
Phylogeographic History of Tomato Chlorosis Virus

 and
and {kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
3. Results
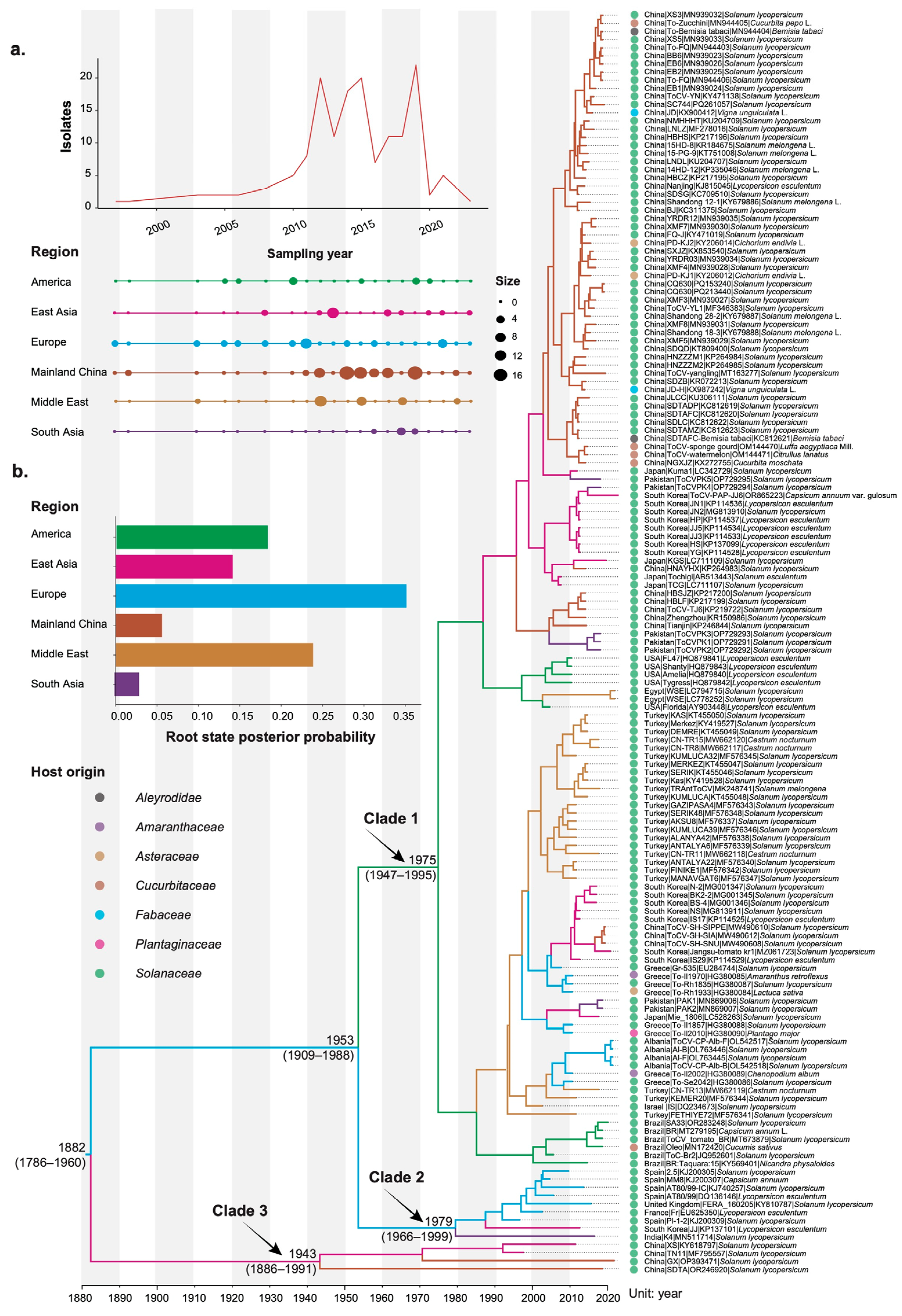
3.1. Temporal Dynamics of Tomato Chlorosis Virus
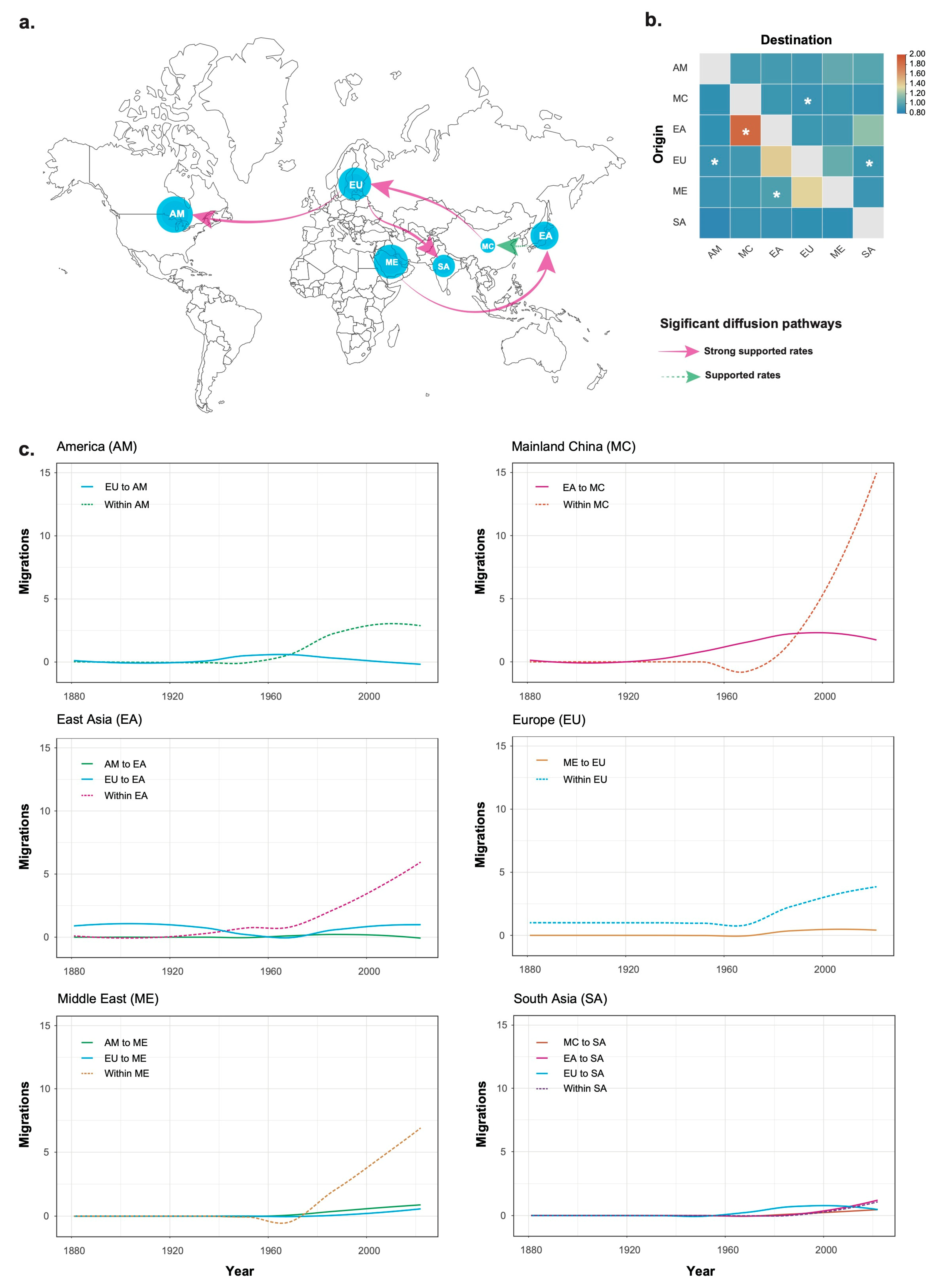
3.2. Phylogeographic History of Tomato Chlorosis Virus
3.3. Temporal Migration Pattern of Tomato Chlorosis Virus
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fuchs, M.; Bar-Joseph, M.; Candresse, T.; Maree, H.J.; Martelli, G.P.; Melzer, M.J.; Menzel, W.; Minafra, A.; Sabanadzovic, S.; ICTV Report Consortium. ICTV Virus Taxonomy Profile: Closteroviridae. J. Gen. Virol. 2020, 101, 364–365. [Google Scholar] [PubMed]
- Fiallo-Olivé, E.; Navas-Castillo, J. Tomato chlorosis virus, an emergent plant virus still expanding its geographical and host ranges. Mol. Plant Pathol. 2019, 20, 1307–1320. [Google Scholar] [CrossRef] [PubMed]
- Fiallo-Olivé, E.; Navas-Castillo, J. Tomato chlorosis virus, a promiscuous virus with multiple host plants and whitefly vectors. Ann. Appl. Biol. 2023, 182, 29–36. [Google Scholar]
- Ontiveros, I.; López-Moya, J.J.; Díaz-Pendón, J.A. Coinfection of tomato plants with tomato yellow leaf curl virus and tomato chlorosis virus affects the interaction with host and whiteflies. Phytopathology 2021, 112, 944–952. [Google Scholar]
- Favara, G.M.; Bampi, D.; Molina, J.P.E.; Rezende, J.A.M. Kinetics of systemic invasion and latent and incubation periods of tomato severe rugose virus and tomato chlorosis virus in single and co-infections in tomato plants. Phytopathology 2019, 109, 480–487. [Google Scholar]
- Davino, S.; Davino, M.; Bellardi, M.G.; Agosteo, G.E. Pepino mosaic virus and Tomato chlorosis virus causing mixed infection in protected tomato crops in Sicily. Phytopathol. Mediterr. 2008, 47, 35–41. [Google Scholar]
