

An Integrative Computational Approach for Identifying Cotton Host Plant MicroRNAs with Potential to Abate CLCuKoV-Bur Infection



Abstract
1. Introduction
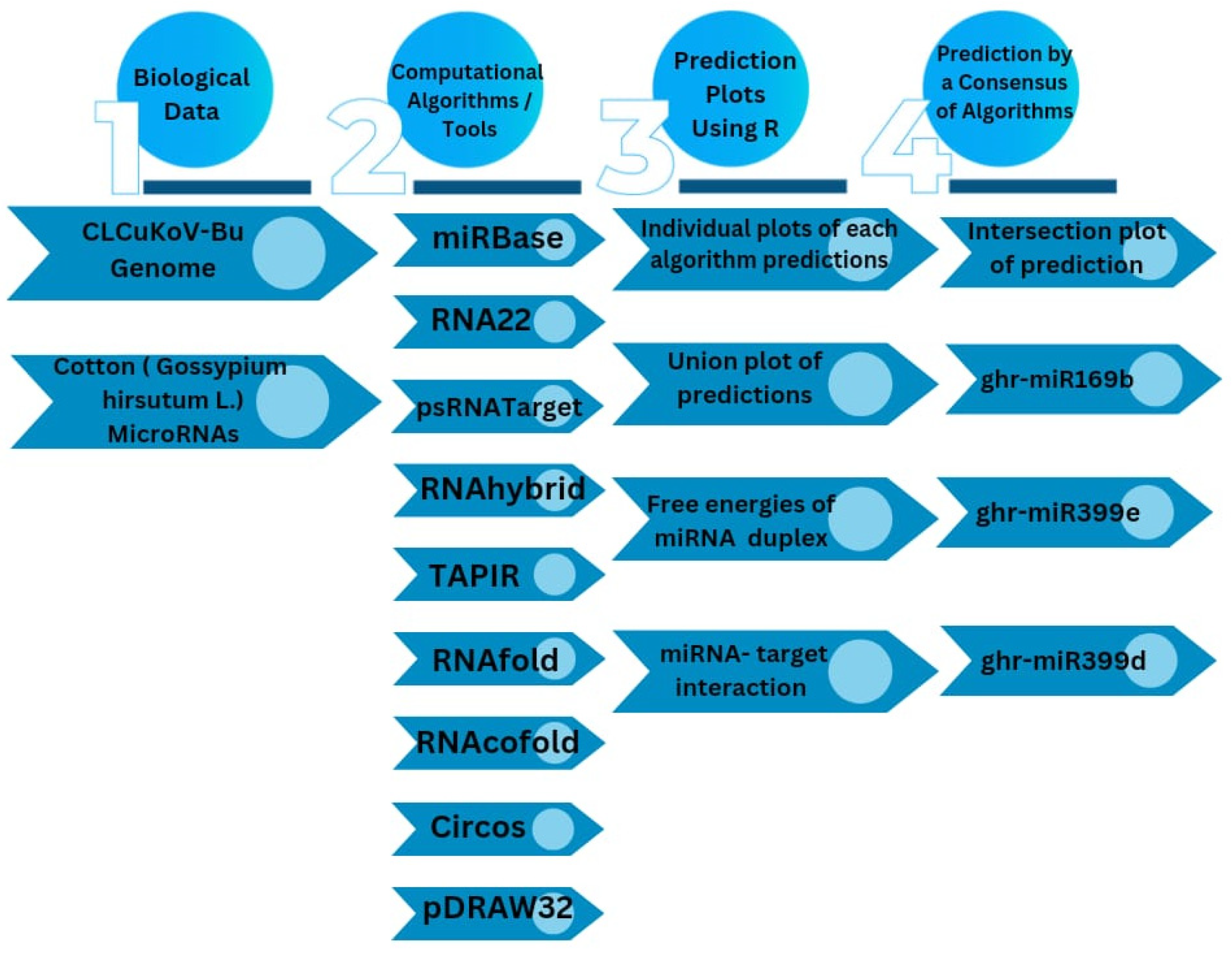
2. Materials and Methods
2.1. Upland Cotton (G. hirsutum) MicroRNAs and CLCuKoV-Bur Genome Retrieval
2.2. RNA22 Algorithm
2.3. psRNATarget Algorithm
2.4. RNAhybrid Algorithm
2.5. TAPIR Algorithm
2.6. RNAfold Algorithm
2.7. RNAcofold Algorithm
2.8. Discovering Cotton Genome-Encoded miRNAs–Target Interaction
2.9. Statistical Analysis
2.10. CLCuKoV-Bur Genome Annotation
3. Results
3.1. High-Probability miRNA Binding Sites in CLCuKoV-Bur Genome
3.2. Coat Protein (CP) of CLCuKoV-Bur Genome
3.3. Predicted Targets for the V2 ORF of CLCuKoV-Bur
3.4. Predicted Targets for C1 ORF of CLCuKoV-Bur
3.5. Predicted Targets for C3 ORF of CLCuKoV-Bur
3.6. Predicted Targets for the C4 ORF of CLCuKoV-Bur
3.7. Large Intergenic Region of CLCuKoV-Bur
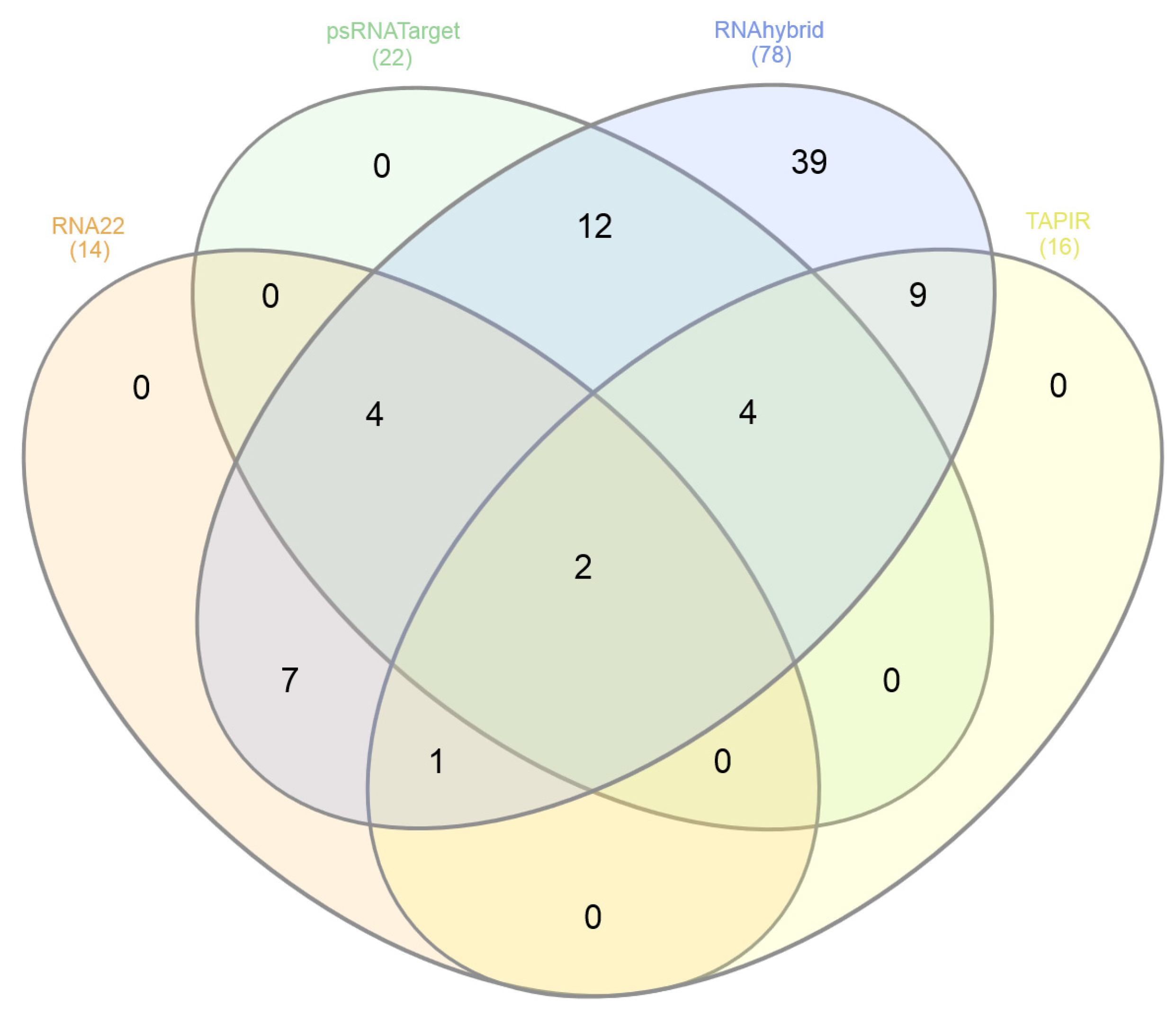
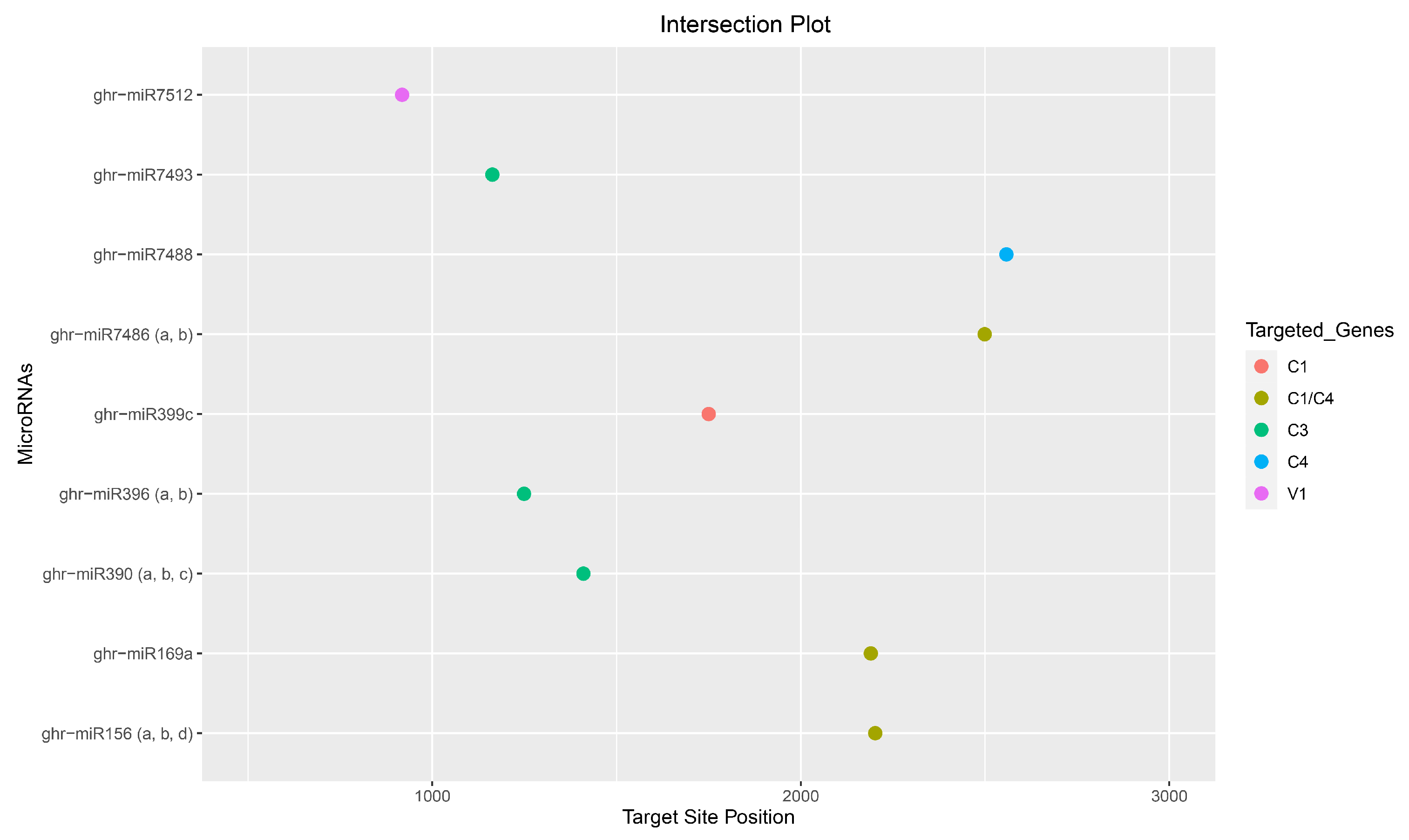
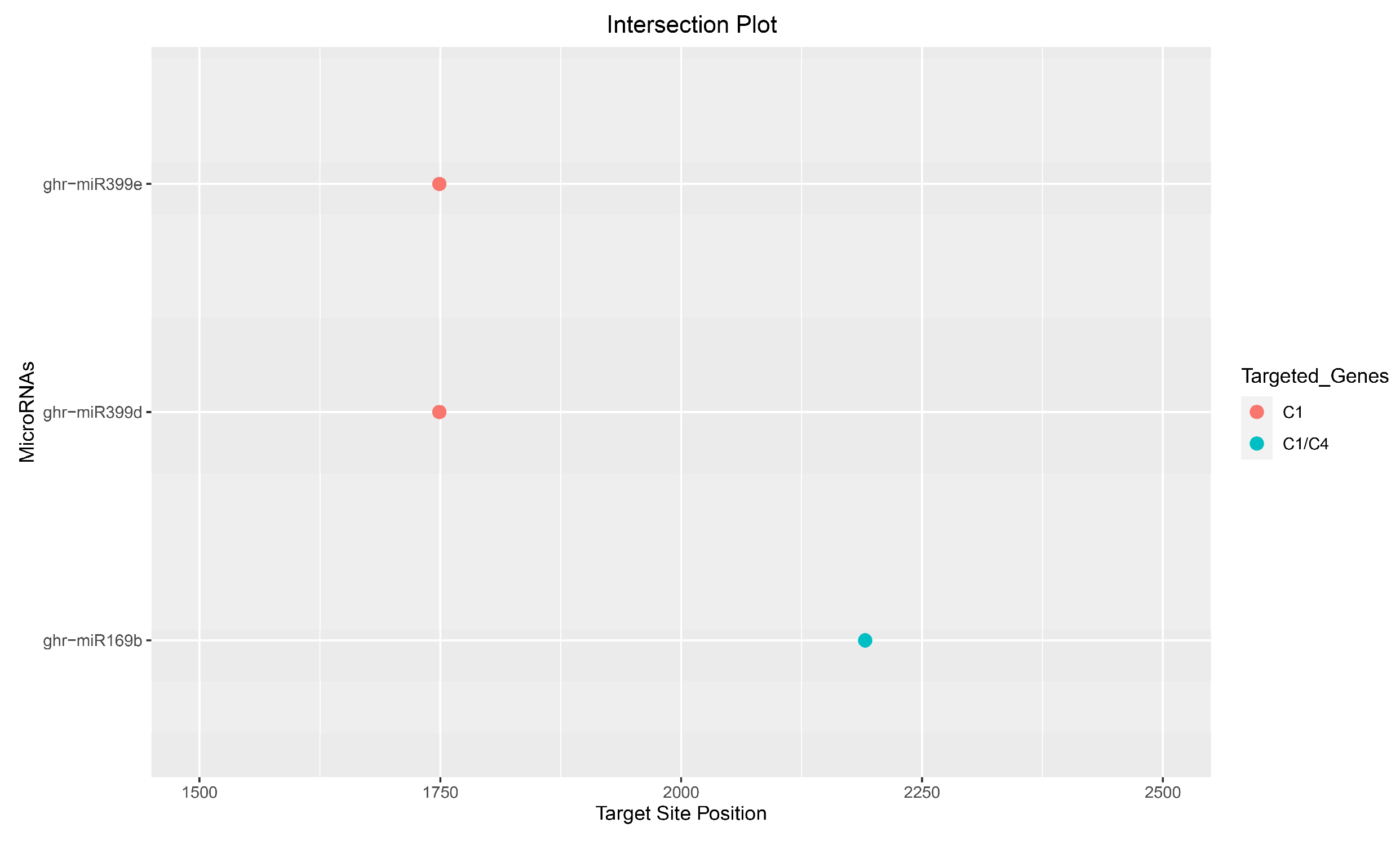
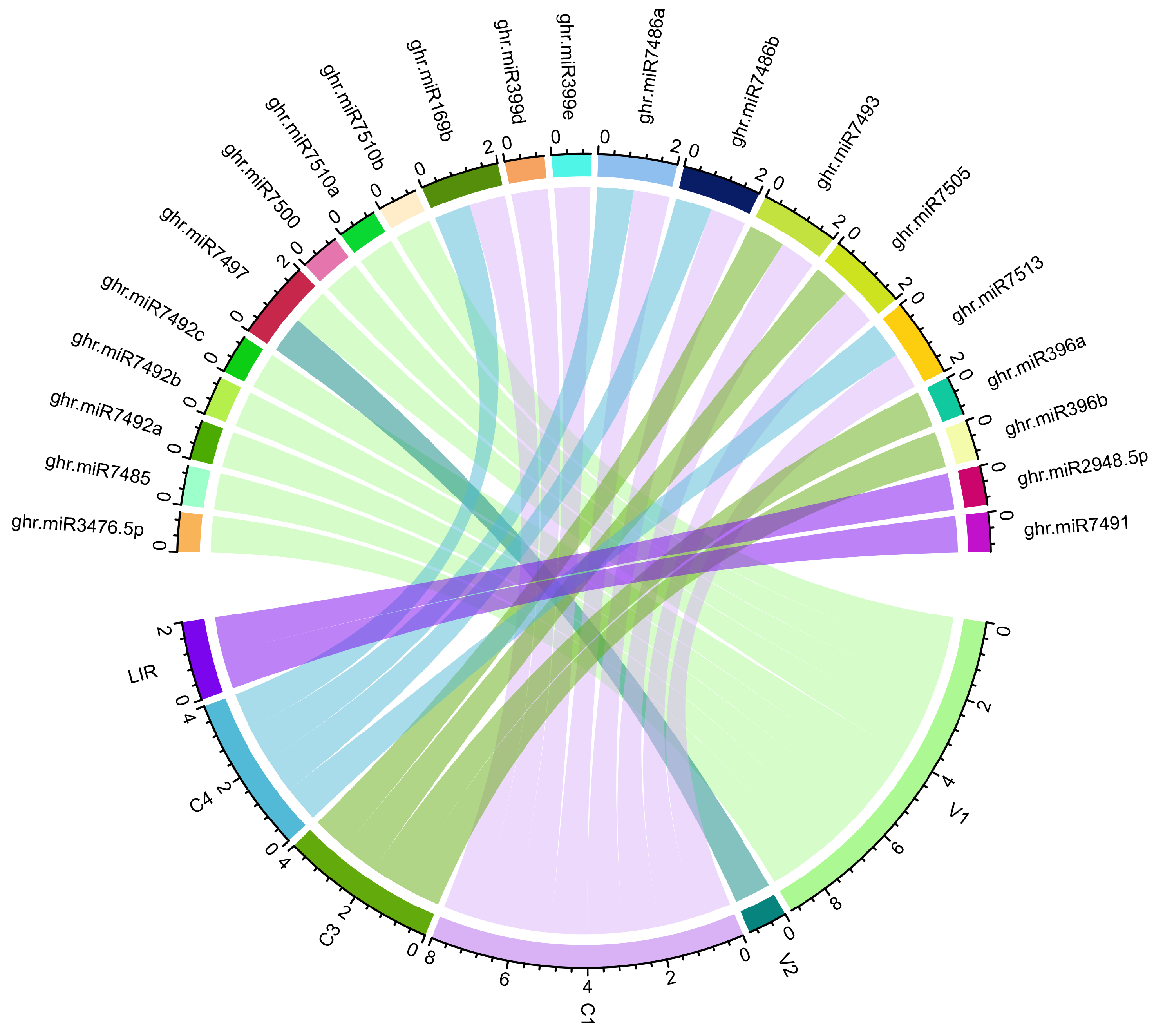
3.8. Consensus miRNAs Predictions
3.9. Predicted miRNA–Target Interaction
3.10. Estimation of the Free Energy (ΔG) of Consensus miRNA–mRNA Pairs
3.10.1. Secondary Structure Predictions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Paterson, A.H.; Wendel, J.F.; Gundlach, H.; Guo, H.; Jenkins, J.; Jin, D.; Llewellyn, D.; Showmaker, K.C.; Shu, S.; Udall, J. Repeated polyploidization of Gossypium genomes and the evolution of spinnable cotton fibres. Nature 2012, 492, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Z.; Zhang, K.; Fang, Y.; Yang, Y.; Cao, X.; Liu, L.; Tian, Y. Systematically and comprehensively understanding the regulation of cotton fiber initiation: A review. Plants 2023, 12, 3771. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Hu, Y.; Jiang, W.; Fang, L.; Guan, X.; Chen, J.; Zhang, J.; Saski, C.A.; Scheffler, B.E.; Stelly, D.M. Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement. Nat. Biotechnol. 2015, 33, 531–537. [Google Scholar] [CrossRef]
- Sattar, M.N.; Kvarnheden, A.; Saeed, M.; Briddon, R.W. Cotton leaf curl disease–an emerging threat to cotton production worldwide. J. Gen. Virol. 2013, 94, 695–710. [Google Scholar] [CrossRef] [PubMed]
- Nadeem, S.; Riaz Ahmed, S.; Luqman, T.; Tan, D.K.; Maryum, Z.; Akhtar, K.P.; Muhy Ud Din Khan, S.; Tariq, M.S.; Muhammad, N.; Khan, M.K.R. A comprehensive review on Gossypium hirsutum resistance against cotton leaf curl virus. Front. Genet. 2024, 15, 1306469. [Google Scholar] [CrossRef]
- Zubair, M.; Zaidi, S.S.-e.-A.; Shakir, S.; Farooq, M.; Amin, I.; Scheffler, J.A.; Scheffler, B.E.; Mansoor, S. Multiple begomoviruses found associated with cotton leaf curl disease in Pakistan in early 1990 are back in cultivated cotton. Sci. Rep. 2017, 7, 680. [Google Scholar] [CrossRef]
- Amrao, L.; Amin, I.; Shahid, M.S.; Briddon, R.W.; Mansoor, S. Cotton leaf curl disease in resistant cotton is associated with a single begomovirus that lacks an intact transcriptional activator protein. Virus. Res. 2010, 152, 153–163. [Google Scholar] [CrossRef]
- Iqbal, M.J.; Zia-Ur-Rehman, M.; Ilyas, M.; Hameed, U.; Herrmann, H.W.; Chingandu, N.; Manzoor, M.T.; Haider, M.S.; Brown, J.K. Sentinel plot surveillance of cotton leaf curl disease in Pakistan—A case study at the cultivated cotton-wild host plant interface. Virus Res. 2023, 333, 199144. [Google Scholar] [CrossRef]
- Mahmood, M.A.; Ahmed, N.; Hussain, A.; Naqvi, R.Z.; Amin, I.; Mansoor, S. Dominance of cotton leaf curl Multan virus-Rajasthan strain associated with third epidemic of cotton leaf curl disease in Pakistan. Sci. Rep. 2024, 14, 13532. [Google Scholar] [CrossRef]
