
HTLV-1 p13 Protein Hijacks Macrophage Polarization and Promotes T-Cell Recruitment
 ,
,  , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Plasmid and Cell Lines
2.2. Immunoblotting
2.3. Immunofluorescence
2.4. Proliferation and Viability Assay
2.5. Seahorse Metabolic Flux Analysis
2.6. Efferocytosis Assay
2.7. M1 and M2 Macrophage Differentiation
2.8. Measuring Mitochondrial ROS
2.9. Luminex
2.10. Migration Assay
2.11. Statistical Analysis
3. Results
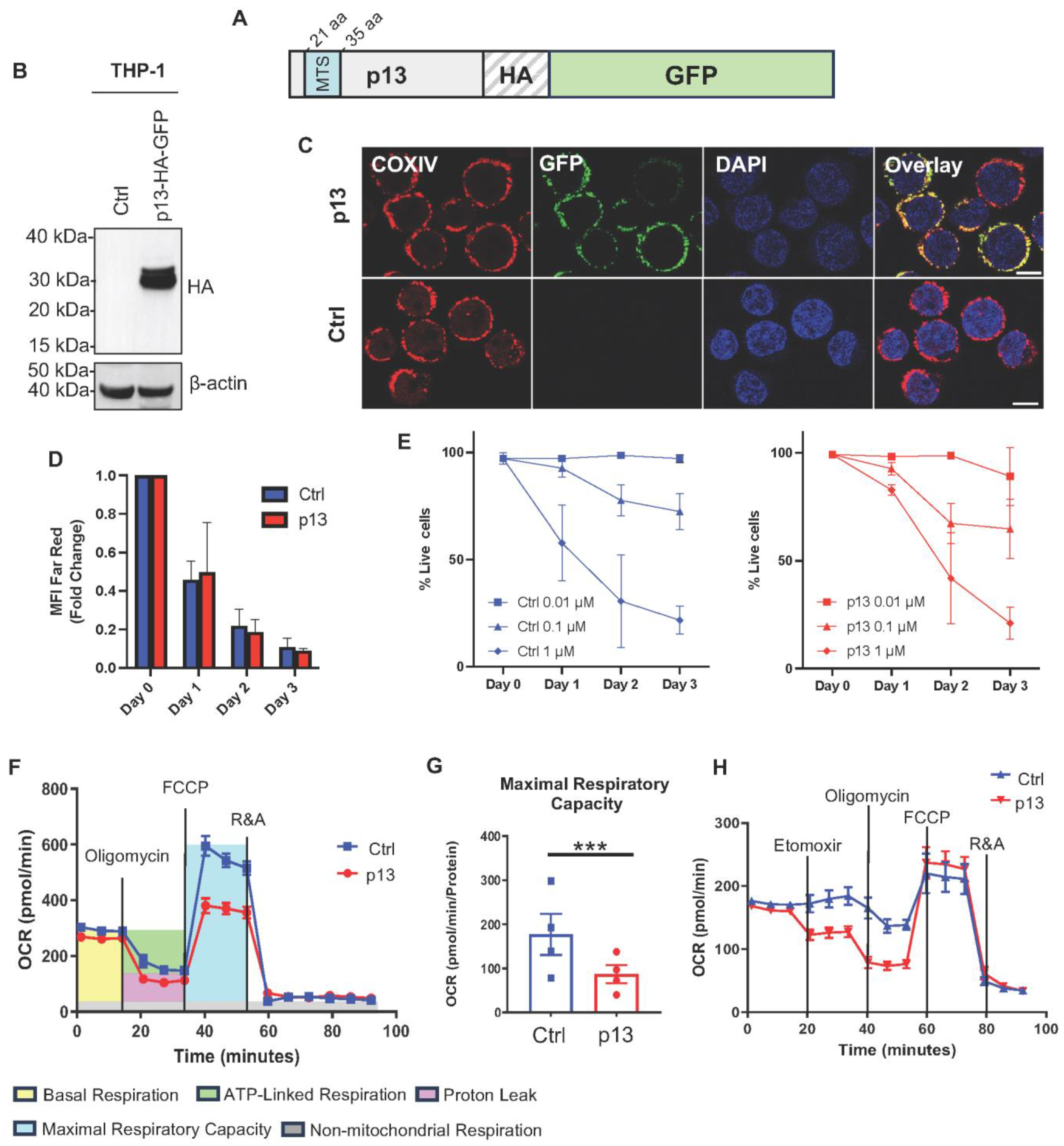
3.1. p13 Localizes to Mitochondria and Changes Monocyte Metabolism
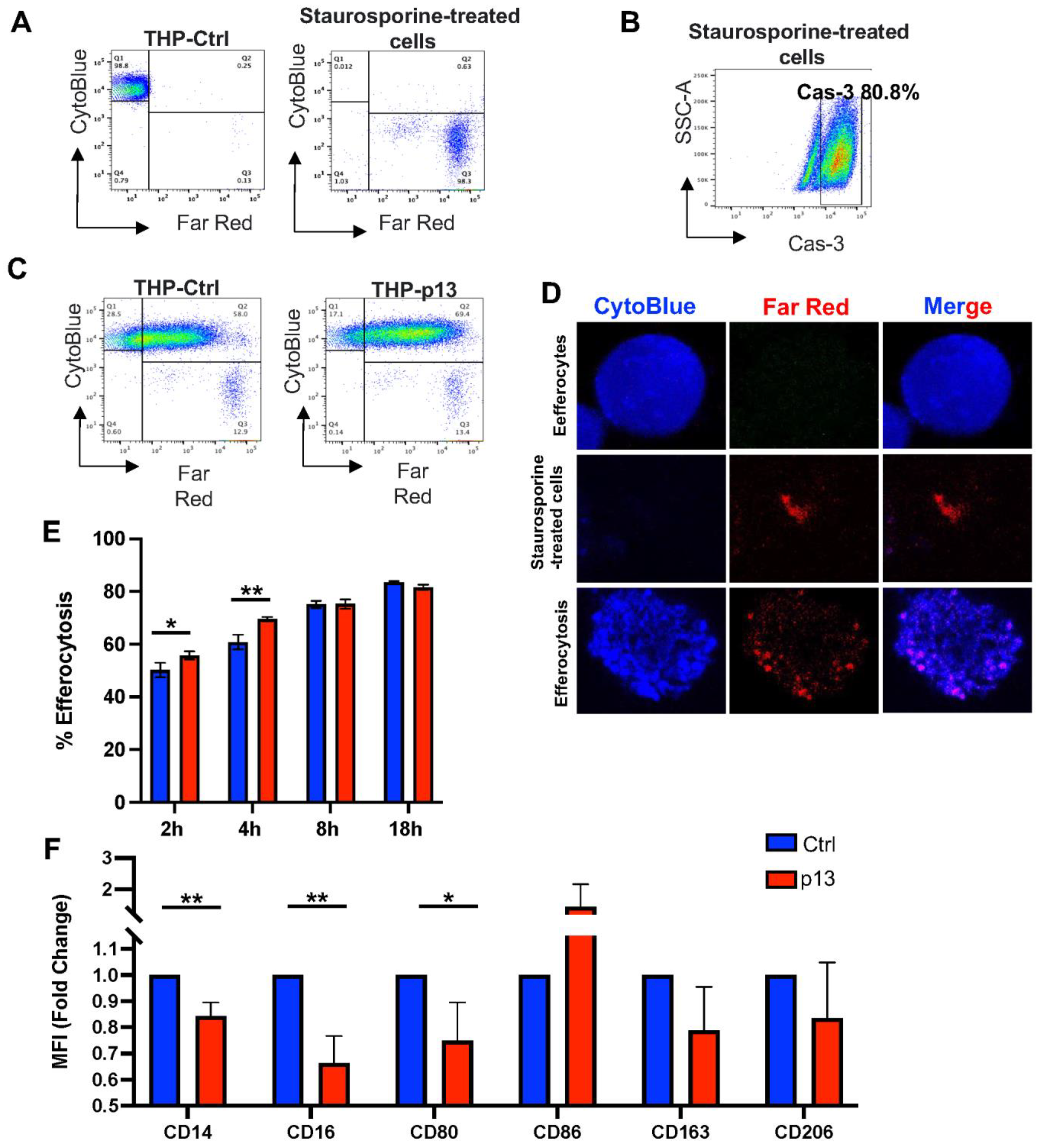
3.2. The Effect of p13 Expression on Efferocytosis and Differentiation
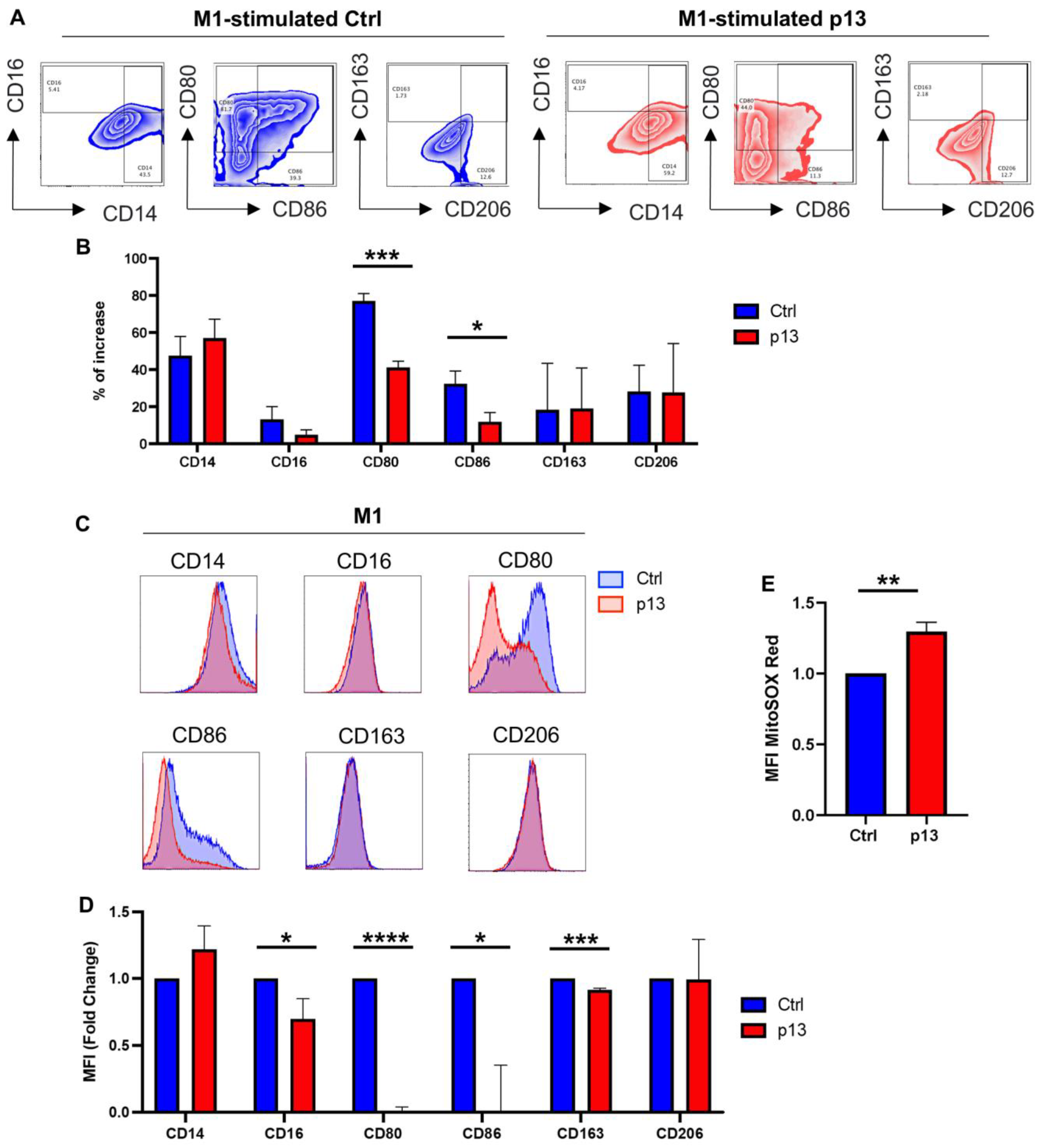
3.3. p13 Influences M1 and M2 Macrophage Polarization
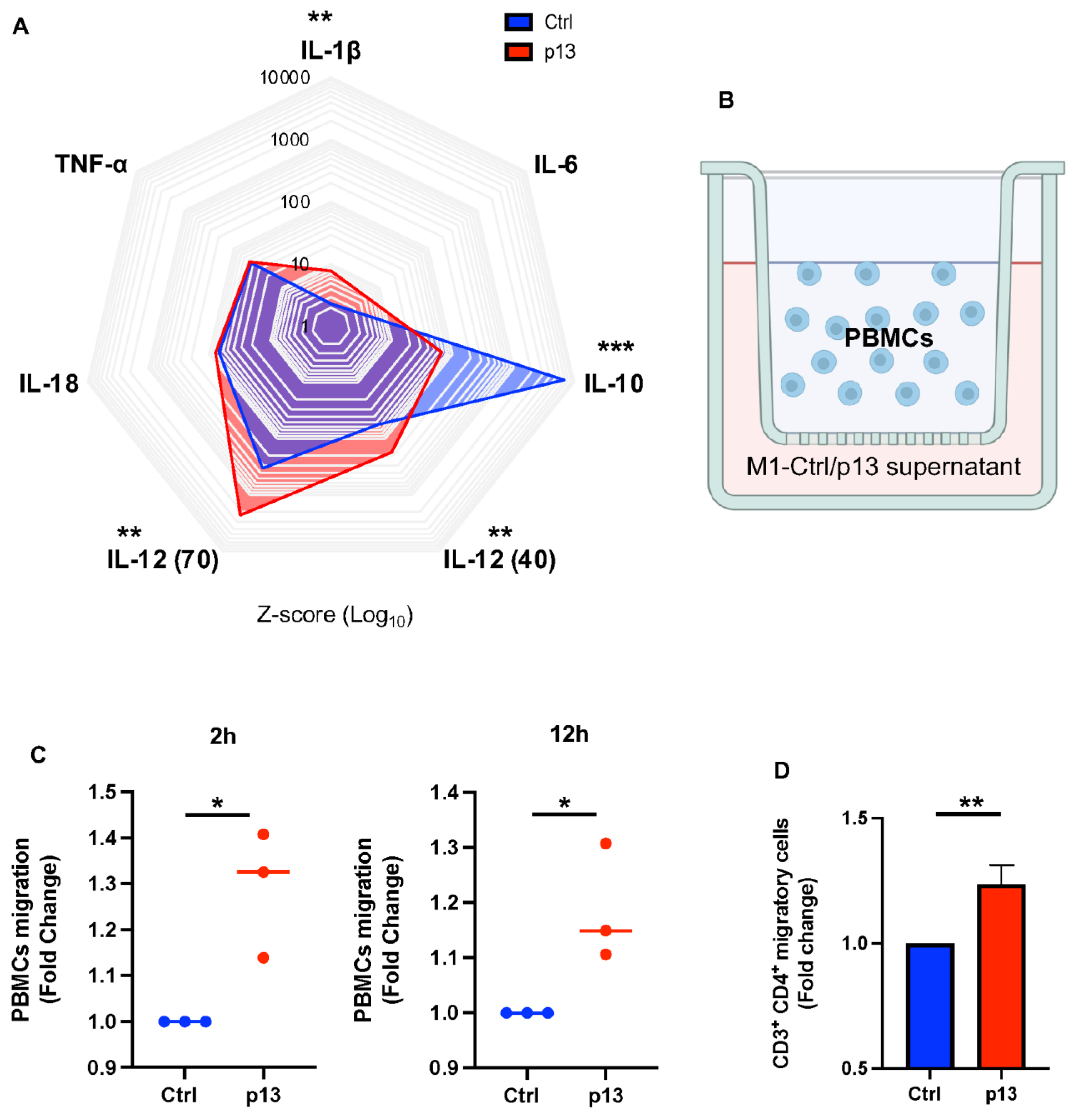
3.4. p13 M1 Like Macrophages Induced Migration of CD4+ T Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Disclaimer
References
- Poiesz, B.J.; Ruscetti, F.W.; Gazdar, A.F.; Bunn, P.A.; Minna, J.D.; Gallo, R.C. Detection and isolation of type C retrovirus particles from fresh and cultured lymphocytes of a patient with cutaneous T-cell lymphoma. Proc. Natl. Acad. Sci. USA 1980, 77, 7415–7419. [Google Scholar] [CrossRef]
- Takatsuki, K. Discovery of adult T-cell leukemia. Retrovirology 2005, 2, 16. [Google Scholar] [CrossRef] [PubMed]
- Gessain, A.; Barin, F.; Vernant, J.C.; Gout, O.; Maurs, L.; Calender, A.; de The, G. Antibodies to human T-lymphotropic virus type-I in patients with tropical spastic paraparesis. Lancet 1985, 2, 407–410. [Google Scholar] [CrossRef]
- Osame, M.; Usuku, K.; Izumo, S.; Ijichi, N.; Amitani, H.; Igata, A.; Matsumoto, M.; Tara, M. HTLV-I associated myelopathy, a new clinical entity. Lancet 1986, 1, 1031–1032. [Google Scholar] [CrossRef] [PubMed]
- Kamoi, K.; Mochizuki, M. HTLV-1 uveitis. Front. Microbiol. 2012, 3, 270. [Google Scholar] [CrossRef]
- Nishioka, K.; Maruyama, I.; Sato, K.; Kitajima, I.; Nakajima, Y.; Osame, M. Chronic inflammatory arthropathy associated with HTLV-I. Lancet 1989, 1, 441. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, K.; Matsuoka, N.; Ida, H.; Nakashima, M.; Sakai, M.; Sakito, S.; Kawakami, A.; Terada, K.; Shimada, H.; Kawabe, Y.; et al. Primary Sjogren’s syndrome with antibodies to HTLV-I: Clinical and laboratory features. Ann. Rheum. Dis. 1992, 51, 769–776. [Google Scholar] [CrossRef]
