

Evolutionary Studies on the Coxsackievirus A-24 Variants Causing Acute Hemorrhagic Conjunctivitis with Emphasis on the Recent Outbreak of 2023 in India

 , , , , ,
, , , , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and Identification of EV
2.2. Virus Isolation
2.3. Confirmation of Isolates by Real-Time q-Reverse Transcriptase PCR (qRT-PCR)
2.4. Whole-Genome Sequencing Using Next-Generation Sequencing (NGS)
2.5. Genome Assembly and Annotation
2.6. Sequence Data Collection
2.7. Sequences, Phylogenetic and Phylodynamic Analysis
2.8. Mutation Analysis Based on Whole-Genome Sequencing
3. Results
3.1. Molecular Characterization of Enterovirus-Positive Samples
3.2. Isolation of CV-A24v
3.3. Confirmation of Isolates by q-PCR
3.4. Whole-Genome Sequencing by NGS
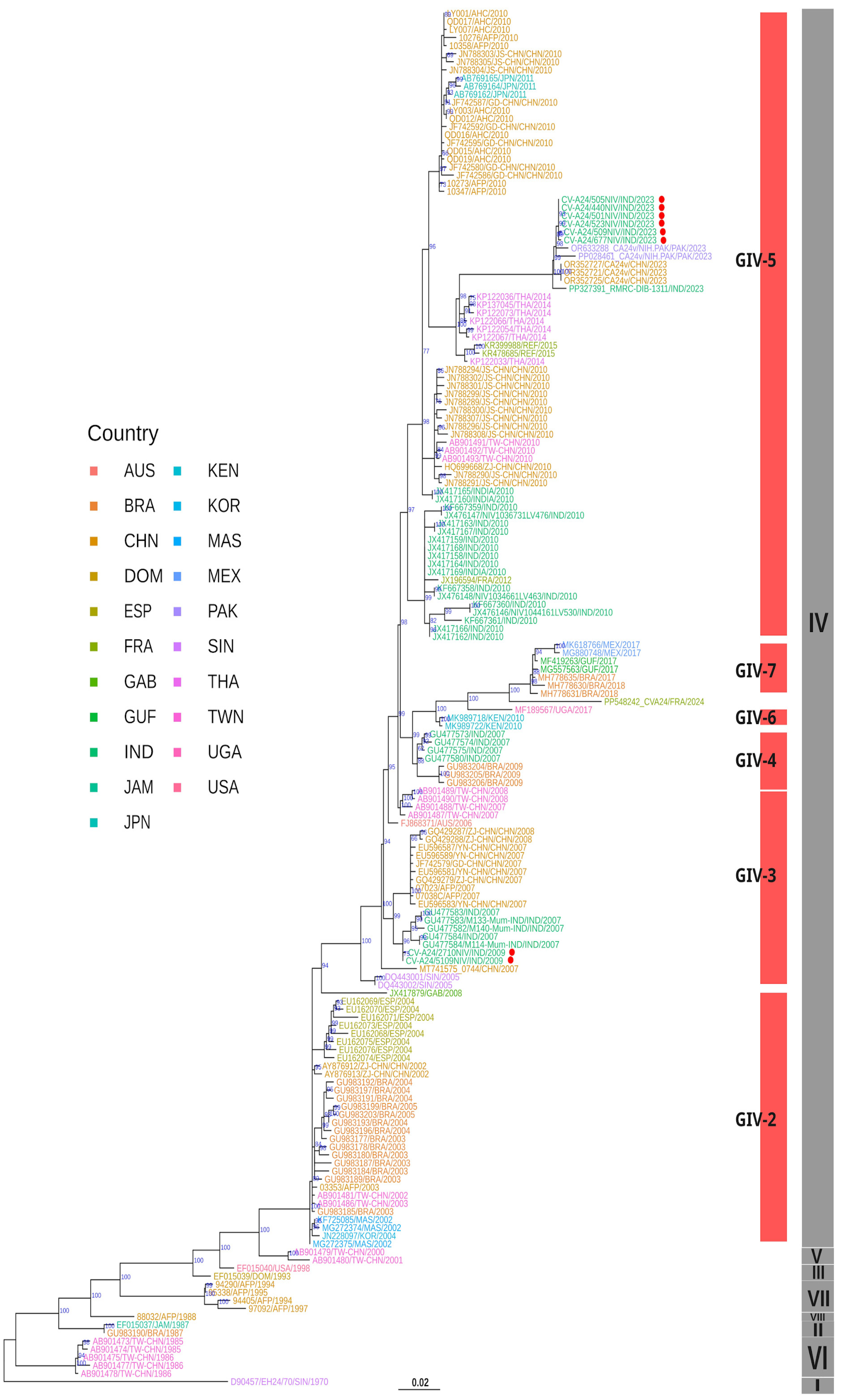
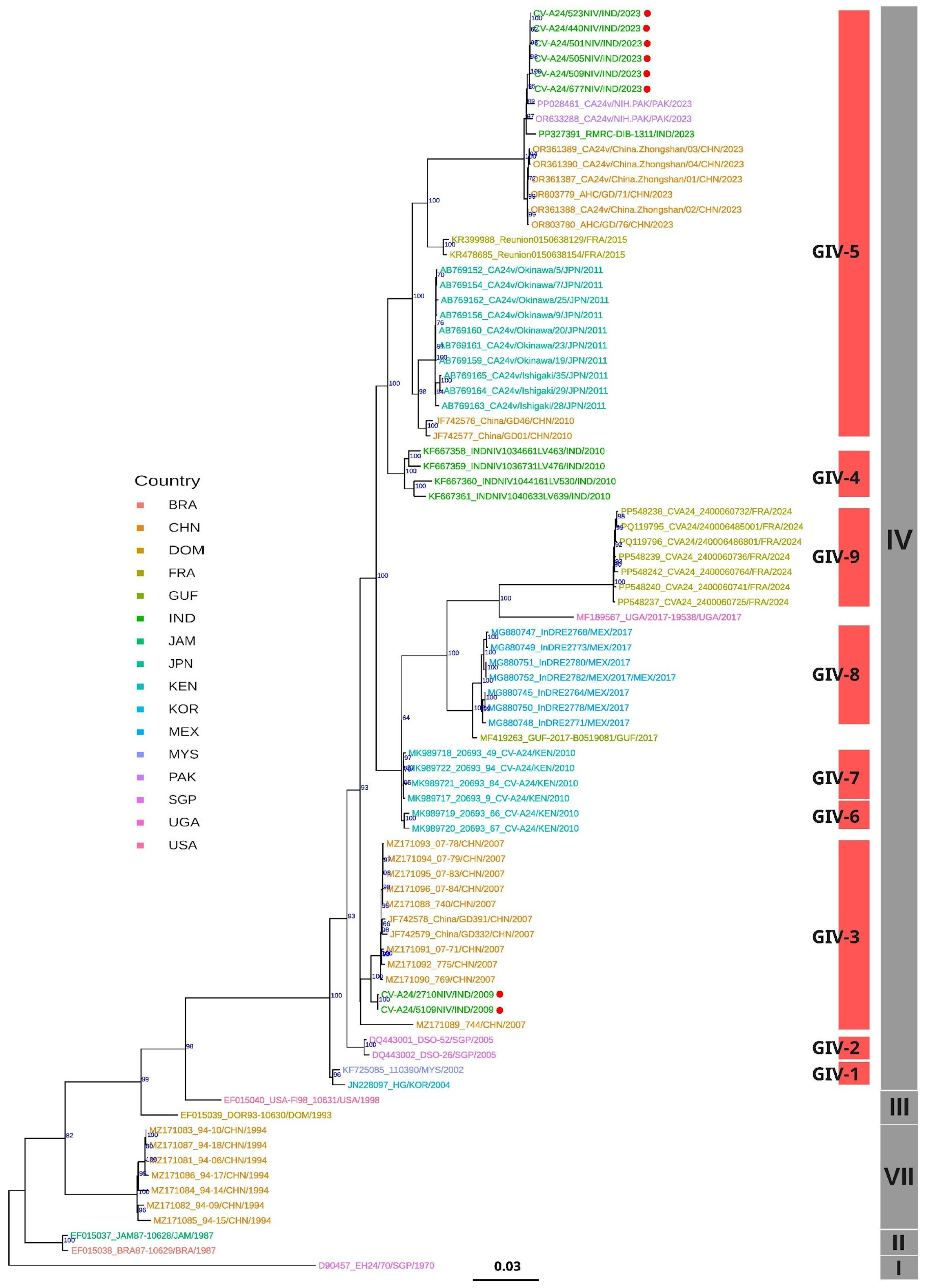
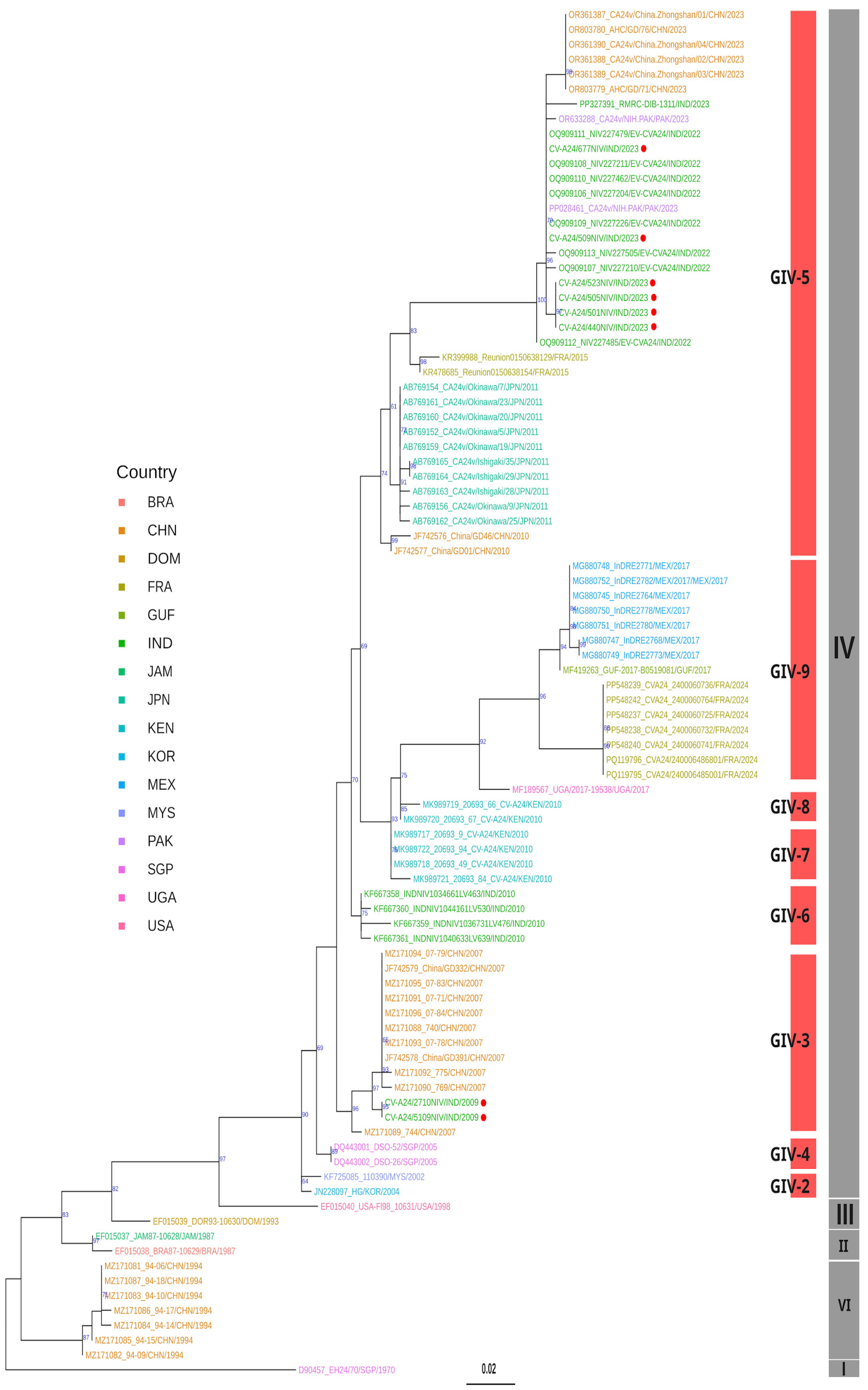
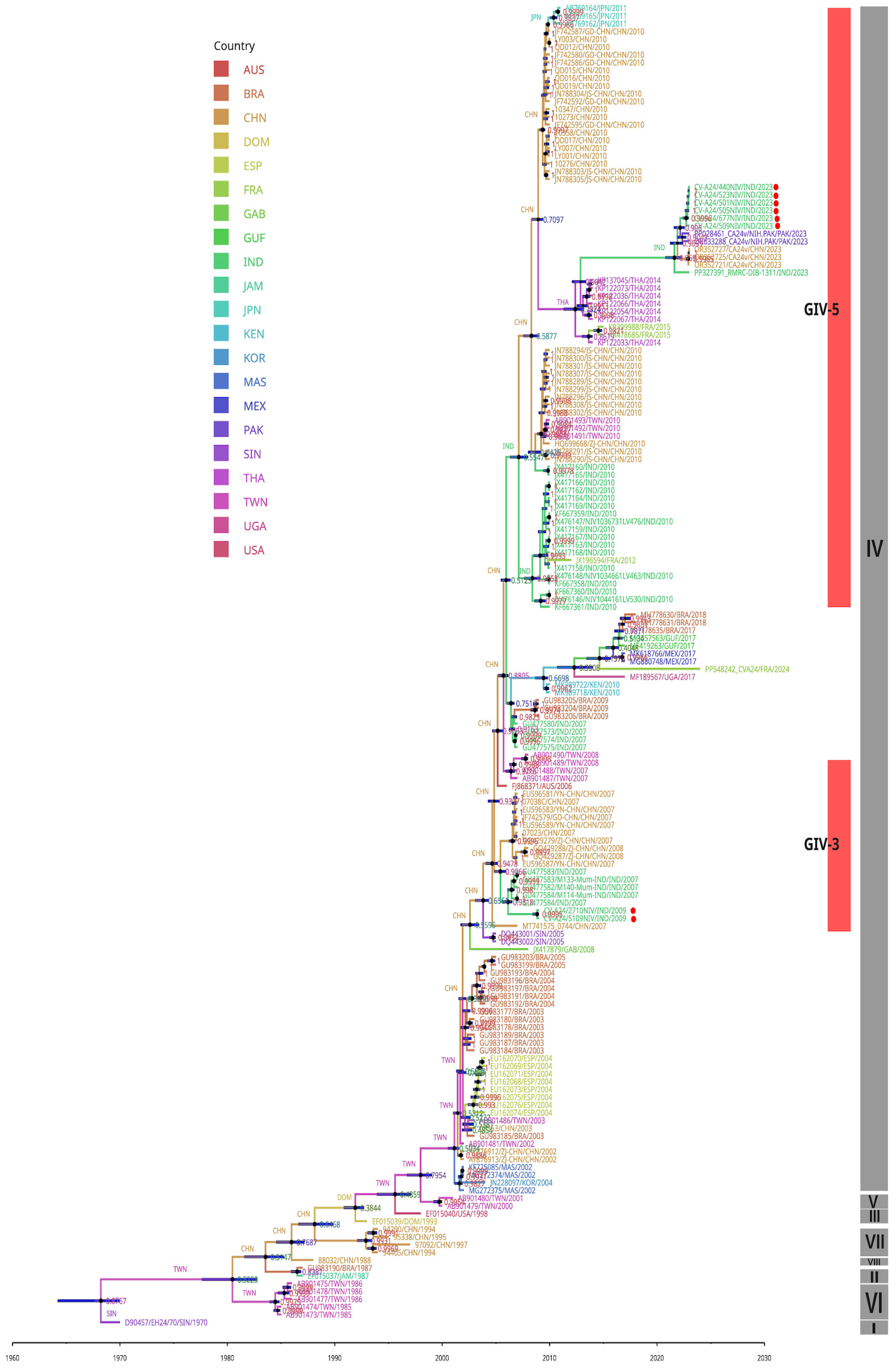
3.5. Phylogenetic Analysis
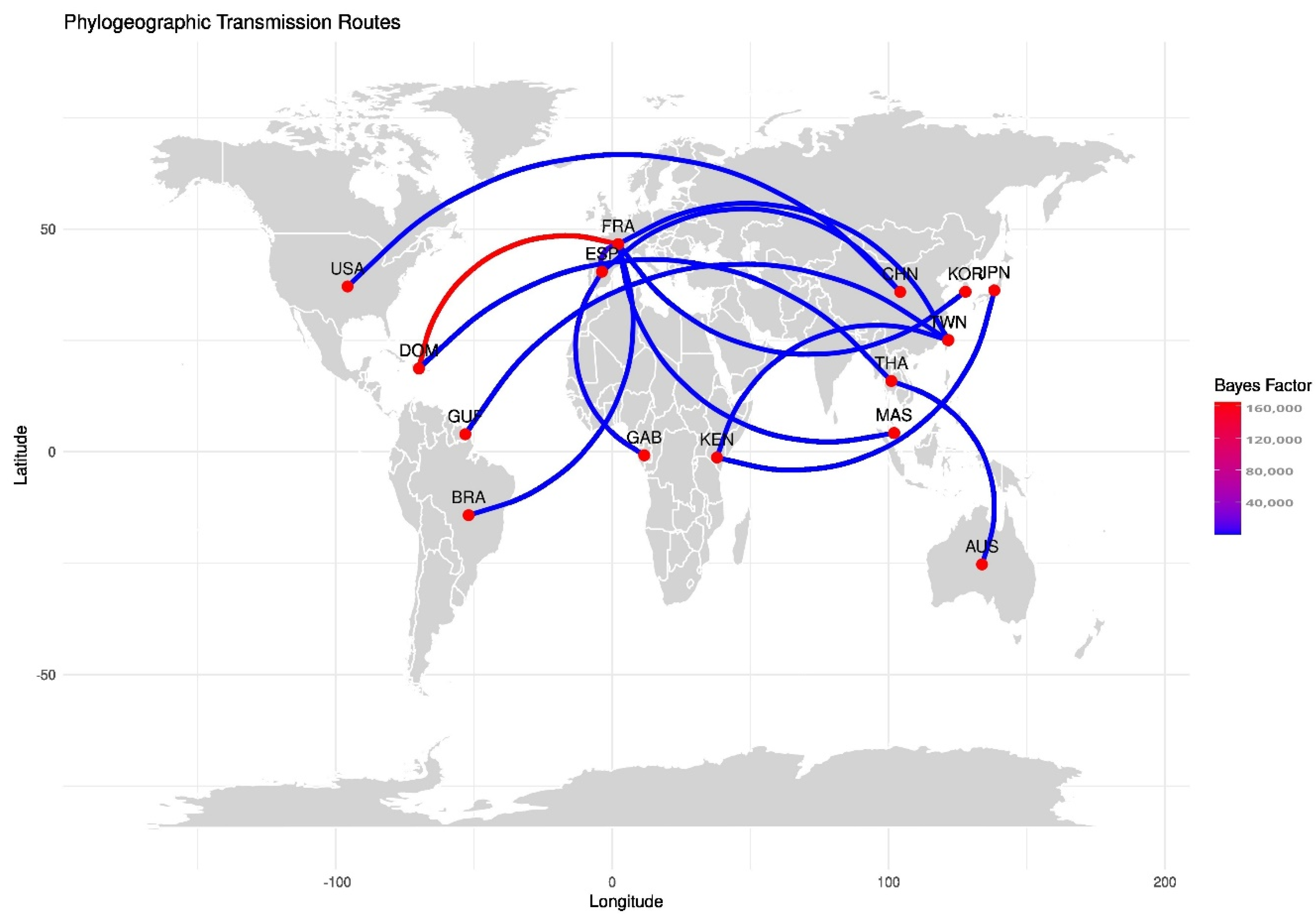
3.6. Molecular Clock and Phylogeography Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abdelkhalek, I.; Seghier, M.; Yahia, A.B.; Touzi, H.; Meddeb, Z.; Triki, H.; Rezig, D. Molecular epidemiology of coxsackievirus type B1. Arch. Virol. 2015, 160, 2815–2821. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Aoki, K.; Kitaichi, N.; Ohno, S. Acute hemorrhagic conjunctivitis. Jpn. J. Clin. Ophthalmol. 2010, 64, 18–22. [Google Scholar]
- Aubry, C.; Gautret, P.; Nougairede, A.; Dussouil, A.S.; Botelho-Nevers, E.; Zandotti, C.; de Lamballerie, X.; Brouqui, P.; Parola, P.; Parola, P. 2012 outbreak of acute haemorrhagic conjunctivitis in Indian Ocean Islands: Identification of Coxsackievirus A24 in a returned traveller. Eurosurveillance 2012, 17, 20185. [Google Scholar] [CrossRef] [PubMed]