- Ashraf, M.A.; Shahid, A.A.; Mohamed, B.B.; Dahab, A.A.; Bajwa, K.S.; Rao, A.Q.; Khan, M.A.U.; Ilyas, M.; Haider, M.S.; Husnain, T. Molecular characterization and phylogenetic analysis of a variant of highly infectious cotton leaf curl Burewala virus associated with CLCuD from Pakistan. Aust. J. Crop Sci. 2013, 7, 1113–1122. [Google Scholar]
- Pawar, T.; Sharma, S.; Arora, R.K.; Biswas, K.K. Distribution of cotton leaf curl virus species/strain and characterization of associated satellite molecules in Gossypium hirsutum. Indian Phytopathol. 2024, 77, 815–823. [Google Scholar] [CrossRef]
- Afzal, M.; Saeed, S.; Riaz, H.; Ishtiaq, M.; Habib ur Rahman, M. Transmission efficiency of cotton leaf curl Khokhran virus/Cotton leaf curl Multan betasatellite complex by two whitefly cryptic species in Pakistan. Int. J. Trop. Insect Sci. 2023, 43, 819–830. [Google Scholar] [CrossRef]
- Shuja, M.N.; Briddon, R.W.; Tahir, M. Identification of a distinct strain of cotton leaf curl Burewala virus. Arch. Virol. 2014, 159, 2787–2790. [Google Scholar] [CrossRef]
- Idrees, M.A.; Abbas, A.; Saddam, B.; Bashir, M.H.; Naveed, H.; Khan, A.K.; Dara, M.Z.N. A comprehensive review: Persistence, circulative transmission of begomovirus by whitefly vectors. Int. J. Trop. Insect Sci. 2024, 44, 405–417. [Google Scholar] [CrossRef]
- de Moya, R.S.; Brown, J.K.; Sweet, A.D.; Walden, K.K.; Paredes-Montero, J.R.; Waterhouse, R.M.; Johnson, K.P. Nuclear orthologs derived from whole genome sequencing indicate cryptic diversity in the Bemisia tabaci (Insecta: Aleyrodidae) complex of whiteflies. Diversity 2019, 11, 151. [Google Scholar] [CrossRef]
- Shah, S.H.J.; Paredes-Montero, J.R.; Malik, A.H.; Brown, J.K.; Qazi, J. Distribution of Bemisia tabaci (Gennadius) (Hemiptera: Aleyrodidae) mitotypes in commercial cotton fields in the Punjab province of Pakistan. Fla. Entomol. 2020, 103, 41–47. [Google Scholar] [CrossRef]
- Chen, T.; Saeed, Q.; He, Z.; Lu, L. Transmission efficiency of Cotton leaf curl Multan virus by three cryptic species of Bemisia tabaci complex in cotton cultivars. PeerJ 2019, 7, e7788. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.K.; Zerbini, F.M.; Navas-Castillo, J.; Moriones, E.; Ramos-Sobrinho, R.; Silva, J.C.; Fiallo-Olivé, E.; Briddon, R.W.; Hernández-Zepeda, C.; Idris, A. Revision of Begomovirus taxonomy based on pairwise sequence comparisons. Arch. Virol. 2015, 160, 1593–1619. [Google Scholar] [CrossRef]
- Ashraf, M.A.; Shahid, A.A.; Rao, A.Q.; Bajwa, K.S.; Husnain, T. Functional characterization of a bidirectional plant promoter from cotton leaf curl Burewala virus using an Agrobacterium-mediated transient assay. Viruses 2014, 6, 223–242. [Google Scholar] [CrossRef]
- Ashraf, M.A.; Shahid, A.A.; Rao, A.Q.; Brown, J.K.; Husnain, T. Development and evaluation of the cotton leaf curl Kokhran virus-Burewala bidirectional promoter for enhanced Cry1Ac endotoxin expression in Bt transgenic cotton. Appl. Sci. 2022, 12, 11275. [Google Scholar] [CrossRef]
- Koeppe, S.; Kawchuk, L.; Kalischuk, M. RNA interference past and future applications in plants. Int. J. Mol. Sci. 2023, 24, 9755. [Google Scholar] [CrossRef] [PubMed]
- Akbar, S.; Wei, Y.; Zhang, M.-Q. RNA interference: Promising approach to combat plant viruses. Int. J. Mol. Sci. 2022, 23, 5312. [Google Scholar] [CrossRef] [PubMed]
- Shang, R.; Lee, S.; Senavirathne, G.; Lai, E.C. microRNAs in action: Biogenesis, function and regulation. Nat. Rev. Genet. 2023, 24, 816–833. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Zhu, Y.; Lin, Z.; Lei, J.; Li, L.; Zhu, W.; Wu, D. Evidence of cross-kingdom gene regulation by plant MicroRNAs and possible reasons for inconsistencies. J. Agric. Food. Chem. 2024, 72, 4564–4573. [Google Scholar] [CrossRef]
- Bajczyk, M.; Jarmolowski, A.; Jozwiak, M.; Pacak, A.; Pietrykowska, H.; Sierocka, I.; Swida-Barteczka, A.; Szewc, L.; Szweykowska-Kulinska, Z. Recent insights into plant miRNA biogenesis: Multiple layers of miRNA level regulation. Plants 2023, 12, 342. [Google Scholar] [CrossRef]
