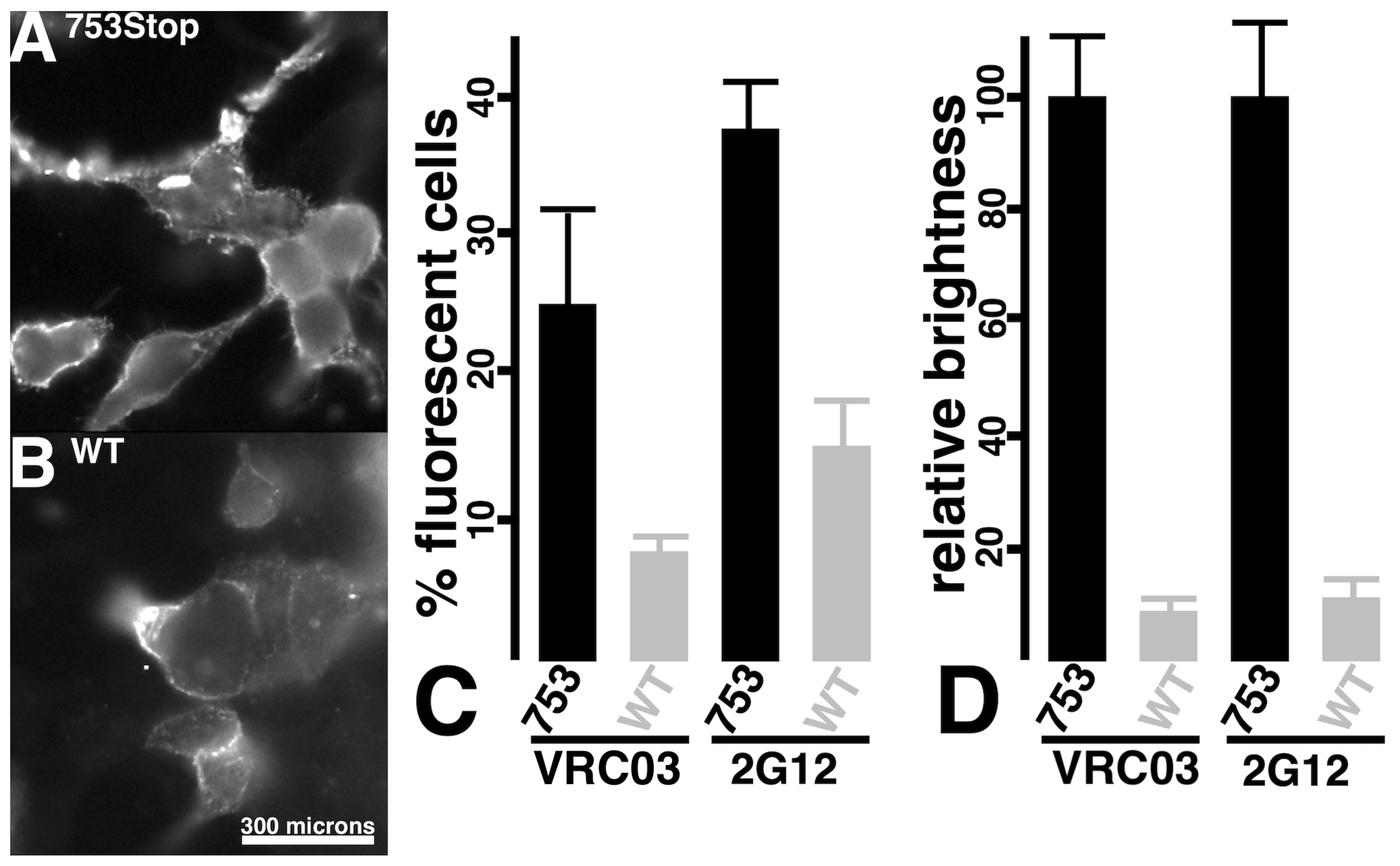
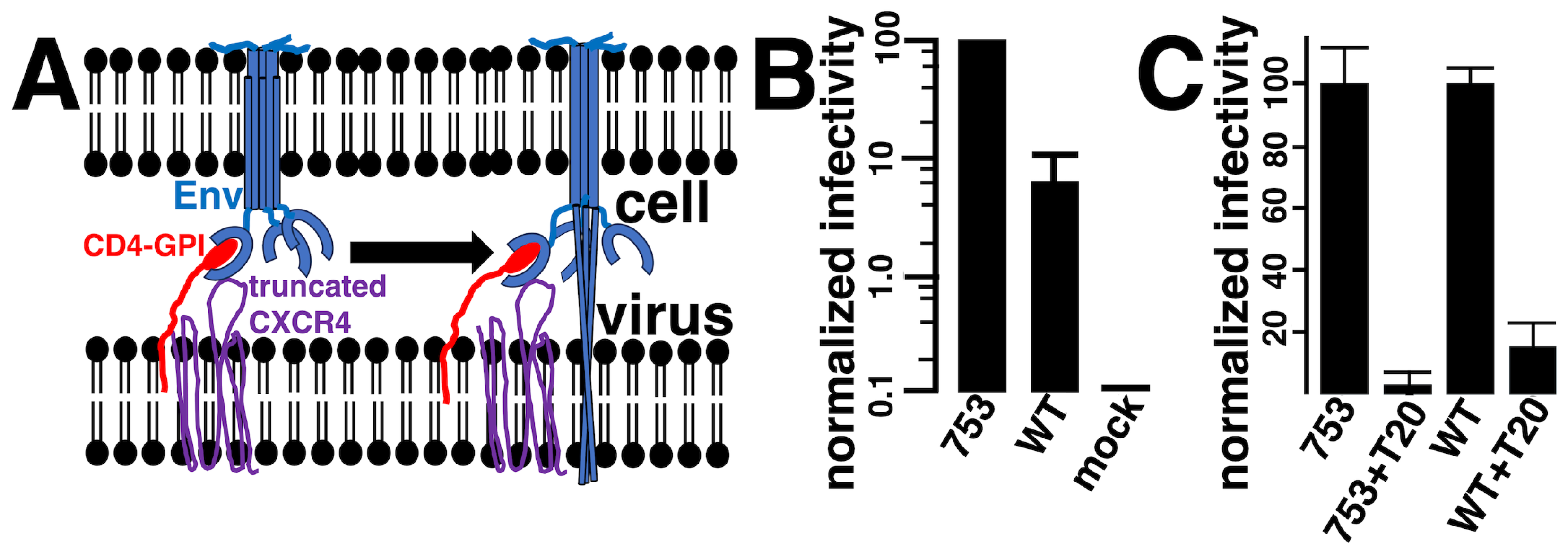
3. Results
3.1. Analysis of Env-Mediated Cell–Cell Fusion
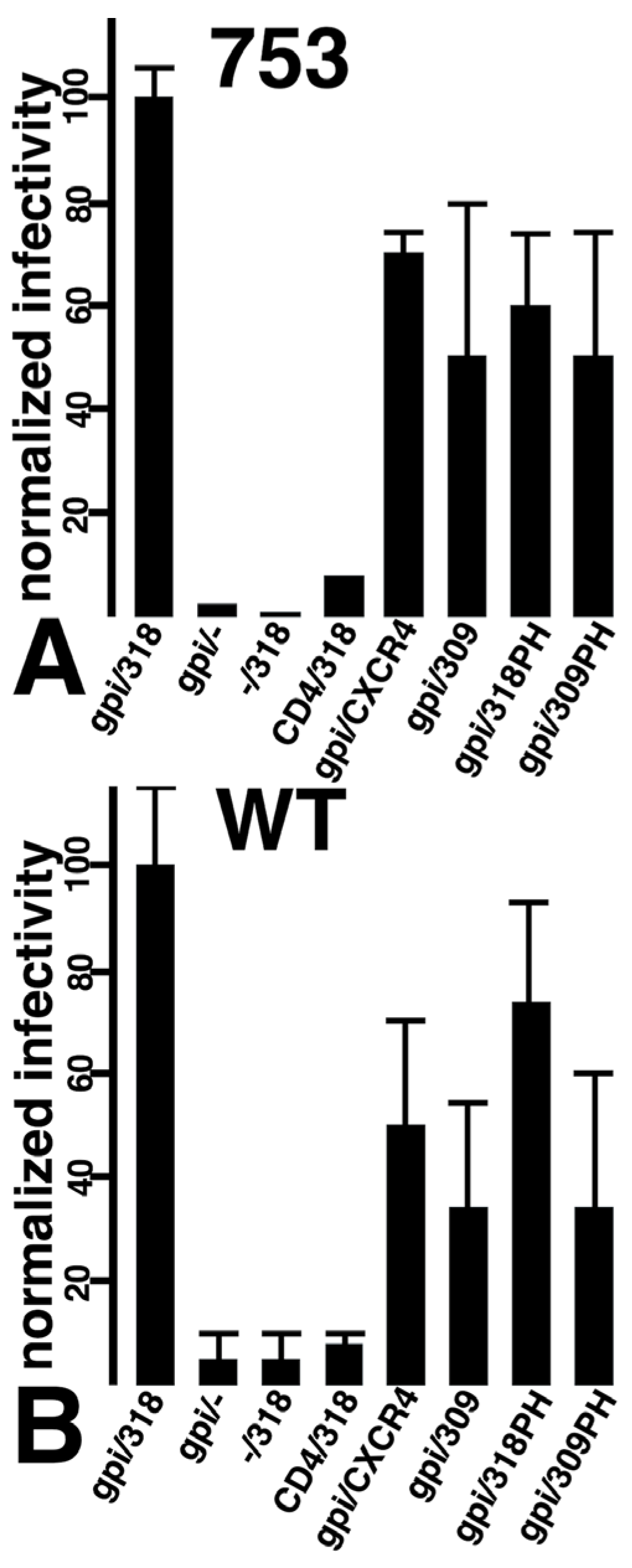
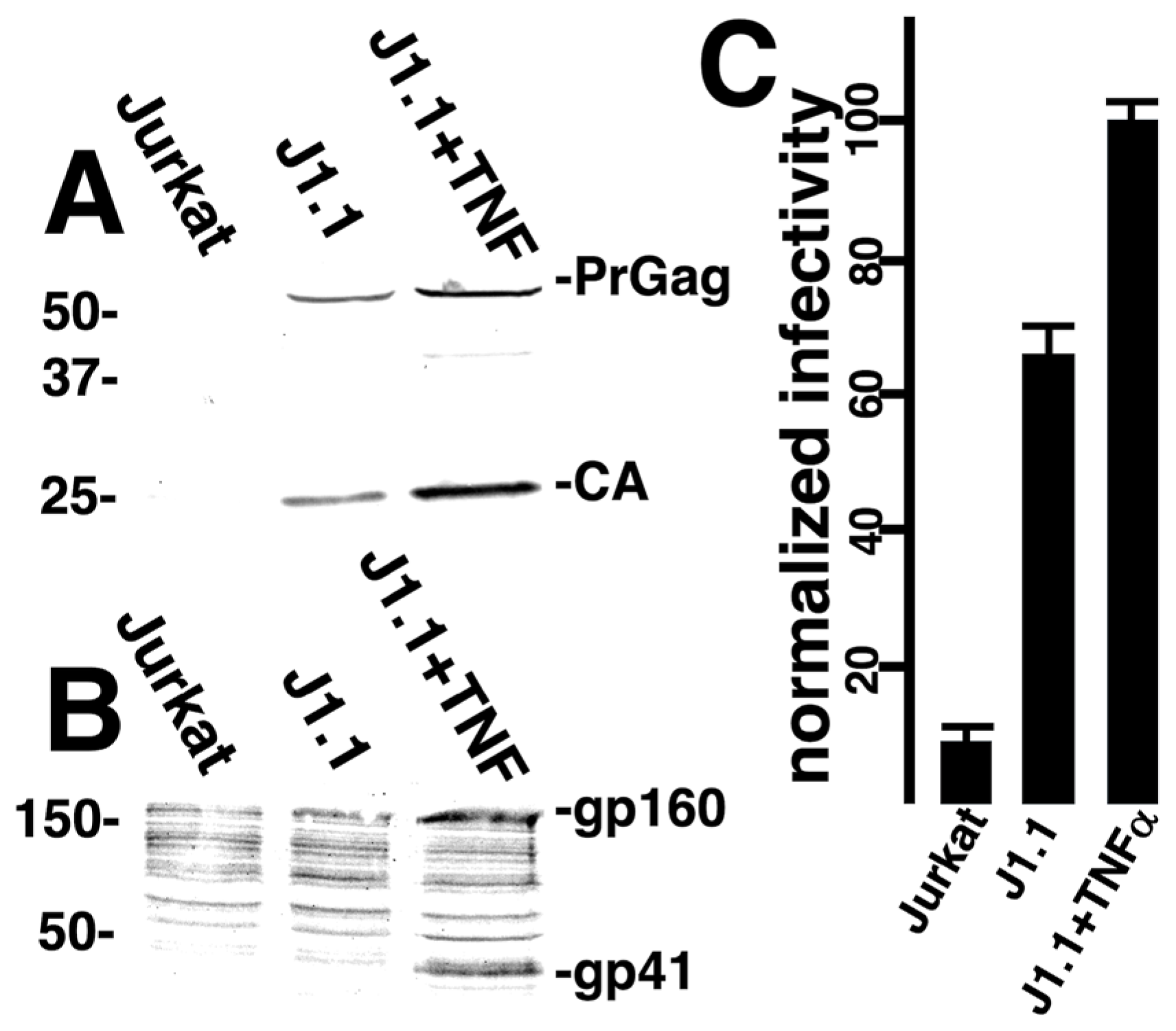
3.2. Viral Infection via Fusion in Reverse
3.3. Targeted Transduction of HIV-1 Infected Cells
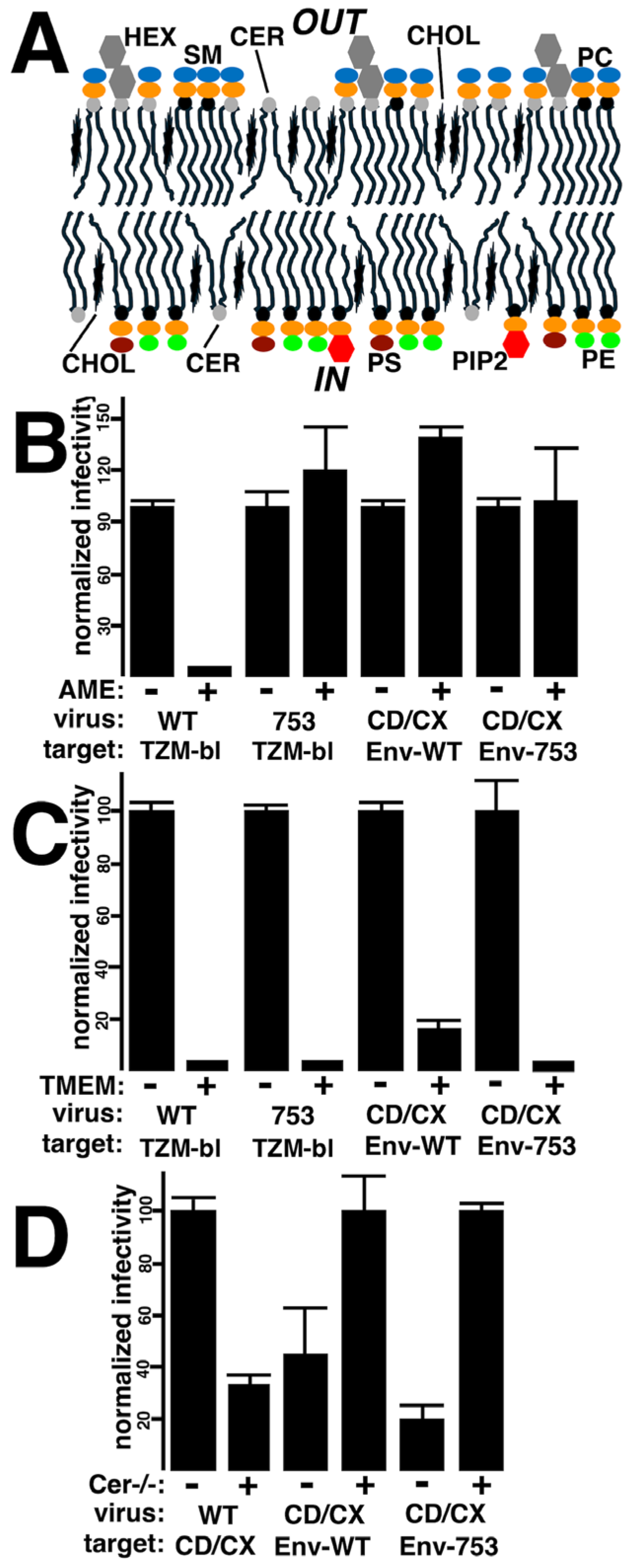
3.4. Effects of Membrane Lipid Compositions
4. Discussion
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AME | amphotericin B methyl ester |
| β-gal | β-galactosidase |
| Cer | ceramide |
| CerS2 | ceramide synthase 2 |
| CT | cytoplasmic tail |
| Env | envelope protein |
| GFP | green fluorescent protein |
| GPI | glycosylphosphatidylinositol |
| HEX | hexosylceramide |
| MA | matrix |
| PC | phosphatidylcholine |
| PE | phosphatidylethanolamine |
| PH | Pleckstrin homology |
| PI(4,5)P2 | phosphatitylinositol-(4,5)-bisphosphate |
| PS | phosphatidylserine |
| SL | sphingolipid |
| SM | sphingomyelin |
References
- Checkley, M.; Luttge, B.; Freed, E. HIV-1 envelope glycoprotein biosynthesis, trafficking, and incorporation. J. Mol. Biol. 2011, 410, 582–608. [Google Scholar] [CrossRef] [PubMed]
- Ohno, H.; Aguilar, R.; Fournier, M.; Hennecke, S.; Cosson, P.; Bonifacino, J. Interaction of endocytic signals from the HIV-1 envelope glycoprotein complex with members of the adaptor medium chain family. Virology 1997, 238, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Boge, M.; Wyss, S.; Bonifacino, J.; Thali, M. A membrane-proximal tyrosine-based signal mediates internalization of the HIV-1 envelope glycoprotein via interaction with the AP-2 clathrin adaptor. J. Biol. Chem. 1998, 273, 15773–15778. [Google Scholar] [CrossRef]
- Wyss, S.; Berlioz-Torrent, C.; Boge, M.; Blot, G.; Höning, S.; Benarous, R.; Thali, M. The highly conserved C-terminal dileucine motif in the cytosolic domain of the human immunodeficiency virus type 1 envelope glycoprotein is critical for its association with the AP-1 clathrin adaptor. J. Virol. 2001, 75, 2982–2992. [Google Scholar] [CrossRef] [PubMed]
- Bhakta, S.; Shang, L.; Prince, D.; Hunter, E. Mutagenesis of tyrosine and di-leucine motifs in the HIV-1 envelope cytoplasmic domain results in a loss of Env-mediated fusion and infectivity. Retrovirology 2011, 8, 37. [Google Scholar] [CrossRef]
- Zhang, S.; Nguyen, H.T.; Ding, H.; Wang, J.; Zou, S.; Liu, L.; Guha, D.; Gabuzda, D.; Ho, D.D.; Kappes, J.C.; et al. Dual Pathways of Human Immunodeficiency Virus Type 1 Envelope Glycoprotein Trafficking Modulate the Selective Exclusion of Uncleaved Oligomers from Virions. J. Virol. 2021, 95, e01369-20. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Qi, M.; Williams, J.; Chu, H.; Chen, X.; Wang, J.; Ding, L.; Akhirome, E.; Wen, X.; Lapierre, L.; Goldenring, J.; et al. Rab11-FIP1C and Rab14 direct plasma membrane sorting and particle incorporation of the HIV-1 envelope glycoprotein complex. PLoS Pathog. 2013, 9, e1003278. [Google Scholar] [CrossRef]
- Qi, M.; Chu, H.; Chen, X.; Choi, J.; Wen, X.; Hammonds, J.; Ding, L.; Hunter, E.; Spearman, E. A tyrosine-based motif in the HIV-1 envelope glycoprotein tail mediates cell-type- and Rab11-FIP1C-dependent incorporation into virions. Proc. Natl. Acad. Sci. USA 2015, 112, 7575–7580. [Google Scholar] [CrossRef]
- Kirschman, J.; Qi, M.; Ding, L.; Hammonds, J.; Dienger-Stambaugh, K.; Wang, J.; Lapierre, L.; Goldenring, J.; Spearman, P. HIV-1 envelope glycoprotein trafficking through the endosomal recycling compartment is required for particle incorporation. J. Virol. 2018, 92, e01893-17. [Google Scholar] [CrossRef]