- Azari, A.A.; Barney, N.P. Conjunctivitis: A systematic review of diagnosis and treatment. JAMA 2013, 310, 1721–1729. [Google Scholar] [CrossRef]
- Balaban, M.; Moshiri, N.; Mai, U.; Jia, X.; Mirarab, S. TreeCluster: Clustering biological sequences using phylogenetic trees. PLoS ONE 2019, 14, e0221068. [Google Scholar] [CrossRef]
- Bielejec, F.; Baele, G.; Vrancken, B.; Suchard, M.A.; Rambaut, A.; Lemey, P. SpreaD3: Interactive Visualization of Spatiotemporal History and Trait Evolutionary Processes. Mol. Biol. Evol. 2016, 33, 2167–2169. [Google Scholar] [CrossRef] [PubMed]
- Boro, P.; Gongo, T.; Ori, K.; Kamki, Y.; Ete, N.; Jini, M.; Borkakoty, B. An outbreak of acute hemorrhagic conjunctivitis due to Coxsackievirus A24 in a residential school, Naharlagun, Arunachal Pradesh: July 2023. Indian J. Med. Microbiol. 2024, 48, 100549. [Google Scholar] [CrossRef] [PubMed]
- Broor, S.; Kishore, J.; Dogra, V.; Satapathy, G.; Seth, P. An epidemic of acute hemorrhagic conjunctivitis caused by coxsackie A24 variant. Indian J. Med. Res. 1992, 95, 253–255. [Google Scholar] [PubMed]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Chansaenroj, J.; Vongpunsawad, S.; Puenpa, J.; Theamboonlers, A.; Vuthitanachot, V.; Chattakul, P.; Areechokchai, D.; Poovorawan, Y. Epidemic outbreak of acute haemorrhagic conjunctivitis caused by coxsackievirus A24 in Thailand, 2014. Epidemiol. Infect. 2015, 143, 3087–3093. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Quarcoopome, C.O.; Apenteng, A. Unusual type of epidemic conjunctivitis in Ghana. Br. J. Opthal. 1970, 54, 628. [Google Scholar] [CrossRef]