- Luo, C.; Bashir, N.H.; Li, Z.; Liu, C.; Shi, Y.; Chu, H. Plant microRNAs regulate the defense response against pathogens. Front. Microbiol. 2024, 15, 1434798. [Google Scholar] [CrossRef]
- Yang, X.; Zhang, L.; Yang, Y.; Schmid, M.; Wang, Y. miRNA mediated regulation and interaction between plants and pathogens. Int. J. Mol. Sci. 2021, 22, 2913. [Google Scholar] [CrossRef]
- Al-Roshdi, M.R.; Ammara, U.; Khan, J.; Al-Sadi, A.M.; Shahid, M.S. Artificial microRNA-mediated resistance against Oman strain of tomato yellow leaf curl virus. Front. Plant Sci. 2023, 14, 1150. [Google Scholar] [CrossRef]
- Khalid, A.; Zhang, X.; Ji, H.; Yasir, M.; Farooq, T.; Dai, X.; Li, F. Large artificial microRNA cluster genes confer effective resistance against multiple tomato yellow leaf curl viruses in transgenic tomato. Plants 2023, 12, 2179. [Google Scholar] [CrossRef]
- Teotia, S.; Wang, X.; Zhou, N.; Wang, M.; Liu, H.; Qin, J.; Han, D.; Li, C.; Li, C.E.; Pan, S. A high-efficiency gene silencing in plants using two-hit asymmetrical artificial MicroRNAs. Plant Biotechnol. J. 2023, 21, 1799–1811. [Google Scholar] [CrossRef]
- Ma, Z.; Wang, J.; Li, C. Research Progress on miRNAs and artificial miRNAs in insect and disease resistance and breeding in plants. Genes 2024, 15, 1200. [Google Scholar] [CrossRef] [PubMed]
- Cisneros, A.E.; Carbonell, A. Artificial small RNA-based silencing tools for antiviral resistance in plants. Plants 2020, 9, 669. [Google Scholar] [CrossRef]
- Cisneros, A.E.; Martín-García, T.; Primc, A.; Kuziuta, W.; Sánchez-Vicente, J.; Aragonés, V.; Daròs, J.-A.; Carbonell, A. Transgene-free, virus-based gene silencing in plants by artificial microRNAs derived from minimal precursors. Nucleic Acids Res. 2023, 51, 10719–10736. [Google Scholar] [CrossRef] [PubMed]
- Niu, Q.-W.; Lin, S.-S.; Reyes, J.L.; Chen, K.-C.; Wu, H.-W.; Yeh, S.-D.; Chua, N.-H. Expression of artificial microRNAs in transgenic Arabidopsis thaliana confers virus resistance. Nat. Biotechnol. 2006, 24, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Hao, J.; Li, J.; Baker, B.; Luo, L. Artificial microRNA-mediated resistance to cucumber green mottle mosaic virus in Nicotiana benthamiana. Planta 2019, 250, 1591–1601. [Google Scholar] [CrossRef]
- Miao, S.; Liang, C.; Li, J.; Baker, B.; Luo, L. Polycistronic artificial microRNA-mediated resistance to cucumber green mottle mosaic virus in cucumber. Int. J. Mol. Sci. 2021, 22, 12237. [Google Scholar] [CrossRef]
- Zhou, L.; Yuan, Q.; Ai, X.; Chen, J.; Lu, Y.; Yan, F. Transgenic Rice plants expressing artificial miRNA targeting the Rice stripe virus MP gene are highly resistant to the virus. Biology 2022, 11, 332. [Google Scholar] [CrossRef]
- Petchthai, U.; Yee, C.S.L.; Wong, S.-M. Resistance to CymMV and ORSV in artificial microRNA transgenic Nicotiana benthamiana plants. Sci. Rep. 2018, 8, 9958. [Google Scholar] [CrossRef]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. miRBase: From microRNA sequences to function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef]
- Li, J.; Hull, J.J.; Liang, S.; Wang, Q.; Chen, L.; Zhang, Q.; Wang, M.; Mansoor, S.; Zhang, X.; Jin, S. Genome-wide analysis of cotton miRNAs during whitefly infestation offers new insights into plant-herbivore interaction. Int. J. Mol. Sci. 2019, 20, 5357. [Google Scholar] [CrossRef]
- Wang, W.; Liu, D.; Chen, D.; Cheng, Y.; Zhang, X.; Song, L.; Hu, M.; Dong, J.; Shen, F. MicroRNA414c affects salt tolerance of cotton by regulating reactive oxygen species metabolism under salinity stress. RNA Biol. 2019, 16, 362–375. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Zhang, J.; Zhu, Q.; Zhao, L.; Sui, S.; Li, Z.; Zhang, Y.; Wang, H.; Tian, D.; Zhao, Y. Identification of microRNAs involved in drought stress responses in early-maturing cotton by high-throughput sequencing. Genes Genom. 2018, 40, 305–314. [Google Scholar] [CrossRef]