- Koralnik, I.J.; Gessain, A.; Klotman, M.E.; Lo Monico, A.; Berneman, Z.N.; Franchini, G. Protein isoforms encoded by the pX region of human T-cell leukemia/lymphotropic virus type I. Proc. Natl. Acad. Sci. USA 1992, 89, 8813–8817. [Google Scholar] [CrossRef]
- Ciminale, V.; Pavlakis, G.N.; Derse, D.; Cunningham, C.P.; Felber, B.K. Complex splicing in the human T-cell leukemia virus (HTLV) family of retroviruses: Novel mRNAs and proteins produced by HTLV type I. J. Virol. 1992, 66, 1737–1745. [Google Scholar] [CrossRef]
- Omsland, M.; Silic-Benussi, M.; Moles, R.; Sarkis, S.; Purcell, D.F.J.; Yurick, D.; Khoury, G.; D’Agostino, D.M.; Ciminale, V.; Franchini, G. Functional properties and sequence variation of HTLV-1 p13. Retrovirology 2020, 17, 11. [Google Scholar] [CrossRef]
- Silic-Benussi, M.; Biasiotto, R.; Andresen, V.; Franchini, G.; D’Agostino, D.M.; Ciminale, V. HTLV-1 p13, a small protein with a busy agenda. Mol. Asp. Med. 2010, 31, 350–358. [Google Scholar] [CrossRef]
- Silic-Benussi, M.; Cannizzaro, E.; Venerando, A.; Cavallari, I.; Petronilli, V.; La Rocca, N.; Marin, O.; Chieco-Bianchi, L.; Di Lisa, F.; D’Agostino, D.M.; et al. Modulation of mitochondrial K(+) permeability and reactive oxygen species production by the p13 protein of human T-cell leukemia virus type 1. Biochim. Biophys. Acta 2009, 1787, 947–954. [Google Scholar] [CrossRef]
- Hiraragi, H.; Michael, B.; Nair, A.; Silic-Benussi, M.; Ciminale, V.; Lairmore, M. Human T-lymphotropic virus type 1 mitochondrion-localizing protein p13II sensitizes Jurkat T cells to Ras-mediated apoptosis. J. Virol. 2005, 79, 9449–9457. [Google Scholar] [CrossRef]
- Andresen, V.; Pise-Masison, C.A.; Sinha-Datta, U.; Bellon, M.; Valeri, V.; Washington Parks, R.; Cecchinato, V.; Fukumoto, R.; Nicot, C.; Franchini, G. Suppression of HTLV-1 replication by Tax-mediated rerouting of the p13 viral protein to nuclear speckles. Blood 2011, 118, 1549–1559. [Google Scholar] [CrossRef]
- Derse, D.; Crise, B.; Li, Y.; Princler, G.; Lum, N.; Stewart, C.; McGrath, C.F.; Hughes, S.H.; Munroe, D.J.; Wu, X. Human T-cell leukemia virus type 1 integration target sites in the human genome: Comparison with those of other retroviruses. J. Virol. 2007, 81, 6731–6741. [Google Scholar] [CrossRef]
- Yoshida, M.; Miyoshi, I.; Hinuma, Y. Isolation and characterization of retrovirus from cell lines of human adult T-cell leukemia and its implication in the disease. Proc. Natl. Acad. Sci. USA 1982, 79, 2031–2035. [Google Scholar] [CrossRef]
