
Phylogenetic Analysis of Chikungunya Virus Eastern/Central/South African-Indian Ocean Epidemic Strains, 2004–2019

 , , , , ,
, , , , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. CHIKV Sequence Dataset
2.2. Phylogenetic Analysis
2.3. Evolutionary Rate Estimate, Time-Scaled Phylogeny Reconstruction, and Bayesian Phylogeography
2.4. CHIKV E1 Mutations
2.5. CHIKV E1 Evolutionary Demographic Reconstruction (Bayesian Skyline Plot)
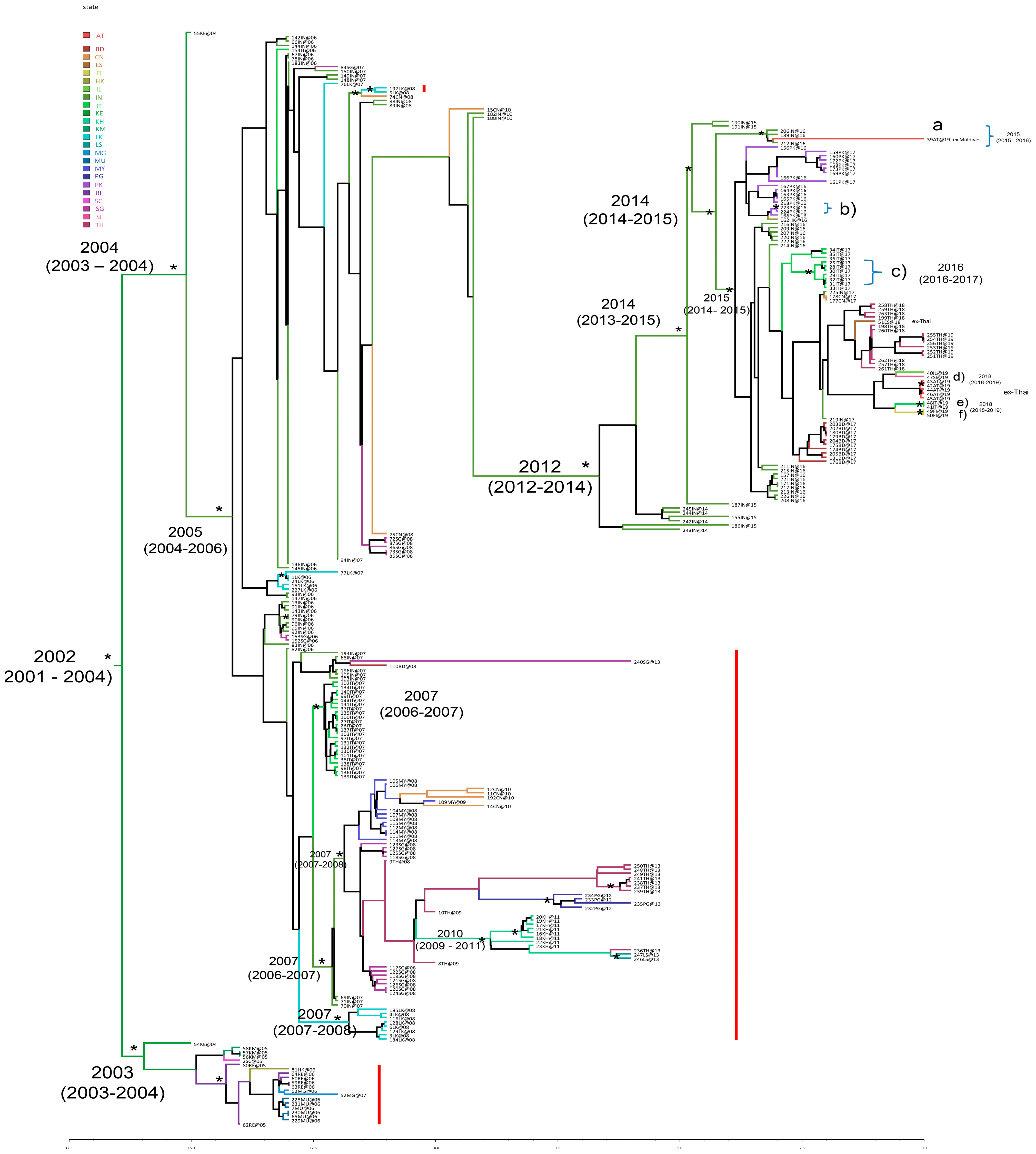
3. Results
3.1. Likelihood Mapping
3.2. Estimated Rate of CHIKV E1 Gene Evolution, Time-Scaled Phylogeny Reconstruction, and Bayesian Phylogeography
- -
- Further expansion in India (years 2014–2015)
- -
- Spread from India to Pakistan (approximately May 2016)
- -
- Spread from Pakistan to Hong Kong (September 2016)
- -
- Spread from India to Thailand (December 2017–September 2019, with multiple cases acquired in Thailand and detected as imported cases in several non-endemic countries, such as Austria, Spain, Italy, Israel, Finland, and Slovenia).