- Mei, J.; Wu, Y.; Niu, Q.; Miao, M.; Zhang, D.; Zhao, Y.; Cai, F.; Yu, D.; Ke, L.; Feng, H. Integrative analysis of expression profiles of mRNA and microRNA provides insights of cotton response to Verticillium dahliae. Int. J. Mol. Sci. 2022, 23, 4702. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Feng, C.; Zhang, W.; Sun, S.; Yue, D.; Zhang, X.; Yang, X. Comprehensive non-coding RNA analysis reveals specific lncRNA/circRNA–miRNA–mRNA regulatory networks in the cotton response to drought stress. Int. J. Biol. Macromol. 2023, 253, 126558. [Google Scholar] [CrossRef] [PubMed]
- Zhan, J.; Diao, Y.; Yin, G.; Sajjad, M.; Wei, X.; Lu, Z.; Wang, Y. Integration of mRNA and miRNA analysis reveals the molecular mechanism of cotton response to salt stress. Front. Plant Sci. 2021, 12, 767984. [Google Scholar] [CrossRef]
- Sayers, E.W.; Beck, J.; Bolton, E.E.; Bourexis, D.; Brister, J.R.; Canese, K.; Comeau, D.C.; Funk, K.; Kim, S.; Klimke, W. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2021, 49, D10. [Google Scholar] [CrossRef]
- Miranda, K.C.; Huynh, T.; Tay, Y.; Ang, Y.-S.; Tam, W.-L.; Thomson, A.M.; Lim, B.; Rigoutsos, I. A pattern-based method for the identification of microRNA binding sites and their corresponding heteroduplexes. Cell 2006, 126, 1203–1217. [Google Scholar] [CrossRef]
- Loher, P.; Rigoutsos, I. Interactive exploration of RNA22 microRNA target predictions. Bioinformatics 2012, 28, 3322–3323. [Google Scholar] [CrossRef]
- Dai, X.; Zhao, P.X. psRNATarget: A plant small RNA target analysis server. Nucleic Acids Res. 2011, 39, W155–W159. [Google Scholar] [CrossRef]
- Dai, X.; Zhuang, Z.; Zhao, P.X. psRNATarget: A plant small RNA target analysis server (2017 release). Nucleic Acids Res. 2018, 46, W49–W54. [Google Scholar] [CrossRef]
- Krüger, J.; Rehmsmeier, M. RNAhybrid: microRNA target prediction easy, fast and flexible. Nucleic Acids Res. 2006, 34, W451–W454. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, E.; He, Y.; Billiau, K.; Van de Peer, Y. TAPIR, a web server for the prediction of plant microRNA targets, including target mimics. Bioinformatics 2010, 26, 1566–1568. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, R.; Bernhart, S.; Siederdissen, C.; Tafer, H.; Flamm, C.; Stadler, P.; Hofacker, I. ViennaRNA package 2.0. Algorithms Mol. Biol. 2011, 6, 26. [Google Scholar] [CrossRef]
- Bernhart, S.H.; Tafer, H.; Mückstein, U.; Flamm, C.; Stadler, P.F.; Hofacker, I.L. Partition function and base pairing probabilities of RNA heterodimers. Algorithms Mol. Biol. 2006, 1, 3. [Google Scholar] [CrossRef]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef]
- Gandrud, C. Reproducible Research with R and RStudio; Chapman and Hall/CRC: Boca Raton, FL, USA, 2018. [Google Scholar]
- Poornima Priyadarshini, C.; Ambika, M.; Tippeswamy, R.; Savithri, H. Functional characterization of coat protein and V2 involved in cell to cell movement of Cotton leaf curl Kokhran virus-Dabawali. PLoS ONE 2011, 6, e26929. [Google Scholar] [CrossRef]
- Rasool, G.; Yousaf, S.; Ammara, U.e.; Iqbal, A.; Saeed, M.; Amin, I.; Mansoor, S. Transgenic expression of synthetic coat protein and synthetic replication associated protein produces mild symptoms and reduce begomovirus-betasatellite accumulation in Nicotiana benthamiana. Front. Agron. 2021, 3, 676820. [Google Scholar] [CrossRef]
- Breves, S.S.; Silva, F.A.; Euclydes, N.C.; Saia, T.F.; Jean-Baptiste, J.; Andrade Neto, E.R.; Fontes, E.P. Begomovirus–host interactions: Viral proteins orchestrating intra and intercellular transport of viral DNA while suppressing host defense mechanisms. Viruses 2023, 15, 1593. [Google Scholar] [CrossRef]
- Bahari, A.; Castillo, A.G.; Safaie, N.; Bejarano, E.R.; Luna, A.P.; Shams-Bakhsh, M. Functional analysis of V2 protein of Beet curly top Iran virus. Plants 2022, 11, 3351. [Google Scholar] [CrossRef]