- Yoshida, M.; Seiki, M.; Yamaguchi, K.; Takatsuki, K. Monoclonal integration of human T-cell leukemia provirus in all primary tumors of adult T-cell leukemia suggests causative role of human T-cell leukemia virus in the disease. Proc. Natl. Acad. Sci. USA 1984, 81, 2534–2537. [Google Scholar] [CrossRef] [PubMed]
- Cavrois, M.; Gessain, A.; Gout, O.; Wain-Hobson, S.; Wattel, E. Common human T cell leukemia virus type 1 (HTLV-1) integration sites in cerebrospinal fluid and blood lymphocytes of patients with HTLV-1-associated myelopathy/tropical spastic paraparesis indicate that HTLV-1 crosses the blood-brain barrier via clonal HTLV-1-infected cells. J. Infect. Dis. 2000, 182, 1044–1050. [Google Scholar] [CrossRef] [PubMed]
- Chiyoda, S.; Kinoshita, T.; Miwa, M. Monoclonal integration of HTLV-1 in pleural effusion cells in a seropositive patient with tuberculous pleuritis. Int. J. Hematol. 1995, 61, 35–38. [Google Scholar] [CrossRef]
- Cook, L.B.; Melamed, A.; Niederer, H.; Valganon, M.; Laydon, D.; Foroni, L.; Taylor, G.P.; Matsuoka, M.; Bangham, C.R. The role of HTLV-1 clonality, proviral structure, and genomic integration site in adult T-cell leukemia/lymphoma. Blood 2014, 123, 3925–3931. [Google Scholar] [CrossRef]
- Fang, J.; Kushida, S.; Feng, R.; Tanaka, M.; Kikukawa, H.; Kawamura, T.; Uchida, K.; Miwa, M. Integration of HTLV-1 provirus into mouse transforming growth factor-alpha gene. Biochem. Biophys. Res. Commun. 1997, 233, 792–795. [Google Scholar] [CrossRef] [PubMed]
- Firouzi, S.; Lopez, Y.; Suzuki, Y.; Nakai, K.; Sugano, S.; Yamochi, T.; Watanabe, T. Development and validation of a new high-throughput method to investigate the clonality of HTLV-1-infected cells based on provirus integration sites. Genome Med. 2014, 6, 46. [Google Scholar] [CrossRef]
- Hashikura, Y.; Umeki, K.; Umekita, K.; Nomura, H.; Yamamoto, I.; Hasegawa, H.; Yanagihara, K.; Okayama, A. The diversity of the structure and genomic integration sites of HTLV-1 provirus in MT-2 cell lines. Hum. Cell 2016, 29, 122–129. [Google Scholar] [CrossRef]
- Enose-Akahata, Y.; Oh, U.; Grant, C.; Jacobson, S. Retrovirally induced CTL degranulation mediated by IL-15 expression and infection of mononuclear phagocytes in patients with HTLV-I-associated neurologic disease. Blood 2008, 112, 2400–2410. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.S.; Petrow-Sadowski, C.; Huang, Y.K.; Bertolette, D.C.; Ruscetti, F.W. Cell-free HTLV-1 infects dendritic cells leading to transmission and transformation of CD4(+) T cells. Nat. Med. 2008, 14, 429–436. [Google Scholar] [CrossRef]