- Chen, M.; He, S.; Yan, Q.; Xu, X.; Wu, W.; Ge, S.; Zhang, S.; Chen, M.; Xia, N. Severe hand, foot and mouth disease associated with Coxsackievirus A10 infections in Xiamen, China in 2015. J. Clin. Virol. 2017, 93, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Lin, X.-J.; Ji, F.; Li, Y.; Wang, S.-T.; Liu, Y.; Tao, Z.-X.; Xu, A.-Q. Evolutionary phylogeography reveals novel genotypes of coxsackievirus A24 variant and updates the spatiotemporal dynamics in the population with acute hemorrhagic conjunctivitis. Int. J. Infect. Dis. 2022, 124, 227–239. [Google Scholar] [CrossRef]
- Fragoso-Fonseca, D.E.; Escobar-Escamilla, N.; Rodríguez-Maldonado, A.P.; Barrera-Badillo, G.; Garcés-Ayala, F.; Mendieta-Condado, E.; González-Durán, E.; Puerto, F.I.; Hernández-Rivas, L.; López-Martínez, I.; et al. Complete genome sequence of a coxsackievirus type A24 variant causing an outbreak of acute haemorrhagic conjunctivitis in southeastern Mexico in 2017. Arch. Virol. 2020, 165, 1015–1018. [Google Scholar] [CrossRef] [PubMed]
- Gopalkrishna, V.; Patil, P.R.; Kolhapure, R.M.; Bilaiya, H.; Fulmali, P.V.; Deolankar, R.P. Outbreak of acute hemorrhagic conjunctivitis in Maharashtra and Gujarat states of India, caused by coxsackie virus A-24 variant. J. Med. Virol. 2007, 79, 748–753. [Google Scholar] [CrossRef]
- Haider, A.S.; Jamal, Z.; Ammar, M.; Hakim, R.; Salman, M.; Umair, M. Genomic Insights into the 2023 Outbreak of Acute Hemorrhagic Conjunctivitis in Pakistan: Identification of Coxsackievirus A24 Variant through Next Generation Sequencing. medRxiv 2023. [Google Scholar] [CrossRef]
- Hashmi, M.F.; Gurnani, B.; Benson, S.; Price, K.L. Conjunctivitis (Nursing). In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Henschel, R.; Lieber, M.; Wu, L.-S.; Nista, P.M.; Haas, B.J.; LeDuc, R.D. Trinity RNA-Seq assembler performance optimization. In Proceedings of the 1st Conference of the Extreme Science and Engineering Discovery Environment: Bridging from the eXtreme to the Campus and Beyond, Chicago, IL, USA, 16–20 July 2012; Association for Computing Machinery: New York, NY, USA, 2012; pp. 1–8. [Google Scholar]