- Li, M.; Li, C.; Jiang, K.; Li, K.; Zhang, J.; Sun, M.; Wu, G.; Qing, L. Characterization of pathogenicity-associated V2 protein of tobacco curly shoot virus. Int. J. Mol. Sci. 2021, 22, 923. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, Y.; Gong, Q.; Ismayil, A.; Yuan, Y.; Lian, B.; Jia, Q.; Han, M.; Deng, H.; Hong, Y. Geminiviral V2 protein suppresses transcriptional gene silencing through interaction with AGO4. J. Virol. 2019, 93, e01675-01618. [Google Scholar] [CrossRef]
- Torres-Herrera, S.I.; Romero-Osorio, A.; Moreno-Valenzuela, O.; Pastor-Palacios, G.; Cardenas-Conejo, Y.; Ramírez-Prado, J.H.; Riego-Ruiz, L.; Minero-García, Y.; Ambriz-Granados, S.; Argüello-Astorga, G.R. A lineage of begomoviruses encode Rep and AC4 proteins of enigmatic ancestry: Hints on the evolution of geminiviruses in the New World. Viruses 2019, 11, 644. [Google Scholar] [CrossRef] [PubMed]
- Kamal, H.; Zafar, M.M.; Razzaq, A.; Parvaiz, A.; Ercisli, S.; Qiao, F.; Jiang, X. Functional role of geminivirus encoded proteins in the host: Past and Present. Biotechnol. J. 2024, 19, 2300736. [Google Scholar] [CrossRef] [PubMed]
- Ascencio-Ibáñez, J.T.; Bobay, B.G. Conserved structural motif identified in peptides that bind to geminivirus replication protein rep. Biochemistry 2021, 60, 2795–2809. [Google Scholar] [CrossRef]
- Sun, M.; Jiang, K.; Li, C.; Du, J.; Li, M.; Ghanem, H.; Wu, G.; Qing, L. Tobacco curly shoot virus C3 protein enhances viral replication and gene expression in Nicotiana benthamiana plants. Virus Res. 2020, 281, 197939. [Google Scholar] [CrossRef] [PubMed]
- Settlage, S.B.; See, R.G.; Hanley-Bowdoin, L. Geminivirus C3 protein: Replication enhancement and protein interactions. J. Virol. 2005, 79, 9885–9895. [Google Scholar] [CrossRef]
- Medina-Puche, L.; Orílio, A.F.; Zerbini, F.M.; Lozano-Durán, R. Small but mighty: Functional landscape of the versatile geminivirus-encoded C4 protein. PLoS Pathog. 2021, 17, e1009915. [Google Scholar] [CrossRef]
- Dai, K.-W.; Tsai, Y.-T.; Wu, C.-Y.; Lai, Y.-C.; Lin, N.-S.; Hu, C.-C. Identification of crucial amino acids in begomovirus C4 proteins involved in the modulation of the severity of leaf curling symptoms. Viruses 2022, 14, 499. [Google Scholar] [CrossRef]
- Zhao, W.; Ji, Y.; Zhou, Y.; Wang, X. Geminivirus C4/AC4 proteins hijack cellular coat protein complex I for chloroplast targeting and viral infections. Plant Physiol. 2024, 196, 1826–1839. [Google Scholar] [CrossRef]
- Sattar, M.N.; Iqbal, Z.; Tahir, M.N.; Ullah, S. The prediction of a new CLCuD epidemic in the Old World. Front. Microbiol. 2017, 8, 631. [Google Scholar] [CrossRef]
- Iqbal, Z.; Masood, M.; Shafiq, M.; Briddon, R.W. Temporal changes in the levels of virus and betasatellite DNA in B. tabaci feeding on CLCuD affected cotton during the growing season. Front. Microbiol. 2024, 15, 1410568. [Google Scholar] [CrossRef] [PubMed]
- Dweep, H.; Sticht, C.; Gretz, N. In-silico algorithms for the screening of possible microRNA binding sites and their interactions. Curr. Genom. 2013, 14, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Ben Or, G.; Veksler-Lublinsky, I. Comprehensive machine-learning-based analysis of microRNA–target interactions reveals variable transferability of interaction rules across species. BMC Bioinform. 2021, 22, 264. [Google Scholar] [CrossRef] [PubMed]
- Riffo-Campos, Á.L.; Riquelme, I.; Brebi-Mieville, P. Tools for sequence-based miRNA target prediction: What to choose? Int. J. Mol. Sci. 2016, 17, 1987. [Google Scholar] [CrossRef]
- Arif, K.T.; Okolicsanyi, R.K.; Haupt, L.M.; Griffiths, L.R. A combinatorial in silico approach for microRNA-target identification: Order out of chaos. Biochimie 2021, 187, 121–130. [Google Scholar] [CrossRef]
- Akhter, Y.; Khan, J.A. Genome wide identification of cotton (Gossypium hirsutum)-encoded microRNA targets against Cotton leaf curl Burewala virus. Gene 2018, 638, 60–65. [Google Scholar]