3.3. CHIKV E1 Mutations
3.4. CHIKV E1 Evolutionary Demographic Reconstruction
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CHIKV | Chikungunya virus |
| ECSA-IOL | Eastern/Central/South African—Indian Ocean Lineage |
| NSP | Non-structural proteins |
| MCMC | Markov chain Monte Carlo |
| BSP | Bayesian skyline plot |
| GMRF | Gaussian Markov Random Field |
| BEAST | Bayesian Evolutionary Analysis Sampling Trees |
| ESS | Effective sample size |
| BSSVS | Bayesian Stochastic Search Variable Selection |
| sp | State probability |
| pp | Posterior probability |
| HPD | Highest posterior density |
References
- Silva, L.A.; Dermody, T.S. Chikungunya virus: Epidemiology, replication, disease mechanisms, and prospective intervention strategies. J. Clin. Investig. 2017, 127, 737–749. [Google Scholar] [CrossRef]
- Rezza, R.; Nicoletti, L.; Angelini, R.; Romi, R.; Finarelli, A.C.; Panning, M.; Cordioli, P.; Fortuna, C.; Boros, S.; Magurano, F.; et al. Infection with chikungunya virus in Italy: An outbreak in a temperate region. Lancet 2007, 370, 1840–1846. [Google Scholar] [CrossRef] [PubMed]
- Venturi, G.; Di Luca, M.; Fortuna, C.; Remoli, M.E.; Riccardo, F.; Severini, F.; Toma, L.; Del Manso, M.; Benedetti, E.; Caporali, M.G.; et al. Detection of a chikungunya outbreak in Central Italy, August to September 2017. Euro Surveill. 2017, 22, 17-00646. [Google Scholar] [CrossRef] [PubMed]
- Riccardo, F.; Venturi, G.; Di Luca, M.; Del Manso, M.; Severini, F.; Andrianou, X.; Fortuna, C.; Remoli, M.E.; Benedetti, E.; Caporali, M.G.; et al. Secondary Autochthonous Outbreak of Chikungunya, Southern Italy, 2017. Emerg. Infect. Dis. 2019, 25, 2093–2095. [Google Scholar] [CrossRef] [PubMed]
- Lindh, E.; Argentini, C.; Remoli, M.E.; Fortuna, C.; Faggioni, G.; Benedetti, E.; Amendola, A.; Marsili, G.; Lista, F.; Rezza, G.; et al. The Italian 2017 Outbreak Chikungunya Virus Belongs to an Emerging Aedes albopictus—Adapted Virus Cluster Introduced From the Indian Subcontinent. Open Forum Infect. Dis. 2018, 6, ofy321. [Google Scholar] [CrossRef]
- Fortuna, C.; Toma, L.; Remoli, M.E.; Amendola, A.; Severini, F.; Boccolini, D.; Romi, R.; Venturi, G.; Rezza, G.; Di Luca, M. Vector competence of Aedes albopictus for the Indian Ocean lineage (IOL) chikungunya viruses of the 2007 and 2017 outbreaks in Italy: A comparison between strains with and without the E1:A226V mutation. Euro Surveill. 2018, 23, 1800246. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Severini, F.; Boccolini, D.; Fortuna, C.; Di Luca, M.; Toma, L.; Amendola, A.; Benedetti, E.; Minelli, G.; Romi, R.; Venturi, G.; et al. Vector competence of Italian Aedes albopictus populations for the chikungunya virus (E1-226V). PLoS Neglected Trop. Dis. 2018, 12, e0006435. [Google Scholar] [CrossRef]
- Kraemer, M.U.G.; Reiner, R.C., Jr.; Brady, O.J.; Messina, J.P.; Gilbert, M.; Pigott, D.M.; Yi, D.; Johnson, K.; Earl, L.; Marczak, L.B.; et al. Past and future spread of the arbovirus vectors Aedes aegypti and Aedes albopictus. Nat. Microbiol. 2019, 4, 854–863. [Google Scholar] [CrossRef]
- Kraemer, M.U.; Sinka, M.E.; Duda, K.A.; Mylne, A.Q.; Shearer, F.M.; Barker, C.M.; Moore, C.G.; Carvalho, R.G.; Coelho, G.E.; Van Bortel, W.; et al. The global distribution of the arbovirus vectors Aedes aegypti and Ae. albopictus. eLife 2015, 4, e08347. [Google Scholar] [CrossRef]