- Higgins, P.G.; Chapman, T.E.D. Coxsackie-virus A24 and acute haemorrhagic conjunctivitis in Sri Lanka. Lancet 1977, 309, 361. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT: Iterative Refinement and Additional Methods. Methods Mol. Biol. 2014, 1079, 131–146. [Google Scholar] [PubMed]
- Kyriakopoulou, Z.; Bletsa, M.; Tsakogiannis, D.; Dimitriou, T.G.; Amoutzias, G.D.; Gartzonika, C.; Levidiotou-Stefanou, S.; Markoulatos, P. Molecular epidemiology and evolutionary dynamics of Echovirus 3 serotype. Infect. Genet. Evol. 2015, 32, 305–312. [Google Scholar] [CrossRef]
- Langford, M.P.; Anders, E.A.; Burch, M.A. Acute hemorrhagic conjunctivitis: Anti-coxsackievirus a24 variant secretory immunoglobulin a in acute and convalescent tear. Clin. Ophthalmol. 2015, 9, 1665. [Google Scholar] [CrossRef]
- Laxmivandana, R.; Yergolkar, P.; Rajeshwari, M.; Chitambar, S.D. Genomic characterization of coxsackievirus type A24 strains associated with acute flaccid paralysis and rarely identified Hopkins syndrome. Arch. Virol. 2014, 159, 3125–3129. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.H.; Yin-Murphy, M. An epidemic of conjunctivitis in Singapore in 1970. Singap. Med. J. 1971, 12, 245–247. [Google Scholar]
- Machado, R.S., Jr.; De Sousa, I.P.; Monteiro, J.C.; Ferreira, J.L.; Santos, A.J.C.; Tavares, F.N. Detection and identification of enteroviruses circulating in children with acute gastroenteritis in Para State, Northern Brazil (2010–2011). Virol. J. 2020, 17, 156. [Google Scholar] [CrossRef]
- Mahmmod, A.R.; Narang, A.T. Diagnosis and Management of Acute Red Eye. Emerg. Med. Clin. Am. 2008, 26, 35–55. [Google Scholar] [CrossRef] [PubMed]