- Zhu, P.; Liu, J.; Bess, J., Jr.; Chertova, E.; Lifson, J.D.; Grisé, H.; Ofek, G.A.; Taylor, K.A.; Roux, K.H. Distribution and three-dimensional structure of AIDS virus envelope spikes. Nature 2006, 441, 847–852. [Google Scholar] [CrossRef] [PubMed]
- Stano, A.; Leaman, D.P.; Kim, A.S.; Zhang, L.; Autin, L.; Ingale, J.; Gift, S.K.; Truong, J.; Wyatt, R.T.; Olson, A.J.; et al. Dense Array of Spikes on HIV-1 Virion Particles. J. Virol. 2017, 91, e00415-17. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mangala Prasad, V.; Leaman, D.P.; Lovendahl, K.N.; Croft, J.T.; Benhaim, M.A.; Hodge, E.A.; Zwick, M.B.; Lee, K.K. Cryo-ET of Env on intact HIV virions reveals structural variation and positioning on the Gag lattice. Cell 2022, 185, 641–653.e17. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Alfadhli, A.; Romanaggi, C.; Barklis, R.L.; Barklis, E. Analysis of HIV-1 envelope cytoplasmic tail effects on viral replication. Virology 2023, 579, 54–66. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Brugger, B.; Glass, B.; Haberkant, P.; Liebrecht, I.; Wieland, F.; Krausslich, H.-G. The HIV lipidome: A raft with an unusual composition. Proc. Natl. Acad. Sci. USA 2006, 103, 2641–2646. [Google Scholar] [CrossRef]
- Chan, R.; Uchil, P.D.; Jin, J.; Shui, G.; Ott, D.E.; Mothes, W.; Wenk, M.R. Retroviruses human immunodeficiency virus and murine leukemia virus are enriched in phosphoinositides. J. Virol. 2008, 82, 11228–11238. [Google Scholar]
- Lorizate, M.; Sachsenheimer, T.; Glass, B.; Habermann, A.; Gerl, M.J.; Krausslich, H.-G.; Brugger, B. Comparative lipidomics analysis of HIV-1 particles and their producer cell membrane in different cell lines. Cell. Microbiol. 2013, 15, 292–304. [Google Scholar]
- Nguyen, D.H.; Hildreth, J.E. Evidence for budding of human immunodeficiency virus type 1 selectively from glycolipid-enriched membrane lipid rafts. J. Virol. 2000, 74, 3264–3272. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ono, A.; Freed, E.O. Plasma membrane rafts play a critical role in HIV-1 assembly and release. Proc. Natl. Acad. Sci. USA 2001, 98, 13925–13930. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ono, A.; Ablan, S.D.; Lockett, S.J.; Nagashima, K.; Freed, E.O. Phosphatidylinositol (4,5) bisphosphate regulates HIV-1 Gag targeting to the plasma membrane. Proc. Natl. Acad. Sci. USA 2004, 101, 14889–14894. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Krementsov, D.N.; Rassam, P.; Margeat, E.; Roy, N.H.; Schneider-Schaulies, J.; Milhiet, P.E.; Thali, M. HIV-1 assembly differentially alters dynamics and partitioning of tetraspanins and raft components. Traffic 2010, 11, 1401–1414. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hogue, I.B.; Grover, J.R.; Soheilian, F.; Nagashima, K.; Ono, A. Gag induces the coalescence of clustered lipid rafts and tetraspanin-enriched microdomains at HIV-1 assembly sites on the plasma membrane. J. Virol. 2011, 85, 9749–9766. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Inlora, J.; Collins, D.R.; Trubin, M.E.; Chung, J.Y.; Ono, A. Membrane binding and subcellular localization of retroviral Gag proteins are differentially regulated by MA interactions with phosphatidylinositol-(4,5)-bisphosphate and RNA. mBio 2014, 5, e02202. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Patil, A.; Gautam, A.; Bhattacharya, J. Evidence that Gag facilitates HIV-1 envelope association both in GPI-enriched plasma membrane and detergent resistant membranes and facilitates envelope incorporation onto virions in primary CD4+ T cells. Virol. J. 2010, 7, 3. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tomishige, N.; Bin Nasim, M.; Murate, M.; Pollet, B.; Didier, P.; Godet, J.; Richert, L.; Sako, Y.; Mély, Y.; Kobayashi, T. HIV-1 Gag targeting to the plasma membrane reorganizes sphingomyelin-rich and cholesterol-rich lipid domains. Nat. Commun. 