- Koralnik, I.J.; Lemp, J.F., Jr.; Gallo, R.C.; Franchini, G. In vitro infection of human macrophages by human T-cell leukemia/lymphotropic virus type I (HTLV-I). AIDS Res. Hum. Retroviruses 1992, 8, 1845–1849. [Google Scholar] [CrossRef] [PubMed]
- Makino, M.; Shimokubo, S.; Wakamatsu, S.I.; Izumo, S.; Baba, M. The role of human T-lymphotropic virus type 1 (HTLV-1)-infected dendritic cells in the development of HTLV-1-associated myelopathy/tropical spastic paraparesis. J. Virol. 1999, 73, 4575–4581. [Google Scholar] [CrossRef]
- de Castro-Amarante, M.F.; Pise-Masison, C.A.; McKinnon, K.; Washington Parks, R.; Galli, V.; Omsland, M.; Andresen, V.; Massoud, R.; Brunetto, G.; Caruso, B.; et al. Human T Cell Leukemia Virus Type 1 Infection of the Three Monocyte Subsets Contributes to Viral Burden in Humans. J. Virol. 2015, 90, 2195–2207. [Google Scholar] [CrossRef]
- Austermann, J.; Roth, J.; Barczyk-Kahlert, K. The Good and the Bad: Monocytes’ and Macrophages’ Diverse Functions in Inflammation. Cells 2022, 11, 1979. [Google Scholar] [CrossRef]
- Jakubzick, C.V.; Randolph, G.J.; Henson, P.M. Monocyte differentiation and antigen-presenting functions. Nat. Rev. Immunol. 2017, 17, 349–362. [Google Scholar] [CrossRef]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell. Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef] [PubMed]
- Kratofil, R.M.; Kubes, P.; Deniset, J.F. Monocyte Conversion During Inflammation and Injury. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Guilliams, M.; Mildner, A.; Yona, S. Developmental and Functional Heterogeneity of Monocytes. Immunity 2018, 49, 595–613. [Google Scholar] [CrossRef]
- Rocamonde, B.; Carcone, A.; Mahieux, R.; Dutartre, H. HTLV-1 infection of myeloid cells: From transmission to immune alterations. Retrovirology 2019, 16, 45. [Google Scholar] [CrossRef]
- West, A.P.; Shadel, G.S.; Ghosh, S. Mitochondria in innate immune responses. Nat. Rev. Immunol. 2011, 11, 389–402. [Google Scholar] [CrossRef] [PubMed]
- Morgenstern, J.P.; Land, H. Advanced mammalian gene transfer: High titre retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic Acids Res. 1990, 18, 3587–3596. [Google Scholar] [CrossRef]
- Chevalier, S.A.; Durand, S.; Dasgupta, A.; Radonovich, M.; Cimarelli, A.; Brady, J.N.; Mahieux, R.; Pise-Masison, C.A. The transcription profile of Tax-3 is more similar to Tax-1 than Tax-2: Insights into HTLV-3 potential leukemogenic properties. PLoS ONE 2012, 7, e41003. [Google Scholar] [CrossRef]