- McWilliam Leitch, E.C.; Cabrerizo, M.; Cardosa, J.; Harvala, H.; Ivanova, O.E.; Koike, S.; Kroes, A.C.; Lukashev, A.; Perera, D.; Roivainen, M.; et al. The association of recombination events in the founding and emergence of subgenogroup evolutionary lineages of human enterovirus 71. J. Virol. 2012, 86, 2676–2685. [Google Scholar] [CrossRef] [PubMed]
- Mirkovic, R.R.; Schmidt, N.J.; Yin-Murphy, M.; Melnick, J.L. Enterovirus etiology of the 1970 Singapore epidemic of acute conjunctivitis. Intervirology 1974, 4, 119–127. [Google Scholar] [CrossRef]
- Miyamura, K.; Yamashita, K.; Takeda, N.; Ogino, T.; Utagawa, E.; Yamazaki, S.; Shinjo, N. The first epidemic of acute hemorrhagic conjunctivitis due to a coxsackievirus A-24 variant in Okinawa, Japan, in 1985–1986. Jpn. J. Med. Sci. Biol. 1988, 41, 159–174. [Google Scholar] [CrossRef] [PubMed]
- Muto, T.; Imaizumi, S.; Kamoi, K. Viral conjunctivitis. Viruses 2023, 15, 676. [Google Scholar] [CrossRef]
- Nayak, N.; Gupta, S.K.; Murthy, G.V.S.; Satpathy, G.; Mohanty, S. Community-based investigation of an outbreak of acute viral conjunctivitis in urban slums. Trop. Med. Int. Health 1996, 1, 667–671. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Ponnuraj, E.M.; Mukundan, P. Coxsackie virus A 24 variant as the etiological agent of the acute haemorrhagic conjunctivitis epidemic at Vellore, in 1986. Indian J. Med. Res. 1989, 89, 283–286. [Google Scholar] [PubMed]
- Sawyer, L.A.; Hershow, R.C.; Pallansch, M.A.; Fishbein, D.B.; Pinsky, P.F.; Broerman, S.F.; Grimm, B.B.; Anderson, L.J.; Hall, D.B.; Schonberger’ Sawyer, L.B.; et al. An epidemic of Acute Hemorrhagic Conjunctivitis in American Samoa caused by Coxsackievirus A24 variant. Am. J. Epidemiol. 2018, 130, 1187–1198. [Google Scholar] [CrossRef]
- Shean, R.C.; Makhsous, N.; Stoddard, G.D.; Lin, M.J.; Greninger, A.L. VAPiD: A lightweight cross-platform viral annotation pipeline and identification tool to facilitate virus genome submissions to NCBI GenBank. BMC Bioinform. 2019, 20, 48. [Google Scholar] [CrossRef] [PubMed]