2023, 14, 7353. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Barklis, E.; Alfadhli, A.; Kyle, J.E.; Bramer, L.M.; Bloodsworth, K.J.; Barklis, R.L.; Leier, H.C.; Petty, R.M.; Zelnik, I.D.; Metz, T.O.; et al. Ceramide synthase 2 deletion decreases the infectivity of HIV-1. J. Biol. Chem. 2021, 296, 100340. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Waheed, A.; Ablan, S.; Mankowski, M.; Cummins, J.; Ptak, R.; Schaffner, C.; Freed, E. Inhibition of HIV-1 replication by amphotericin B methyl ester. J. Biol. Chem. 2006, 281, 28699–28711. [Google Scholar]
- Waheed, A.; Ablan, S.; Roser, J.; Sowder, R.; Schaffner, C.; Chertova, E.; Freed, E. HIV-1 escape from the entry-inhibiting effects of a cholesterol-binding compound via cleavage of gp41 by the viral protease. Proc. Natl. Acad. Sci. USA 2007, 104, 8467–8471. [Google Scholar]
- Waheed, A.; Ablan, S.; Sowder, R.; Roser, J.; Schaffner, C.; Chertova, E.; Freed, E. Effect of mutations in the human immunodeficiency virus 1 protease on cleavage of the gp41 cytoplasmic tail. J. Virol. 2010, 84, 3121–3126. [Google Scholar]
- Contreras, F.X.; Sánchez-Magraner, L.; Alonso, A.; Goñi, F.M. Transbilayer (flip-flop) lipid motion and lipid scrambling in membranes. FEBS Lett. 2010, 584, 1779–1786. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, J.; Umeda, M.; Sims, P.J.; Nagata, S. Calcium-dependent phospholipid scrambling by TMEM16F. Nature 2010, 468, 834–838. [Google Scholar] [CrossRef] [PubMed]
- Segawa, K.; Suzuki, J.; Nagata, S. Constitutive exposure of phosphatidylserine on viable cells. Proc. Natl. Acad. Sci. USA 2011, 108, 19246–19251. [Google Scholar] [CrossRef] [PubMed]
- Kunzelmann, K.; Nilius, B.; Owsianik, G.; Schreiber, R.; Ousingsawat, J.; Sirianant, L.; Wanitchakool, P.; Bevers, E.M.; Heemskerk, J.W. Molecular functions of anoctamin 6 (TMEM16F): A chloride channel, cation channel, or phospholipid scramblase? Pflugers Arch. 2014, 466, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Arndt, M.; Alvadia, C.; Straub, M.S.; Clerico Mosina, V.; Paulino, C.; Dutzler, R. Structural basis for the activation of the lipid scramblase TMEM16F. Nat Commun. 2022, 13, 6692. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Leonhardt, S.A.; Purdy, M.D.; Grover, J.R.; Yang, Z.; Poulos, S.; McIntire, W.E.; Tatham, E.A.; Erramilli, S.K.; Nosol, K.; Lai, K.K.; et al. Antiviral HIV-1 SERINC restriction factors disrupt virus membrane asymmetry. Nat. Commun. 2023, 14, 4368. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Camerini, D.; Seed, B. A CD4 domain important for HIV-mediated syncytium formation lies outside the virus binding site. Cell 1990, 60, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, M.I.; Koyanagi, Y.; Suzuki, M.; Kobayashi, S.; Yamaguchi, K.; Yamamoto, N. Increased production of human immunodeficiency virus (HIV) in HIV-induced syncytia formation: An efficient infection process. Virus Genes 1992, 6, 63–78. [Google Scholar] [CrossRef] [PubMed]
- Valentin, A.; Trivedi, H.; Lu, W.; Kostrikis, L.G.; Pavlakis, G.N. CXCR4 mediates entry and productive infection of syncytia-inducing (X4) HIV-1 strains in primary macrophages. Virology 2000, 269, 294–304. [Google Scholar] [CrossRef] [PubMed]
- Symeonides, M.; Murooka, T.T.; Bellfy, L.N.; Roy, N.H.; Mempel, T.R.; Thali, M. HIV-1-Induced Small T Cell Syncytia Can Transfer Virus Particles to Target Cells through Transient Contacts. Viruses 2015, 7, 6590–6603. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Starling, T.; Padilla-Parra, S. HIV-1 Induced Cell-to-Cell Fusion or Syncytium Formation. In Syncytia: Origin, Structure, and Functions. Results and Problems in Cell Differentiation; Kloc, M., Uosef, A., Eds.; Springer: Cham, Switzerland, 2024; Volume 71. [Google Scholar] [CrossRef]