- Duan, G.; Zhan, F.; Du, Z.; Ho, S.Y.W.; Gao, F. Europe was a hub for the global spread of potato virus S in the 19th century. Virology 2018, 525, 200–204. [Google Scholar]
- Guan, X.; Yang, C.; Fu, J.; Du, Z.; Ho, S.Y.W.; Gao, F. Rapid evolutionary dynamics of pepper mild mottle virus. Virus Res. 2018, 256, 96–99. [Google Scholar]
- Mbewe, W.; Mukasa, S.; Ochwo-Ssemakula, M.; Sseruwagi, P.; Tairo, F.; Ndunguru, J.; Duffy, S. Cassava brown streak virus evolves with a nucleotide-substitution rate that is typical for the family Potyviridae. Virus Res. 2024, 346, 199397. [Google Scholar]
- Gao, F.; Zou, W.; Xie, L.; Zhan, J. Adaptive evolution and demographic history contribute to the divergent population genetic structure of potato virus Y between China and Japan. Evol. Appl. 2017, 10, 379–390. [Google Scholar]
- Shen, J.; Guo, J.; Chen, X.; Cai, W.; Du, Z.; Zhang, Y. The spatial diffusion of cherry leaf roll virus revealed by a Bayesian phylodynamic analysis. Viruses 2022, 14, 2179. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Fan, G.; Cheng, S.; Zhang, W.; Bai, Y. Evolutionary history and global spatiotemporal pattern of alfalfa mosaic virus. Front. Microbiol. 2022, 13, 1051834. [Google Scholar] [CrossRef] [PubMed]
- Tzanetakis, I.E.; Martin, R.R.; Wintermantel, W. Epidemiology of criniviruses: An emerging problem in world agriculture. Front. Microbiol. 2013, 4, 119. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Xiang, C.-Y.; Gao, F.; Jakovlić, I.; Lei, H.-P.; Hu, Y.; Zhang, H.; Zou, H.; Wang, G.-T.; Zhang, D. Using PhyloSuite for molecular phylogeny and tree-based analyses. iMeta 2023, 2, e87. [Google Scholar] [CrossRef]