- Shukla, D.; Kumar, A.; Srivastava, S.; Dhole, T.N. Molecular identification and phylogenetic study of coxsackievirus A24 variant isolated from an outbreak of acute hemorrhagic conjunctivitis in India in 2010. Arch. Virol. 2013, 158, 679–684. [Google Scholar] [CrossRef]
- Supanaranond, K.; Takeda, N.; Yamazaki, S. The complete nucleotide sequence of a variant of Coxsackievirus A24, an agent causing acute hemorrhagic conjunctivitis. Virus Genes 1992, 6, 149–158. [Google Scholar] [CrossRef]
- Tao, Z.; Wang, H.; Li, Y.; Xu, A.; Zhang, Y.; Song, L.; Yoshida, H.; Xu, Q.; Yang, J.; Zhang, Y.; et al. Cocirculation of two transmission lineages of echovirus 6 in Jinan, China, as revealed by environmental surveillance and sequence analysis. Appl. Environ. Microbiol. 2011, 77, 3786–3792. [Google Scholar] [CrossRef]
- Tavares, F.N.; Campos, R.D.M.; Burlandy, F.M.; Fontella, R.; Melo, M.M.M.D.; Da Costa, E.V.; Da Silva, E.E. Molecular characterization and phylogenetic study of coxsackievirus A24v causing outbreaks of acute hemorrhagic conjunctivitis (AHC) in Brazil. PLoS ONE 2011, 16, e23206. [Google Scholar] [CrossRef] [PubMed]
- Sanjaykumar, T.; Pratik, D.; Nutan, C.; Anita, S.; Pooja, S.; Pragya, Y.; Mallika, L. Emergence of recombinant subclades D3/Y in coxsackievirus A-6 strains in Hand, Foot and Mouth Disease (HFMD) outbreak in India, 2022. Microorganisms 2024, 12, 490. [Google Scholar] [CrossRef] [PubMed]
- Yen, Y.C.; Chu, P.H.; Lu, P.L.; Lin, Y.C.; Shi, Y.Y.; Chou, L.C.; Wang, C.-F.; Lin, Y.-Y.; Su, H.-J.; Lin, C.-C.; et al. Phylodynamic Characterization of an Ocular-Tropism Coxsackievirus A24 Variant. PLoS ONE 2016, 11, e0160672. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T.-Y. ggtree: An r package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Well No. | Fluorescent Dye | Target | Sample ID | Ct Values of Clinical Specimens | CT Values of Isolates |