- Mebatsion, T.; Finke, S.; Weiland, F.; Conzelmann, K.K. A CXCR4/CD4 pseudotype rhabdovirus that selectively infects HIV-1 envelope protein-expressing cells. Cell 1997, 90, 841–847. [Google Scholar] [CrossRef] [PubMed]
- Schnell, M.J.; Johnson, J.E.; Buonocore, L.; Rose, J.K. Construction of a novel virus that targets HIV-1-infected cells and controls HIV-1 infection. Cell 1997, 90, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Endres, M.J.; Jaffer, S.; Haggarty, B.; Turner, J.D.; Doranz, B.J.; O’Brien, P.J.; Kolson, D.L.; Hoxie, J.A. Targeting of HIV- and SIV-infected cells by CD4-chemokine receptor pseudotypes. Science 1997, 278, 1462–1464. [Google Scholar] [CrossRef] [PubMed]
- Somia, N.V.; Miyoshi, H.; Schmitt, M.J.; Verma, I.M. Retroviral vector targeting to human immunodeficiency virus type 1-infected cells by receptor pseudotyping. J. Virol. 2000, 74, 4420–4424. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bittner, A.; Mitnacht-Kraus, R.; Schnierle, B.S. Specific transduction of HIV-1 envelope expressing cells by retroviral vectors pseudotyped with hybrid CD4/CXCR4 receptors. J. Virol. Methods 2002, 104, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Harmison, G.G.; Ragheb, J.A.; Schubert, M. Targeted infection of HIV-1 Env expressing cells by HIV(CD4/CXCR4) vectors reveals a potential new rationale for HIV-1 mediated down-modulation of CD4. Retrovirology 2005, 2, 80. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lee, C.L.; Dang, J.; Joo, K.I.; Wang, P. Engineered lentiviral vectors pseudotyped with a CD4 receptor and a fusogenic protein can target cells expressing HIV-1 envelope proteins. Virus Res. 2011, 160, 340–350. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Peretti, S.; Schiavoni, I.; Pugliese, K.; Federico, M. Selective elimination of HIV-1-infected cells by Env-directed, HIV-1-based virus-like particles. Virology 2006, 345, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Kamiyama, D.; Sekine, S.; Barsi-Rhyne, B.; Hu, J.; Chen, B.; Gilbert, L.A.; Ishikawa, H.; Leonetti, M.D.; Marshall, W.F.; Weissman, J.S.; et al. Versatile protein tagging in cells with split fluorescent protein. Nat. Commun. 2016, 7, 11046. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Stauffer, T.P.; Ahn, S.; Meyer, T. Receptor-induced transient reduction in plasma membrane PtdIns(4,5)P2 concentration monitored in living cells. Curr. Biol. 1998, 8, 343–346. [Google Scholar] [CrossRef] [PubMed]
- Zuffery, R.; Nagy, D.; Mandel, R.; Naldini, L.; Trono, D. Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nat. Biotechnol. 1997, 15, 871–875. [Google Scholar]
- Syed, A.M.; Taha, T.Y.; Tabata, T.; Chen, I.P.; Ciling, A.; Khalid, M.M.; Sreekumar, B.; Chen, P.Y.; Hayashi, J.M.; Soczek, K.M.; et al. Rapid assessment of SARS-CoV-2-evolved variants using virus-like particles. Science 2021, 374, 1626–1632. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sušac, L.; Vuong, M.T.; Thomas, C.; von Bülow, S.; O’Brien-Ball, C.; Santos, A.M.; Fernandes, R.A.; Hummer, G.; Tampé, R.; Davis, S.J. Structure of a fully assembled tumor-specific T cell receptor ligated by pMHC. Cell 2022, 185, 3201–3213.e19. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Heredia, J.D.; Park, J.; Brubaker, R.J.; Szymanski, S.K.; Gill, K.S.; Procko, E. Mapping Interaction Sites on Human Chemokine Receptors by Deep Mutational Scanning. J. Immunol. 2018, 200, 3825–3839. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chesarino, N.M.; McMichael, T.M.; Hach, J.C.; Yount, J.S. Phosphorylation of the antiviral protein interferon-inducible transmembrane protein 3 (IFITM3) dually regulates its endocytosis and ubiquitination. J. Biol. Chem. 2014, 289, 11986–11992. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Alfadhli, A.; Bates, T.A.; Barklis, R.L.; Romanaggi, C.; Tafesse, F.G.; Barklis, E. A nanobody interaction with SARS-COV-2 Spike allows the versatile targeting of lentivirus vectors. J. Virol. 2024, 98, e0079524. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- López, C.S.; Sloan, R.; Cylinder, I.; Kozak, S.L.; Kabat, D.; Barklis, E. RRE-dependent HIV-1 Env RNA effects on Gag protein expression, assembly and release. Virology 2014, 462–463, 126–134. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Adachi, A.; Gendelman, H.E.; Koenig, S.; Folks, T.; Willey, R.; Rabson, A.; Martin, M.A. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J. Virol. 1986, 59, 284–291. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Millán, J.L. Molecular cloning and sequence analysis of human placental alkaline phosphatase. J. Biol. Chem. 1986, 261, 3112–3125, Erratum in J. Biol. Chem. 1991, 266, 4023. [Google Scholar] [CrossRef] [PubMed]
- Mace, M.C.; Hansen, M.; Whiting, S.; Wang, C.T.; Barklis, E. Retroviral envelope protein fusions to secreted and membrane markers. Virology 1992, 188, 869–874. [Google Scholar] [CrossRef] [PubMed]
- Scholz, I.; Still, A.; Dhenub, T.C.; Coday, K.; Webb, M.; Barklis, E. Analysis of human immunodeficiency virus matrix domain replacements. Virology 2008, 371, 322–335. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Barklis, E.; Staubus, A.O.; Mack, A.; Harper, L.; Barklis, R.L.; Alfadhli, A. Lipid biosensor interactions with wild type and matrix deletion HIV-1 Gag proteins. Virology 2018, 518, 264–271. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- DuBridge, R.B.; Tang, P.; Hsia, H.C.; Leong, P.M.; Miller, J.H.; Calos, M.P. Analysis of mutation in human cells by using an Epstein-Barr virus shuttle system. Mol. Cell Biol. 1987, 7, 379–387. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Platt, E.J.; Wehrly, K.; Kuhmann, S.E.; Chesebro, B.; Kabat, D. Effects of CCR5 and CD4 cell surface concentrations on infections by macrophagetropic isolates of human immunodeficiency virus type 1. J. Virol. 1998, 72, 2855–2864. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Derdeyn, C.A.; Decker, J.M.; Sfakianos, J.N.; Wu, X.; O’Brien, W.A.; Ratner, L.; Kappes, J.C.; Shaw, G.M.; Hunter, E. Sensitivity of human immunodeficiency virus type 1 to the fusion inhibitor T-20 is modulated by coreceptor specificity defined by the V3 loop of gp120. J. Virol. 2000, 74, 8358–8367. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wei, X.; Decker, J.M.; Liu, H.; Zhang, Z.; Arani, R.B.; Kilby, J.M.; Saag, M.S.; Wu, X.; Shaw, G.M.; Kappes, J.C. Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob. Agents Chemother. 2002, 46, 1896–1905. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tidhar, R.; Zelnik, I.D.; Volpert, G.; Ben-Dor, S.; Kelly, S.; Merrill, A.H., Jr.; Futerman, A.H. Eleven residues determine the acyl chain specificity of ceramide synthases. J. Biol. Chem. 2018, 293, 9912–9921. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Schneider, U.; Schwenk, H.U.; Bornkamm, G. Characterization of EBV-genome negative “null” and “T” cell lines derived from children with acute lymphoblastic leukemia and leukemic transformed non-Hodgkin lymphoma. Int. J. Cancer 1977, 19, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Rodari, A.; Poli, G.; Van Lint, C. Jurkat-Derived (J-Lat, J1.1, and Jurkat E4) and CEM-Derived T Cell Lines (8E5 and ACH-2) as Models of Reversible Proviral Latency. Methods Mol. Biol. 2022, 2407, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Saayman, S.M.; Lazar, D.C.; Scott, T.A.; Hart, J.R.; Takahashi, M.; Burnett, J.C.; Planelles, V.; Morris, K.V.; Weinberg, M.S. Potent and Targeted Activation of Latent HIV-1 Using the CRISPR/dCas9 Activator Complex. Mol. Ther. 2016, 24, 488–498. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Okutomi, T.; Minakawa, S.; Hirota, R.; Katagiri, K.; Morikawa, Y. HIV Reactivation in Latently Infected Cells With Virological Synapse-Like Cell Contact. Viruses 2020, 12, 417. [Google Scholar] [CrossRef]