- Omsland, M.; Pise-Masison, C.; Fujikawa, D.; Galli, V.; Fenizia, C.; Parks, R.W.; Gjertsen, B.T.; Franchini, G.; Andresen, V. Inhibition of Tunneling Nanotube (TNT) Formation and Human T-cell Leukemia Virus Type 1 (HTLV-1) Transmission by Cytarabine. Sci. Rep. 2018, 8, 11118. [Google Scholar] [CrossRef]
- Takemoto, S.; Mulloy, J.C.; Cereseto, A.; Migone, T.S.; Patel, B.K.; Matsuoka, M.; Yamaguchi, K.; Takatsuki, K.; Kamihira, S.; White, J.D.; et al. Proliferation of adult T cell leukemia/lymphoma cells is associated with the constitutive activation of JAK/STAT proteins. Proc. Natl. Acad. Sci. USA 1997, 94, 13897–13902. [Google Scholar] [CrossRef]
- Tsuchiya, S.; Yamabe, M.; Yamaguchi, Y.; Kobayashi, Y.; Konno, T.; Tada, K. Establishment and characterization of a human acute monocytic leukemia cell line (THP-1). Int. J. Cancer 1980, 26, 171–176. [Google Scholar]
- D’Agostino, D.M.; Silic-Benussi, M.; Hiraragi, H.; Lairmore, M.D.; Ciminale, V. The human T-cell leukemia virus type 1 p13II protein: Effects on mitochondrial function and cell growth. Cell Death Differ. 2005, 12 (Suppl. S1), 905–915. [Google Scholar] [CrossRef]
- Ciminale, V.; Zotti, L.; D’Agostino, D.M.; Ferro, T.; Casareto, L.; Franchini, G.; Bernardi, P.; Chieco-Bianchi, L. Mitochondrial targeting of the p13II protein coded by the x-II ORF of human T-cell leukemia/lymphotropic virus type I (HTLV-I). Oncogene 1999, 18, 4505–4514. [Google Scholar] [CrossRef] [PubMed]
- Kadenbach, B. Complex IV—The regulatory center of mitochondrial oxidative phosphorylation. Mitochondrion 2021, 58, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Koralnik, I.J.; Fullen, J.; Franchini, G. The p12I, p13II, and p30II proteins encoded by human T-cell leukemia/lymphotropic virus type I open reading frames I and II are localized in three different cellular compartments. J. Virol. 1993, 67, 2360–2366. [Google Scholar] [CrossRef]
- Silic-Benussi, M.; Cavallari, I.; Vajente, N.; Vidali, S.; Chieco-Bianchi, L.; Di Lisa, F.; Saggioro, D.; D’Agostino, D.M.; Ciminale, V. Redox regulation of T-cell turnover by the p13 protein of human T-cell leukemia virus type 1: Distinct effects in primary versus transformed cells. Blood 2010, 116, 54–62. [Google Scholar] [CrossRef]