|---|---|---|---|---|---|
| E06 | FAM | PAN EV | NC | - | - |
| * C06 | FAM | PAN EV | CV-A24v/5106 | - | 14.52 |
| * D06 | FAM | PAN EV | CV-A24v/2710 | -- | 17.17 |
| B04 | FAM | PAN EV | CV-A24v/677 | 27.62 | 17.63 |
| D03 | FAM | PAN EV | CV-A24v/505 | 28.40 | 21.93 |
| E01 | FAM | PAN EV | CV-A24v/446 | 26.01 | 24.59 |
| B01 | FAM | PAN EV | CV-A24v/501 | 25.24 | 24.81 |
| D01 | FAM | PAN EV | CV-A24v/523 | 26.76 | 25.18 |
| G02 | FAM | PAN EV | CV-A24v/509 | 26.30 | 26.07 |
| F06 | FAM | PAN EV | PC | 31.08 |
| SL. No. | Sample ID | Total Reads | Avg. Coverage | Relevant Reads | Consensus Length | Genome Retrieved Percentage | Accession Number |
|---|---|---|---|---|---|---|---|
| 1 | CVA-24v/5109 | 3,087,154 | 35,242.36 | 2,067,551 | 7456 | 99.93 | PQ095934 |
| 2 | CVA-24v/2710 | 5,586,990 | 59,825.40 | 3,870,238 | 7457 | 99.95 | PQ095935 |
| 3 | CV A-24v/677 | 161,094 | 536 | 30,156 | 7211 | 96.65 | PQ095936 |
| 4 | CV A-24v/505 | 2,055,898 | 3579.94 | 193,627 | 7455 | 99.92 | PQ095937 |
| 5 | CV A-24v/440 | 1,828,954 | 374.72 | 20,146 | 7303 | 97.88 | PQ095938 |
| 6 | CV A-24v/501 | 1,760,594 | 363.29 | 19,637 | 7228 | 96.88 | PQ095939 |
| 7 | CV A-24v/523 | 2,858,114 | 539.44 | 29,495 | 7327 | 98.20 | PQ095940 |
| 8 | CV A-24v/509 | 2,902,554 | 377.45 | 20,570 | 7233 | 96.94 | PQ095941 |
| S. No. | Gene/ Region | CV-A24 Isolates | Nucleotide Substitution | Amino Acid Substitution Position | Amino Acid Substitution | Substitution Type | |
|---|---|---|---|---|---|---|---|
| 1. | VP3 | CV-A24/501NIV/IND/2023 | 1934 | T→C | 395 | M→T | Non-Synonymous |
| 2. | VP3 | CV-A24/505NIV/IND/2023 | 1940 | tca→ttg | 397 | S→L | Non-Synonymous |
| 3. | VP3 | CV-A24/677NIV/IND/2023 | 2432 | T→C | 561 | V→A | Non-Synonymous |