- Liu, H.; Chen, C.; Liao, S.; Sohaii, D.K.; Cruz, C.R.Y.; Burdo, T.H.; Cradick, T.J.; Mehta, A.; Barrero, C.; Florez, M.; et al. Strategic self-limiting production of infectious HIV particles by CRISPR in permissive cells. Mol. Ther. Nucleic Acids 2023, 32, 1010–1025. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Molnar, S.M.; Kim, Y.; Wieczorek, L.; Williams, A.; Patil, K.A.; Khatkar, P.; Santos, M.F.; Mensah, G.; Lorico, A.; Polonis, V.R.; et al. Extracellular vesicle isolation methods identify distinct HIV-1 particles released from chronically infected T-cells. J. Extracell. Vesicles 2024, 13, e12476. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Longo, P.A.; Kavran, J.M.; Kim, M.S.; Leahy, D.J. Transient mammalian cell transfection with polyethylenimine (PEI). Methods Enzymol. 2013, 529, 227–240. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jones, T.A.; Blaug, G.; Hansen, M.; Barklis, E. Assembly of gag-beta-galactosidase proteins into retrovirus particles. J. Virol. 1990, 64, 2265–2279. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wild, C.; Greenwell, T.; Matthews, T. A synthetic peptide from HIV-1 gp41 is a potent inhibitor of virus-mediated cell-cell fusion. AIDS Res. Hum. Retrovir. 1993, 9, 1051–1053. [Google Scholar] [CrossRef] [PubMed]
- Wild, C.T.; Shugars, D.C.; Greenwell, T.K.; McDanal, C.B.; Matthews, T.J. Peptides corresponding to a predictive alpha-helical domain of human immunodeficiency virus type 1 gp41 are potent inhibitors of virus infection. Proc. Natl. Acad. Sci. USA 1994, 91, 9770–9774. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kilby, J.M.; Hopkins, S.; Venetta, T.M.; DiMassimo, B.; Cloud, G.A.; Lee, J.Y.; Alldredge, L.; Hunter, E.; Lambert, D.; Bolognesi, D.; et al. Potent suppression of HIV-1 replication in humans by T-20, a peptide inhibitor of gp41-mediated virus entry. Nat. Med. 1998, 4, 1302–1307. [Google Scholar] [CrossRef] [PubMed]
- Alfadhli, A.; Romanaggi, C.; Barklis, R.L.; Barklis, E. Second site reversion of HIV-1 envelope protein baseplate mutations maps to the matrix protein. J. Virol. 2024, 98, e0174223. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, J.; Kondo, N.; Long, Y.; Iwamoto, A.; Matsuda, Z. Monitoring of HIV-1 envelope-mediated membrane fusion using modified split green fluorescent proteins. J. Virol. Methods 2009, 161, 216–222. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Diamond, D.C.; Finberg, R.; Chaudhuri, S.; Sleckman, B.P.; Burakoff, S.J. Human immunodeficiency virus infection is efficiently mediated by a glycolipid-anchored form of CD4. Proc. Natl. Acad. Sci. USA 1990, 87, 5001–5005. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Marshall, W.L.; Mittler, E.S.; Avery, P.; Lawrence, J.P.; Finberg, R.W. Glycosylphosphatidylinositol-anchored CD4 supports human immunodeficiency virus type 1 replication, but not cytopathic effect, in T-cell transfectants. J. Virol. 1994, 68, 4039–4042. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kost, T.A.; Kessler, J.A.; Patel, I.R.; Gray, J.G.; Overton, L.K.; Carter, S.G. Human immunodeficiency virus infection and syncytium formation in HeLa cells expressing glycophospholipid-anchored CD4. J. Virol. 1991, 65, 3276–3283. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Busillo, J.M.; Benovic, J.L. Regulation of CXCR4 signaling. Biochim. Biophys. Acta 2007, 1768, 952–963. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Cronshaw, D.G.; Nie, Y.; Waite, J.; Zou, Y.R. An essential role of the cytoplasmic tail of CXCR4 in G-protein signaling and organogenesis. PLoS ONE 2010, 5, e15397. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- García-Cuesta, E.M.; Rodríguez-Frade, J.M.; Gardeta, S.R.; D’Agostino, G.; Martínez, P.; Soler Palacios, B.; Cascio, G.; Wolf, T.; Mateos, N.; Ayala-Bueno, R.; et al. Altered CXCR4 dynamics at the cell membrane impairs directed cell migration in WHIM syndrome patients. Proc. Natl. Acad. Sci. USA 2022, 119, e2119483119. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kang, K.; Ma, R.; Cai, W.; Huang, W.; Paul, C.; Liang, J.; Wang, Y.; Zhao, T.; Kim, H.W.; Xu, M.; et al. Exosomes Secreted from CXCR4 Overexpressing Mesenchymal Stem Cells Promote Cardioprotection via Akt Signaling Pathway following Myocardial Infarction. Stem Cells Int. 2015, 2015, 659890. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Luo, H.; Chen, D.; Li, R.; Li, R.; Teng, Y.; Cao, Y.; Zou, X.; Wang, W.; Zhou, C. Genetically engineered CXCR4-modified exosomes for delivery of miR-126 mimics to macrophages alleviate periodontitis. J. Nanobiotechnol. 2023, 21, 116. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Edwards, T.G.; Wyss, S.; Reeves, J.D.; Zolla-Pazner, S.; Hoxie, J.A.; Doms, R.W.; Baribaud, F. Truncation of the cytoplasmic domain induces exposure of conserved regions in the ectodomain of human immunodeficiency virus type 1 envelope protein. J. Virol. 2002, 76, 2683–2691. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wyma, D.J.; Jiang, J.; Shi, J.; Zhou, J.; Lineberger, J.E.; Miller, M.D.; Aiken, C. Coupling of human immunodeficiency virus type 1 fusion to virion maturation: A novel role of the gp41 cytoplasmic tail. J. Virol. 2004, 78, 3429–3435. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wyss, S.; Dimitrov, A.S.; Baribaud, F.; Edwards, T.G.; Blumenthal, R.; Hoxie, J.A. Regulation of human immunodeficiency virus type 1 envelope glycoprotein fusion by a membrane-interactive domain in the gp41 cytoplasmic tail. J. Virol. 2005, 79, 12231–12241. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chen, J.; Kovacs, J.M.; Peng, H.; Rits-Volloch, S.; Lu, J.; Park, D.; Zablowsky, E.; Seaman, M.S.; Chen, B. HIV-1 ENVELOPE. Effect of the cytoplasmic domain on antigenic characteristics of HIV-1 envelope glycoprotein. Science 2015, 349, 191–195. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Piai, A.; Fu, Q.; Cai, Y.; Ghantous, F.; Xiao, T.; Shaik, M.M.; Peng, H.; Rits-Volloch, S.; Chen, W.; Seaman, M.S.; et al. Structural basis of transmembrane coupling of the HIV-1 envelope glycoprotein. Nat. Commun. 2020, 11, 2317. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Rawat, S.S.; Zimmerman, C.; Johnson, B.T.; Cho, E.; Lockett, S.J.; Blumenthal, R.; Puri, A. Restricted lateral mobility of plasma membrane CD4 impairs HIV-1 envelope glycoprotein mediated fusion. Mol. Membr. Biol. 2008, 25, 83–94. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Benkirane, M.; Jin, D.Y.; Chun, R.F.; Koup, R.A.; Jeang, K.T. Mechanism of transdominant inhibition of CCR5-mediated HIV-1 infection by ccr5delta32. J. Biol. Chem. 1997, 272, 30603–30606. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, S.; Petrovic, A.; Locati, M.; Kim, Y.O.; Weissman, D.; Murphy, P.M. A membrane-proximal basic domain and cysteine cluster in the C-terminal tail of CCR5 constitute a bipartite motif critical for cell surface expression. J. Biol. Chem. 2001, 276, 40133–40145. [Google Scholar] [CrossRef] [PubMed]