- Martin, D.P.; Varsani, A.; Roumagnac, P.; Botha, G.; Maslamoney, S.; Schwab, T.; Kelz, Z.; Kumar, V.; Murrell, B. RDP5: A computer program for analyzing recombination in, and removing signals of recombination from, nucleotide sequence datasets. Virus Evol. 2021, 7, veaa087. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Lam, T.T.; Max Carvalho, L.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef]
- Darriba, D.; Posada, D.; Kozlov, A.M.; Stamatakis, A.; Morel, B.; Flouri, T. ModelTest-NG: A new and scalable tool for the selection of DNA and protein evolutionary models. Mol. Biol. Evol. 2020, 37, 291–294. [Google Scholar] [CrossRef]
- Duchene, S.; Duchene, D.; Holmes, E.C.; Ho, S.Y.W. The performance of the date-randomization test in phylogenetic analyses of time-structured virus data. Mol. Biol. Evol. 2015, 32, 895–1906. [Google Scholar] [CrossRef]
- Zou, L.; Tu, L.; Shen, J.; Du, Z.; Cai, W.; Ji, Y.; Gao, F. The evolutionary dynamics and adaptive evolution of tomato chlorosis virus. Sci. Agric. Sin. 2020, 53, 4791–4801. [Google Scholar]
- Lemey, P.; Rambaut, A.; Drummond, A.J.; Suchard, M.A. Bayesian phylogeography finds its roots. PLoS Comput. Biol. 2009, 5, e1000520. [Google Scholar] [CrossRef]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef] [PubMed]
- Nahata, K.D.; Bielejec, F.; Monetta, J.; Dellicour, S.; Rambaut, A.; Suchard, M.A.; Baele, G.; Lemey, P. SPREAD 4: Online visualisation of pathogen phylogeographic reconstructions. Virus Evol. 2022, 8, veac088. [Google Scholar] [CrossRef]
- Baele, G.; Lemey, P.; Bedford, T.; Rambaut, A.; Suchard, M.A.; Alekseyenko, A.V. Improving the accuracy of demographic and molecular clock model comparison while accommodating phylogenetic uncertainty. Mol. Biol. Evol. 2012, 29, 2157–2167. [Google Scholar] [CrossRef] [PubMed]