- More, P.; Agarwal, P.; Anand, A.; Sanan-Mishra, N.; Agarwal, P.K. Artificial miRNA mediated resistance in tobacco against Jatropha leaf curl Gujarat virus by targeting RNA silencing suppressors. Sci. Rep. 2021, 11, 890. [Google Scholar] [CrossRef]
- Ali, I.; Amin, I.; Briddon, R.W.; Mansoor, S. Artificial microRNA-mediated resistance against the monopartite begomovirus cotton leaf curl Burewala virus. Virol. J. 2013, 10, 231. [Google Scholar] [CrossRef]
- Zubair, M.; Khan, M.Z.; Rauf, I.; Raza, A.; Shah, A.H.; Hassan, I.; Amin, I.; Mansoor, S. Artificial micro RNA (amiRNA)-mediated resistance against whitefly (Bemisia tabaci) targeting three genes. Crop Prot. 2020, 137, 105308. [Google Scholar] [CrossRef]
- Ashraf, F.; Ashraf, M.A.; Hu, X.; Zhang, S. A novel computational approach to the silencing of Sugarcane bacilliform Guadeloupe A virus determines potential host-derived microRNAs in sugarcane (Saccharum officinarum L.). PeerJ 2020, 8, e8359. [Google Scholar] [CrossRef]
- Ashraf, M.A.; Ashraf, F.; Feng, X.; Hu, X.; Shen, L.; Khan, J.; Zhang, S. Potential targets for evaluation of sugarcane yellow leaf virus resistance in sugarcane cultivars: In silico sugarcane miRNA and target network prediction. Biotechnol. Biotechnolo. Equip. 2021, 35, 1980–1991. [Google Scholar] [CrossRef]
- Ashraf, M.A.; Feng, X.; Hu, X.; Ashraf, F.; Shen, L.; Iqbal, M.S.; Zhang, S. In silico identification of sugarcane (Saccharum officinarum L.) genome encoded microRNAs targeting sugarcane bacilliform virus. PLoS ONE 2022, 17, e0261807. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, M.A.; Tariq, H.K.; Hu, X.-W.; Khan, J.; Zou, Z. Computational biology and machine learning approaches identify rubber tree (Hevea brasiliensis Muell. Arg.) genome encoded microRNAs targeting rubber tree virus 1. Appl. Sci. 2022, 12, 12908. [Google Scholar] [CrossRef]
- Ashraf, M.A.; Ali, B.; Brown, J.K.; Shahid, I.; Yu, N. In Silico Identification of cassava genome-encoded microRNAs with predicted potential for targeting the ICMV-Kerala begomoviral pathogen of cassava. Viruses 2023, 15, 486. [Google Scholar] [CrossRef]
- Ashraf, M.A.; Murtaza, N.; Brown, J.K.; Yu, N. In silico apple genome-encoded microRNA target binding sites targeting apple chlorotic leaf spot virus. Horticulturae 2023, 9, 808. [Google Scholar] [CrossRef]
- Wenzhi, W.; Ashraf, M.A.; Ghaffar, H.; Ijaz, Z.; Zaman, W.u.; Mazhar, H.; Zulfqar, M.; Zhang, S. In Silico Identification of sugarcane genome-encoded microRNAs targeting sugarcane mosaic virus. Microbiol. Res. 2024, 15, 273–289. [Google Scholar] [CrossRef]
- Mohamed, N.A.; Ngah, N.M.F.N.C.; Abas, A.; Talip, N.; Sarian, M.N.; Hamezah, H.S.; Harun, S.; Bunawan, H. Candidate miRNAs from Oryza sativa for silencing the rice tungro viruses. Agriculture 2023, 13, 651. [Google Scholar] [CrossRef]
- Gaafar, Y.Z.A.; Ziebell, H. Novel targets for engineering physostegia chlorotic mottle and tomato brown rugose fruit virus-resistant tomatoes: In silico prediction of tomato microRNA targets. PeerJ 2020, 8, e10096. [Google Scholar] [CrossRef]
- Iqbal, M.S.; Jabbar, B.; Sharif, M.N.; Ali, Q.; Husnain, T.; Nasir, I.A. In silico MCMV silencing concludes potential host-derived miRNAs in maize. Front. Plant Sci. 2017, 8, 372. [Google Scholar] [CrossRef]
- Shahid, M.N.; Rasheed, S.; Iqbal, M.S.; Jamal, A.; Khalid, S.; Shamim, Z. In Silico prediction of potential miRNAs to target ZYMV in Cucumis melo. Pak. J. Bot 2022, 54, 1319–1325. [Google Scholar]
- Jabbar, B.; Iqbal, M.S.; Batcho, A.A.; Nasir, I.A.; Rashid, B.; Husnain, T.; Henry, R.J. Target prediction of candidate miRNAs from Oryza sativa for silencing the RYMV genome. Comput. Biol. Chem. 2019, 83, 107127. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.; Pandey, V.; Singh, N.; Marwal, A.; Shahid, M.S.; Gaur, R.K. In silico identification of papaya genome-encoded microRNAs to target begomovirus genes in papaya leaf curl disease. Front. Microbiol. 2024, 15, 1340275. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, M.A.; Brown, J.K.; Iqbal, M.S.; Yu, N. Genome-wide identification of cotton microRNAs predicted for targeting cotton leaf curl Kokhran virus-Lucknow. Microbiol. Res. 2023, 15, 1–19. [Google Scholar] [CrossRef]