| 4. | VP1 | CV-A24/2710NIV/IND/2009 | 2800 | aag→gaa | 684 | K→E | Non-Synonymous |
| 5. | 2C | CV-A24/501NIV/IND/2023 | 4872 | C→G | 1374 | F→L | Non-Synonymous |
| 6. | 2C | CV-A24/501NIV/IND/2023 | 4873 | gcc→cct | 1375 | A→P | Non-Synonymous |
| 7. | 3D | CV-A24/505NIV/IND/2023 | 6056 | gtt→gcg | 1769 | V→A | Non-Synonymous |
| 8. | 3D | CV-A24/2710NIV/IND/2009 | 6333 | G→T | 1861 | E→D | Non-Synonymous |
| 9. | 3D | CV-A24/5109NIV/IND/2009 | 6333 | G→T | 1861 | E→D | Non-Synonymous |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tikute, S.; Boro, J.; Sharma, V.; Shete, A.; Ashraf, A.F.; Raju, R.M.; Cherian, S.; Lavania, M. Evolutionary Studies on the Coxsackievirus A-24 Variants Causing Acute Hemorrhagic Conjunctivitis with Emphasis on the Recent Outbreak of 2023 in India. Viruses 2025, 17, 371. https://doi.org/10.3390/v17030371
Tikute S, Boro J, Sharma V, Shete A, Ashraf AF, Raju RM, Cherian S, Lavania M. Evolutionary Studies on the Coxsackievirus A-24 Variants Causing Acute Hemorrhagic Conjunctivitis with Emphasis on the Recent Outbreak of 2023 in India. Viruses. 2025; 17(3):371. https://doi.org/10.3390/v17030371
Chicago/Turabian StyleTikute, Sanjaykumar, Jahnabee Boro, Vikas Sharma, Anita Shete, Alfia Fathima Ashraf, Ranjana Mariyam Raju, Sarah Cherian, and Mallika Lavania. 2025. "Evolutionary Studies on the Coxsackievirus A-24 Variants Causing Acute Hemorrhagic Conjunctivitis with Emphasis on the Recent Outbreak of 2023 in India" Viruses 17, no. 3: 371. https://doi.org/10.3390/v17030371
APA StyleTikute, S., Boro, J., Sharma, V., Shete, A., Ashraf, A. F., Raju, R. M., Cherian, S., & Lavania, M. (2025). Evolutionary Studies on the Coxsackievirus A-24 Variants Causing Acute Hemorrhagic Conjunctivitis with Emphasis on the Recent Outbreak of 2023 in India. Viruses, 17(3), 371. https://doi.org/10.3390/v17030371