- Minin, V.N.; Suchard, M.A. Counting labeled transitions in continuous-time Markov models of evolution. J. Math. Biol. 2008, 56, 391–412. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Cepas, J.; Serra, F.; Bork, P. ETE 3: Reconstruction, analysis, and visualization of phylogenomic data. Mol. Biol. Evol. 2016, 33, 1635–1638. [Google Scholar] [CrossRef]
- Brynildsrud, O.B.; Pepperell, C.S.; Suffys, P.; Grandjean, L.; Monteserin, J.; Debech, N.; Bohlin, J.; Alfsnes, K.; Pettersson, J.O.H.; Kirkeleite, I.; et al. Global expansion of Mycobacterium tuberculosis lineage 4 shaped by colonial migration and local adaptation. Sci. Adv. 2018, 4, eaat5869. [Google Scholar] [CrossRef]
- Möller, S.; du Plessis, L.; Stadler, T. Impact of the tree prior on estimating clock rates during epidemic outbreaks. Proc. Nat. Acad. Sci. USA 2018, 115, 4200–4205. [Google Scholar] [CrossRef]
- Wisler, G.C.; Duffus, J.E.; Liu, H.-Y.; Li, R.H. Ecology and epidemiology of whitefly-transmitted closteroviruses. Plant Dis. 1998, 82, 270–280. [Google Scholar] [CrossRef]
- Navas-Castillo, J.; Fiallo-Olivé, E.; Sánchez-Campos, S. Emerging virus diseases transmitted by whiteflies. Annu. Rev. Phytopathol. 2011, 49, 219–248. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, K.; Zhang, S.; Huang, W.; Du, Z.; Gao, F.; Guan, X. Phylogeographic History of Tomato Chlorosis Virus. Viruses 2025, 17, 457. https://doi.org/10.3390/v17040457
Wu K, Zhang S, Huang W, Du Z, Gao F, Guan X. Phylogeographic History of Tomato Chlorosis Virus. Viruses. 2025; 17(4):457. https://doi.org/10.3390/v17040457
Chicago/Turabian StyleWu, Kangcheng, Shiwei Zhang, Wende Huang, Zhenguo Du, Fangluan Gao, and Xiayu Guan. 2025. "Phylogeographic History of Tomato Chlorosis Virus" Viruses 17, no. 4: 457. https://doi.org/10.3390/v17040457
APA StyleWu, K., Zhang, S., Huang, W., Du, Z., Gao, F., & Guan, X. (2025). Phylogeographic History of Tomato Chlorosis Virus. Viruses, 17(4), 457. https://doi.org/10.3390/v17040457