- Ramiro-Cortes, Y.; Guemez-Gamboa, A.; Moran, J. Reactive oxygen species participate in the p38-mediated apoptosis induced by potassium deprivation and staurosporine in cerebellar granule neurons. Int. J. Biochem. Cell Biol. 2011, 43, 1373–1382. [Google Scholar] [CrossRef] [PubMed]
- Doran, A.C.; Yurdagul, A., Jr.; Tabas, I. Efferocytosis in health and disease. Nat. Rev. Immunol. 2020, 20, 254–267. [Google Scholar] [CrossRef]
- Martin, W.F.; Tielens, A.G.M.; Mentel, M.; Garg, S.G.; Gould, S.B. The Physiology of Phagocytosis in the Context of Mitochondrial Origin. Microbiol. Mol. Biol. Rev. 2017, 81, e00008-17. [Google Scholar] [CrossRef]
- Zhang, S.; Weinberg, S.; DeBerge, M.; Gainullina, A.; Schipma, M.; Kinchen, J.M.; Ben-Sahra, I.; Gius, D.R.; Yvan-Charvet, L.; Chandel, N.S.; et al. Efferocytosis Fuels Requirements of Fatty Acid Oxidation and the Electron Transport Chain to Polarize Macrophages for Tissue Repair. Cell Metab. 2019, 29, 443–456.e445. [Google Scholar] [CrossRef]
- Schwende, H.; Fitzke, E.; Ambs, P.; Dieter, P. Differences in the state of differentiation of THP-1 cells induced by phorbol ester and 1,25-dihydroxyvitamin D3. J. Leukoc. Biol. 1996, 59, 555–561. [Google Scholar]
- Shi, C.; Pamer, E.G. Monocyte recruitment during infection and inflammation. Nat. Rev. Immunol. 2011, 11, 762–774. [Google Scholar] [CrossRef] [PubMed]
- Nikitina, E.; Larionova, I.; Choinzonov, E.; Kzhyshkowska, J. Monocytes and Macrophages as Viral Targets and Reservoirs. Int. J. Mol. Sci. 2018, 19, 2821. [Google Scholar] [CrossRef]
- Italiani, P.; Boraschi, D. From Monocytes to M1/M2 Macrophages: Phenotypical vs. Functional Differentiation. Front. Immunol. 2014, 5, 514. [Google Scholar] [CrossRef] [PubMed]
- Genin, M.; Clement, F.; Fattaccioli, A.; Raes, M.; Michiels, C. M1 and M2 macrophages derived from THP-1 cells differentially modulate the response of cancer cells to etoposide. BMC Cancer 2015, 15, 577. [Google Scholar] [CrossRef]
- Covarrubias, A.; Byles, V.; Horng, T. ROS sets the stage for macrophage differentiation. Cell Res. 2013, 23, 984–985. [Google Scholar] [CrossRef]
- Kany, S.; Vollrath, J.T.; Relja, B. Cytokines in Inflammatory Disease. Int. J. Mol. Sci. 2019, 20, 6008. [Google Scholar] [CrossRef]
- Sokol, C.L.; Luster, A.D. The chemokine system in innate immunity. Cold Spring Harb. Perspect. Biol. 2015, 7, a016303. [Google Scholar] [CrossRef] [PubMed]