- Pandey, V.; Srivastava, A.; Ali, A.; Gupta, R.; Shahid, M.S.; Gaur, R.K. Predicting candidate miRNAs for targeting begomovirus to induce sequence-specific gene silencing in chilli plants. Front. Plant Sci. 2024, 15, 1460540. [Google Scholar] [CrossRef]
- PANDEY, V.; Srivastava, A.; Gupta, R.; Shafiq Shahid, M.; Gaur, R.K. In Silico identification of chilli genome encoded microRNAs targeting the 16S rRNA and secA genes of ‘Candidatus Phytoplasma trifolii’. Front. Bioinform. 2025, 4, 1493712. [Google Scholar] [CrossRef]
- Homberg, N.; Galvão Ferrarini, M.; Gaspin, C.; Sagot, M.-F. MicroRNA target identification: Revisiting accessibility and seed anchoring. Genes 2023, 14, 664. [Google Scholar] [CrossRef]
- Peterson, S.M.; Thompson, J.A.; Ufkin, M.L.; Sathyanarayana, P.; Liaw, L.; Congdon, C.B. Common features of microRNA target prediction tools. Front. Genet. 2014, 5, 23. [Google Scholar] [CrossRef]
- Deng, F.; Zhang, X.; Wang, W.; Yuan, R.; Shen, F. Identification of Gossypium hirsutum long non-coding RNAs (lncRNAs) under salt stress. BMC Plant Biol. 2018, 18, 23. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Dong, J.; Deng, F.; Wang, W.; Cheng, Y.; Song, L.; Hu, M.; Shen, J.; Xu, Q.; Shen, F. The long non-coding RNA lncRNA973 is involved in cotton response to salt stress. BMC Plant Biol. 2019, 19, 459. [Google Scholar] [CrossRef]
- Quillet, A.; Anouar, Y.; Lecroq, T.; Dubessy, C. Prediction methods for microRNA targets in bilaterian animals: Toward a better understanding by biologists. Comput. Struct. Biotechnol. J. 2021, 19, 5811–5825. [Google Scholar] [CrossRef]
- Oliveira, A.C.; Bovolenta, L.A.; Nachtigall, P.G.; Herkenhoff, M.E.; Lemke, N.; Pinhal, D. Combining results from distinct microRNA target prediction tools enhances the performance of analyses. Front. Genet. 2017, 8, 59. [Google Scholar] [CrossRef] [PubMed]
- Min, H.; Yoon, S. Got target?: Computational methods for microRNA target prediction and their extension. Exp. Mol. Med. 2010, 42, 233–244. [Google Scholar] [CrossRef]
- Tsatsakis, A.M.; Nawaz, M.A.; Kouretas, D.; Balias, G.; Savolainen, K.; Tutelyan, V.A.; Golokhvast, K.S.; Lee, J.D.; Yang, S.H.; Chung, G. Environmental impacts of genetically modified plants: A review. Environ. Res. 2017, 156, 818–833. [Google Scholar] [CrossRef] [PubMed]
- Bauer-Panskus, A.; Miyazaki, J.; Kawall, K.; Then, C. Risk assessment of genetically engineered plants that can persist and propagate in the environment. Environ. Sci. Eur. 2020, 32, 32. [Google Scholar] [CrossRef]
- Hokanson, K.E.; Ellstrand, N.C.; Dixon, A.G.; Kulembeka, H.P.; Olsen, K.M.; Raybould, A. Risk assessment of gene flow from genetically engineered virus resistant cassava to wild relatives in Africa: An expert panel report. Transgenic Res. 2016, 25, 71–81. [Google Scholar] [CrossRef]
- Tepfer, M.; Jacquemond, M.; García-Arenal, F. A critical evaluation of whether recombination in virus-resistant transgenic plants will lead to the emergence of novel viral diseases. New Phytol. 2015, 207, 536–541. [Google Scholar] [CrossRef]
- Ossowski, S.; Schwab, R.; Weigel, D. Gene silencing in plants using artificial microRNAs and other small RNAs. Plant J. 2008, 53, 674–690. [Google Scholar] [CrossRef] [PubMed]
- Carbonell, A.; Fahlgren, N.; Mitchell, S.; Cox, K.L., Jr.; Reilly, K.C.; Mockler, T.C.; Carrington, J.C. Highly specific gene silencing in a monocot species by artificial microRNAs derived from chimeric miRNA precursors. Plant J. 2015, 82, 1061–1075. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Protein | RNA22 (Sites) | psRNATarget (miRNAs) | RNAhybrid (miRNAs) | TAPIR (miRNAs) |