- Shaik, M.M.; Peng, H.; Lu, J.; Rits-Volloch, S.; Xu, C.; Liao, M.; Chen, B. Structural basis of coreceptor recognition by HIV-1 envelope spike. Nature 2019, 565, 318–323. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Montefiori, D.C.; Roederer, M.; Morris, L.; Seaman, M.S. Neutralization tiers of HIV-1. Curr. Opin. HIV AIDS 2018, 13, 128–136. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Weiss, R.A.; Verrips, C.T. Nanobodies that Neutralize HIV. Vaccines 2019, 7, 77. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liao, Z.; Cimakasky, L.M.; Hampton, R.; Nguyen, D.H.; Hildreth, J.E. Lipid rafts and HIV pathogenesis: Host membrane cholesterol is required for infection by HIV type 1. AIDS Res. Hum. Retrovir. 2001, 17, 1009–1019. [Google Scholar] [CrossRef] [PubMed]
- Liao, Z.; Graham, D.R.; Hildreth, J.E. Lipid rafts and HIV pathogenesis: Virion-associated cholesterol is required for fusion and infection of susceptible cells. AIDS Res. Hum. Retrovir. 2003, 19, 675–687. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.Y.; Aliyari, R.; Chikere, K.; Li, G.; Marsden, M.D.; Smith, J.K.; Pernet, O.; Guo, H.; Nusbaum, R.; Zack, J.A.; et al. Interferon-inducible cholesterol-25-hydroxylase broadly inhibits viral entry by production of 25-hydroxycholesterol. Immunity 2013, 38, 92–105. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhang, J.; Zhu, Y.; Wang, X.; Wang, J. 25-hydroxycholesterol: An integrator of antiviral ability and signaling. Front. Immunol. 2023, 14, 1268104. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Nieto-Garai, J.A.; Arboleya, A.; Otaegi, S.; Chojnacki, J.; Casas, J.; Fabriàs, G.; Contreras, F.X.; Kräusslich, H.G.; Lorizate, M. Cholesterol in the Viral Membrane is a Molecular Switch Governing HIV-1 Env Clustering. Adv. Sci. 2020, 8, 2003468. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Whitlock, J.M.; Chernomordik, L.V. Flagging fusion: Phosphatidylserine signaling in cell-cell fusion. J. Biol. Chem. 2021, 296, 100411. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Braga, L.; Ali, H.; Secco, I.; Chiavacci, E.; Neves, G.; Goldhill, D.; Penn, R.; Jimenez-Guardeño, J.M.; Ortega-Prieto, A.M.; Bussani, R.; et al. Drugs that inhibit TMEM16 proteins block SARS-CoV-2 spike-induced syncytia. Nature 2021, 594, 88–93. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Finnegan, C.M.; Rawat, S.S.; Cho, E.H.; Guiffre, D.L.; Lockett, S.; Merrill, A.H., Jr.; Blumenthal, R. Sphingomyelinase restricts the lateral diffusion of CD4 and inhibits human immunodeficiency virus fusion. J. Virol. 2007, 81, 5294–5304. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Vieira, C.R.; Munoz-Olaya, J.M.; Sot, J.; Jiménez-Baranda, S.; Izquierdo-Useros, N.; Abad, J.L.; Apellániz, B.; Delgado, R.; Martinez-Picado, J.; Alonso, A.; et al. Dihydrosphingomyelin impairs HIV-1 infection by rigidifying liquid-ordered membrane domains. Chem. Biol. 2010, 17, 766–775. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alfadhli, A.; Barklis, R.L.; Tafesse, F.G.; Barklis, E. Analysis of Factors That Regulate HIV-1 Fusion in Reverse. Viruses 2025, 17, 472. https://doi.org/10.3390/v17040472
Alfadhli A, Barklis RL, Tafesse FG, Barklis E. Analysis of Factors That Regulate HIV-1 Fusion in Reverse. Viruses. 2025; 17(4):472. https://doi.org/10.3390/v17040472
Chicago/Turabian StyleAlfadhli, Ayna, Robin Lid Barklis, Fikadu G. Tafesse, and Eric Barklis. 2025. "Analysis of Factors That Regulate HIV-1 Fusion in Reverse" Viruses 17, no. 4: 472. https://doi.org/10.3390/v17040472
APA StyleAlfadhli, A., Barklis, R. L., Tafesse, F. G., & Barklis, E. (2025). Analysis of Factors That Regulate HIV-1 Fusion in Reverse. Viruses, 17(4), 472. https://doi.org/10.3390/v17040472