- Tan, B.J.; Sugata, K.; Reda, O.; Matsuo, M.; Uchiyama, K.; Miyazato, P.; Hahaut, V.; Yamagishi, M.; Uchimaru, K.; Suzuki, Y.; et al. HTLV-1 infection promotes excessive T cell activation and transformation into adult T cell leukemia/lymphoma. J. Clin. Investig. 2021, 131, e150472. [Google Scholar] [CrossRef]
- Silic-Benussi, M.; Cavallari, I.; Zorzan, T.; Rossi, E.; Hiraragi, H.; Rosato, A.; Horie, K.; Saggioro, D.; Lairmore, M.D.; Willems, L.; et al. Suppression of tumor growth and cell proliferation by p13II, a mitochondrial protein of human T cell leukemia virus type 1. Proc. Natl. Acad. Sci. USA 2004, 101, 6629–6634. [Google Scholar] [CrossRef]
- Maryanovich, M.; Gross, A. A ROS rheostat for cell fate regulation. Trends Cell Biol. 2013, 23, 129–134. [Google Scholar] [CrossRef]
- Rustin, P. Mitochondria, from cell death to proliferation. Nat. Genet. 2002, 30, 352–353. [Google Scholar] [CrossRef]
- Macatonia, S.E.; Cruickshank, J.K.; Rudge, P.; Knight, S.C. Dendritic cells from patients with tropical spastic paraparesis are infected with HTLV-1 and stimulate autologous lymphocyte proliferation. AIDS Res. Hum. Retroviruses 1992, 8, 1699–1706. [Google Scholar] [CrossRef] [PubMed]
- Makino, M.; Wakamatsu, S.; Shimokubo, S.; Arima, N.; Baba, M. Production of functionally deficient dendritic cells from HTLV-I-infected monocytes: Implications for the dendritic cell defect in adult T cell leukemia. Virology 2000, 274, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Nascimento, C.R.; Lima, M.A.; de Andrada Serpa, M.J.; Espindola, O.; Leite, A.C.; Echevarria-Lima, J. Monocytes from HTLV-1-infected patients are unable to fully mature into dendritic cells. Blood 2011, 117, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, H.; Takahashi, M.; Norose, Y.; Takeshita, T.; Fukunaga, Y.; Takahashi, H. Transformation of breast milk macrophages by HTLV-I: Implications for HTLV-I transmission via breastfeeding. Biomed. Res. 2010, 31, 53–61. [Google Scholar] [CrossRef]
- Wang, Y.; Li, N.; Zhang, X.; Horng, T. Mitochondrial metabolism regulates macrophage biology. J. Biol. Chem. 2021, 297, 100904. [Google Scholar] [CrossRef]
- Kulkarni, R.A.; Worth, A.J.; Zengeya, T.T.; Shrimp, J.H.; Garlick, J.M.; Roberts, A.M.; Montgomery, D.C.; Sourbier, C.; Gibbs, B.K.; Mesaros, C.; et al. Discovering Targets of Non-enzymatic Acylation by Thioester Reactivity Profiling. Cell Chem. Biol. 2017, 24, 231–242. [Google Scholar] [CrossRef]
- Sang, Y.; Miller, L.C.; Blecha, F. Macrophage Polarization in Virus-Host Interactions. J. Clin. Cell Immunol. 2015, 6, 311. [Google Scholar] [CrossRef]