|---|---|---|---|---|---|
| V1 | Coat | 3 | 8 | 12 | 4 |
| V2 | Pre-Coat | 0 | 1 | 5 | 0 |
| C1 | Rep | 5 | 4 | 18 | 2 |
| C3 | REn | 5 | 3 | 14 | 4 |
| C4 | C4 | 5 | 5 | 19 | 6 |
| LIR | - | 0 | 2 | 10 | 0 |
| Cotton miRNA | Site/ORF RNA22 | Site/ORF psRNATarget | Site/ORF RNAhybrid | Site/ORF TAPIR | MFE * RNA22 | Expectation psRNATarget | MFE ** RNAhybrid | MFE Ratio TAPIR |
|---|---|---|---|---|---|---|---|---|
| ghr-miR156 (a, b, d) | 2203 (C1/C4) | 2202 (C1/C4) | −23.50 | 0.51 | ||||
| ghr-miR169a | 692 (V1) | 2190 (C1/C4) | 2191 (C1/C4) | −16.70 | −28.90 | 0.69 | ||
| ghr-miR169b | 692 (V1) | 2190 (C1/C4) | 2190 (C1/C4) | 2191 (C1/C4) | −15.40 | 6.00 | −31.90 | 0.64 |
| ghr-miR390 (a, b, c) | 1410 (C3) | 1410 (C3) | −26.80 | −33.40 | ||||
| ghr-miR396 (a, b) | 1249 (C3) | 1225 (C3) | 1249 (C3) | 6.00 | −22.50 | 0.58 | ||
| ghr-miR399c | 1750 (C1) | 1752 (C1) | −19.00 | −24.70 | ||||
| ghr-miR3999d | 1749 (C1) | 1749 (C1) | 1747 (C1) | 1749 (C1) | −16.30 | 6.50 | −22.50 | 0.49 |
| ghr-miR399e | 1747 (C1) | 1749 (C1) | 1747 (C1) | −17.80 | 6.50 | −23.90 | ||
| ghr-miR7486 (a, b) | 2499 (C1/C4) | 2499 (C1/C4) | 850 (V1) | −21.30 | 5.00 | −30.70 | ||
| ghr-miR7488 | 2558 (C4) | 1443 (C3) | 2558 (C4) | 5.50 | −24.80 | 0.42 | ||
| ghr-miR7493 | 1163 (C3) | 1351 (C3) | 1163 (C3) | 6.00 | −22.20 | 0.52 | ||
| ghr-miR7512 | 918 (V1) | 918 (V1) | −16.70 | −23.50 |
| MicroRNAs | RNA22 | psRNATarget | RNAhybrid | TAPIR |
|---|---|---|---|---|
| Folding Energy (p-Value) | Expectation | Minimum Free Energy | MFE Ratio | |
| ghr-miR169b | 6.00 | −31.90 | 0.64 | |
| ghr-miR399d | −16.30 (0.319) | 6.50 | −22.50 | 0.49 |
| ghr-miR399e | −17.80 (0.319) | 6.50 | −23.90 | - |
| miRNA ID | Accession ID | Mature Sequence (5′–3′) | Target ORF(s) | Genomic Target (nt) | Mode of Inhibition |
|---|---|---|---|---|---|
| ghr-miR169b | MIMAT0029157 | CAGCCAAGGAUGAUUUGCCGG | C1/C4 | 2190–2212 | Cleavage |
| ghr-miR399d | MIMAT0014350 | UGCCAAAGGAGAUUUGCCCUG | C1 | 1747–1769 | Cleavage |
| ghr-miR399e | MIMAT0025840 | UGCCAAAGGAGAUUUGCCCCG | C1 | 1747–1767 | Cleavage |
| miRNA ID | miRNA–mRNA Sequence (5′–3′) | ΔG Duplex (Kcal/mol) | ΔG Binding (Kcal/mol) |
|---|---|---|---|
| ghr-miR169b | 5′ CAGCCAAGGAUGAUUUGCCGG 3′ 5′ GCGGCGTAAGCGTCGTTGGCTGT 3′ | −27.00 | −19.15 |
| ghr-miR399d | 5′ UGCCAAAGGAGAUUUGCCCUG 3′ 5′ TGGACTGCCAGTCTCTTTGGGCC 3′ | −18.20 | −12.81 |
| ghr-miR399e | 5′ UGCCAAAGGAGAUUUGCCCCG 3′ 5′ TGGACTGCCAGTCTCTTTGGGCC 3′ | −19.40 | −14.88 |
| miRNA ID | Accession ID | Length Precursor | MFE */Kcal/mol | AMFE ** | MFEI *** | (G + C)% |
|---|---|---|---|---|---|---|
| ghr-MIR169b | MI0024199 | 210 nt | −61.80 | −29.42 | −0.817 | 36.00 |
| ghr-MIR399d | MI0013557 | 98 nt | −47.00 | −47.95 | −1.169 | 41.00 |
| ghr-MIR399e | MI0022547 | 157 nt | −69.10 | −44.01 | −0.880 | 50.00 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ashraf, M.A.; Shahid, I.; Brown, J.K.; Yu, N. An Integrative Computational Approach for Identifying Cotton Host Plant MicroRNAs with Potential to Abate CLCuKoV-Bur Infection. Viruses 2025, 17, 399. https://doi.org/10.3390/v17030399
Ashraf MA, Shahid I, Brown JK, Yu N. An Integrative Computational Approach for Identifying Cotton Host Plant MicroRNAs with Potential to Abate CLCuKoV-Bur Infection. Viruses. 2025; 17(3):399. https://doi.org/10.3390/v17030399
Chicago/Turabian StyleAshraf, Muhammad Aleem, Imran Shahid, Judith K. Brown, and Naitong Yu. 2025. "An Integrative Computational Approach for Identifying Cotton Host Plant MicroRNAs with Potential to Abate CLCuKoV-Bur Infection" Viruses 17, no. 3: 399. https://doi.org/10.3390/v17030399
APA StyleAshraf, M. A., Shahid, I., Brown, J. K., & Yu, N. (2025). An Integrative Computational Approach for Identifying Cotton Host Plant MicroRNAs with Potential to Abate CLCuKoV-Bur Infection. Viruses, 17(3), 399. https://doi.org/10.3390/v17030399