- Brown, J.E.; Evans, B.R.; Zheng, W.; Obas, V.; Barrera-Martinez, L.; Egizi, A.; Zhao, H.; Caccone, A.; Powell, J.R. Human impacts have shaped historical and recent evolution in Aedes aegypti, the dengue and yellow fever mosquito. Evolution 2014, 68, 514–525. [Google Scholar] [CrossRef]
- Izri, A.; Bitam, I.; Charrel, R.N. First entomological documentation of Aedes (Stegomyia) albopictus (Skuse, 1894) in Algeria. Clin. Microbiol. Infect. 2011, 17, 1116–1118. [Google Scholar] [CrossRef]
- Delatte, H.; Gimonneau, G.; Triboire, A.; Fontenille, D. Influence of temperature on immature development, survival, longevity, fecundity, and gonotrophic cycles of Aedes albopictus, vector of chikungunya and dengue in the Indian Ocean. J. Med. Entomol. 2009, 46, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Laras, K.; Sukri, N.C.; Larasati, R.P.; Bangs, M.J.; Kosim, R.; Djauzi; Wandra, T.; Master, J.; Kosasih, H.; Hartati, S.; et al. Tracking the reemergence of epidemic chikungunya virus in Indonesia. Trans. R. Soc. Trop. Med. Hyg. 2005, 99, 128–141. [Google Scholar] [CrossRef]
- Centers for Disease Control. Chikungunya fever among U.S. Peace Corps volunteers—Republic of the Philippines. MMWR Morb. Mortal. Wkly. Rep. 1986, 35, 573–574. [Google Scholar]
- Hasebe, F.; Parquet, M.C.; Pandey, B.D.; Mathenge, E.G.M.; Morita, K.; Balasubramaniam, V.; Saat, Z.; Yusop, A.; Sinniah, M.; Natkunam, S.; et al. Combined detection and genotyping of Chikungunya virus by a specific reverse transcription-polymerase chain reaction. Med. Virol. 2002, 67, 370–374. [Google Scholar] [CrossRef] [PubMed]
- Pavri, K. Disappearance of Chikungunya virus from India and South East Asia. Trans. R. Soc. Trop. Med. Hyg. 1986, 80, 491. [Google Scholar] [CrossRef]
- Volk, S.M.; Chen, R.; Tsetsarkin, K.A.; Adams, A.P.; Garcia, T.I.; Sall, A.A.; Nasar, F.; Schuh, A.J.; Holmes, E.C.; Higgs, S.; et al. Genome-scale phylogenetic analyses of chikungunya virus reveal independent emergences of recent epidemics and various evolutionary rates. J. Virol. 2010, 84, 6497–6504. [Google Scholar] [CrossRef]
- Weaver, S.C.; Lecuit, M. Chikungunya virus and the global spread of a mosquito-borne disease. N. Engl. J. Med. 2015, 372, 1231–1239. [Google Scholar] [CrossRef]
- Thiberville, S.D.; Moyen, N.; Dupuis-Maguiraga, L.; Nougairede, N.; Gould, E.A.; Roques, P.; de Lamballerie, X. Chikungunya fever: Epidemiology, clinical syndrome, pathogenesis and therapy. Antivir. Res. 2013, 99, 345–370. [Google Scholar] [CrossRef]
- Powers, A.M.; Logue, C.H. Changing patterns of chikungunya virus: Re-emergence of a zoonotic arbovirus. J. Gen. Virol. 2007, 88 Pt 9, 2363–2377. [Google Scholar] [CrossRef]
- Schwartz, O.; Albert, M.L. Biology and pathogenesis of chikungunya virus. Nat. Rev. Microbiol. 2010, 8, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Grandadam, M.; Caro, V.; Plumet, S.; Thiberge, J.-M.; Souarès, Y.; Failloux, A.-B.; Tolou, H.J.; Budelot, M.; Cosserat, D.; Leparc-Goffart, I.; et al. Chikungunya virus, southeastern France. Emerg. Infect. Dis. 2011, 17, 910–913. [Google Scholar] [CrossRef] [PubMed]
- Tsetsarkin, K.A.; Vanlandingham, D.L.; McGee, C.E.; Higgs, S. A single mutation in chikungunya virus affects vector specificity and epidemic potential. PLoS Pathog. 2007, 3, e201. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Galaxy Platform. Homepage. Available online: https://galaxyproject.org (accessed on 17 June 2020).