- Amorim, C.F.; Souza, A.S.; Diniz, A.G.; Carvalho, N.B.; Santos, S.B.; Carvalho, E.M. Functional activity of monocytes and macrophages in HTLV-1 infected subjects. PLoS Negl. Trop. Dis. 2014, 8, e3399. [Google Scholar] [CrossRef]
- Hoffman, P.M.; Dhib-Jalbut, S.; Mikovits, J.A.; Robbins, D.S.; Wolf, A.L.; Bergey, G.K.; Lohrey, N.C.; Weislow, O.S.; Ruscetti, F.W. Human T-cell leukemia virus type I infection of monocytes and microglial cells in primary human cultures. Proc. Natl. Acad. Sci. USA 1992, 89, 11784–11788. [Google Scholar] [CrossRef]
- Rocamonde, B.; Futsch, N.; Orii, N.; Allatif, O.; Penalva de Oliveira, A.C.; Mahieux, R.; Casseb, J.; Dutartre, H. Immunoprofiling of fresh HAM/TSP blood samples shows altered innate cell responsiveness. PLoS Negl. Trop. Dis. 2021, 15, e0009940. [Google Scholar] [CrossRef] [PubMed]
- Balistrieri, G.; Barrios, C.; Castillo, L.; Umunakwe, T.C.; Giam, C.Z.; Zhi, H.; Beilke, M.A. Induction of CC-chemokines with antiviral function in macrophages by the human T lymphotropic virus type 2 transactivating protein, Tax2. Viral Immunol. 2013, 26, 3–12. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moles, R.; Omsland, M.; Pise-Masison, C.A.; Subleski, J.J.; McVicar, D.W.; Sarkis, S.; Gutowska, A.; Schifanella, L.; Doster, M.; Washington-Parks, R.; et al. HTLV-1 p13 Protein Hijacks Macrophage Polarization and Promotes T-Cell Recruitment. Viruses 2025, 17, 471. https://doi.org/10.3390/v17040471
Moles R, Omsland M, Pise-Masison CA, Subleski JJ, McVicar DW, Sarkis S, Gutowska A, Schifanella L, Doster M, Washington-Parks R, et al. HTLV-1 p13 Protein Hijacks Macrophage Polarization and Promotes T-Cell Recruitment. Viruses. 2025; 17(4):471. https://doi.org/10.3390/v17040471
Chicago/Turabian StyleMoles, Ramona, Maria Omsland, Cynthia A. Pise-Masison, Jeffrey J. Subleski, Daniel W. McVicar, Sarkis Sarkis, Anna Gutowska, Luca Schifanella, Melvin Doster, Robyn Washington-Parks, and et al. 2025. "HTLV-1 p13 Protein Hijacks Macrophage Polarization and Promotes T-Cell Recruitment" Viruses 17, no. 4: 471. https://doi.org/10.3390/v17040471
APA StyleMoles, R., Omsland, M., Pise-Masison, C. A., Subleski, J. J., McVicar, D. W., Sarkis, S., Gutowska, A., Schifanella, L., Doster, M., Washington-Parks, R., Ciminale, V., & Franchini, G. (2025). HTLV-1 p13 Protein Hijacks Macrophage Polarization and Promotes T-Cell Recruitment. Viruses, 17(4), 471. https://doi.org/10.3390/v17040471