- Afgan, E.; Baker, D.; Batut, B.; van den Beek, M.; Cech, M.; Chilton, J.; Clements, D.; Coraor, N.; Gruning, B.A.; Guerler, A.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef]
- Hall, T.A. BioEdit: A User-Friendly Biological Sequence Alignment Editor and Analysis Program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Posada, D. jModelTest: Phylogenetic model averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef]
- Strimmer, K.; von Haeseler, A. Likelihood-mapping: A simple method to visualize phylogenetic content of a sequence alignment. Proc. Natl. Acad. Sci. USA 1997, 94, 6815–6819. [Google Scholar] [CrossRef]
- Schmidt, H.A.; Strimmer, K.; Vingron, M.; von Haeseler, A. TREE-PUZZLE: Maximum likelihood phylogenetic analysis using quartets and parallel computing. Bioinformatics 2002, 18, 502–504. [Google Scholar] [CrossRef]
- Rambaut, A.; Lam, T.T.; Carvalho, L.M.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef] [PubMed]
- BEAST. Available online: http://beast.bio.ed.ac.uk (accessed on 13 November 2020).
- Drummond, A.J.; Rambaut, A.; Shapiro, B.; Pybus, O.G. Bayesian Coalescent Inference of Past Population Dynamics from Molecular Sequences. Mol. Biol. Evol. 2005, 22, 1185–1192. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef] [PubMed]
- Kass, R.E.; Raftery, A.E. Bayes Factors. J. Am. Stat. Assoc. 1995, 90, 773–795. [Google Scholar] [CrossRef]
- Bartholomeeusen, K.; Daniel, M.; LaBeaud, D.A.; Gasque, P.; Peeling, R.W.; Stephenson, K.E.; Ng, L.F.P.; Ariën, K.K. Chikungunya fever. Nat. Rev. Dis. Primers 2023, 9, 17. [Google Scholar] [CrossRef]
- Schuffenecher, I.; Iteman, I.; Michault, A.; Murri, S.; Frangeul, L.; Vaney, M.C.; Lavenir, R.; Pardigon, N.; Reynes, J.M.; Pettinelli, F.; et al. Genome microevolution of chikungunya viruses causing the Indian Ocean outbreak. PLoS Med. 2006, 3, e263. [Google Scholar] [CrossRef]
- Mohan, A.; Kiran, D.H.N.; Manohar, I.C.; Kumar, D.P. Epidemiology, clinical manifestations, and diagnosis of Chikungunya fever: Lessons learned from the re-emerging epidemic. Indian J. Dermatol. 2010, 55, 54–63. [Google Scholar] [CrossRef]
- Robinson, M.C. An epidemic of virus disease in Southern Province, Tanganyika Territory, in 1952–1953. I. Clinical features. Trans. R. Soc. Trop. Med. Hyg. 1955, 49, 28–32. [Google Scholar] [CrossRef]
- Deeba, F.; Haider, M.S.H.; Ahmed, A.; Tazeen, A.; Faizan, M.I.; Salam, N.; Hussain, T.; Alamery, S.F.; Parveen, S. Global transmission and evolutionary dynamics of the Chikungunya virus. Epidemiol. Infect. 2020, 148, e63. [Google Scholar] [CrossRef] [PubMed]
- Ciccozzi, M.; Lo Presti, A.; Cella, E.; Giovanetti, M.; Lai, A.; El-Sawaf, G.; Faggioni, G.; Vescio, F.; Al Ameri, R.; De Santis, R.; et al. Phylogeny of Dengue and Chikungunya viruses in Al Hudayda governorate, Yemen. Infect. Genet. Evol. 2014, 27, 395–401. [Google Scholar] [CrossRef]
- Lo Presti, A.; Ciccozzi, M.; Cella, E.; Lai, A.; Simonetti, F.R.; Galli, M.; Zehender, G.; Rezza, G. Origin, evolution, and phylogeography of recent epidemic CHIKV strains. Infect. Genet. Evol. 2012, 12, 392–398. [Google Scholar] [CrossRef] [PubMed]
- Epicentro—L’Epidemiologia per la Sanità Pubblica—Chikungunya—Notiziario—27 Settembre 2007—Ecdc: L’Aggiornamento Sull’Epidemia in Italia. Available online: https://www.epicentro.iss.it/chikungunya/notiziario27-9-07#:~:text=Continuano%20gli%20sforzi%20per%20gestire,attesa%20della%20conferma%20di%20laboratorio (accessed on 9 November 2021).
- Angelini, R.; Finarelli, A.; Angelini, P.; Po, C.; Petropulacos, C.; Silvi, G.; Macini, P.; Fortuna, C.; Venturi, F.; Magurano, F.; et al. Chikungunya in north-eastern Italy: A summing up of the outbreak. Euro Surveill. 2007, 12, E071122. [Google Scholar] [CrossRef] [PubMed]
- Berry, I.M.; Eyase, F.; Pollett, S.; Konongoi, S.L.; Joyce, M.G.; Figueroa, K.; Ofula, V.; Koka, H.; Koskei, E.; Nyunja, A.; et al. Global Outbreaks and Origins of a Chikungunya Virus Variant Carrying Mutations Which May Increase Fitness for Aedes aegypti: Revelations from the 2016 Mandera, Kenya Outbreak. Am. J. Trop. Med. Hyg. 2019, 100, 1249–1257. [Google Scholar] [CrossRef] [PubMed]
- Shrinet, J.; Jain, S.; Sharma, A.; Singh, S.S.; Mathus, K.; Rana, V.; Bhatnagar, R.K.; Gupta, B.; Gaind, R.; Deb, M.; et al. Genetic characterization of Chikungunya virus from New Delhi reveal emergence of a new molecular signature in Indian isolates. Virol. J. 2012, 9, 100. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| E1—a.a. Change | n | % |
|---|---|---|
| A226V | 109 | 42.2 |
| D284E | 257 | 99.6 |
| K211E | 97 | 37.6 |
| K211N | 3 | 1.2 |
| A98T | / | / |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lo Presti, A.; Argentini, C.; Marsili, G.; Fortuna, C.; Amendola, A.; Fiorentini, C.; Venturi, G. Phylogenetic Analysis of Chikungunya Virus Eastern/Central/South African-Indian Ocean Epidemic Strains, 2004–2019. Viruses 2025, 17, 430. https://doi.org/10.3390/v17030430
Lo Presti A, Argentini C, Marsili G, Fortuna C, Amendola A, Fiorentini C, Venturi G. Phylogenetic Analysis of Chikungunya Virus Eastern/Central/South African-Indian Ocean Epidemic Strains, 2004–2019. Viruses. 2025; 17(3):430. https://doi.org/10.3390/v17030430
Chicago/Turabian StyleLo Presti, Alessandra, Claudio Argentini, Giulia Marsili, Claudia Fortuna, Antonello Amendola, Cristiano Fiorentini, and Giulietta Venturi. 2025. "Phylogenetic Analysis of Chikungunya Virus Eastern/Central/South African-Indian Ocean Epidemic Strains, 2004–2019" Viruses 17, no. 3: 430. https://doi.org/10.3390/v17030430
APA StyleLo Presti, A., Argentini, C., Marsili, G., Fortuna, C., Amendola, A., Fiorentini, C., & Venturi, G. (2025). Phylogenetic Analysis of Chikungunya Virus Eastern/Central/South African-Indian Ocean Epidemic Strains, 2004–2019. Viruses, 17(3), 430. https://doi.org/10.3390/v17030430