
Impact of Viral Co-Detection on the Within-Host Viral Diversity of Influenza Patients
 , ,
, ,
Abstract
1. Introduction
2. Methods
2.1. Study Design
2.2. Sample Collection and Virus Identification
2.3. Library Preparation and Whole-Genome Sequencing
2.4. Phylogenetic Analysis
2.5. Identification of Intra-Host Single-Nucleotide Variants (iSNVs)
2.6. Statistical Analysis
3. Results
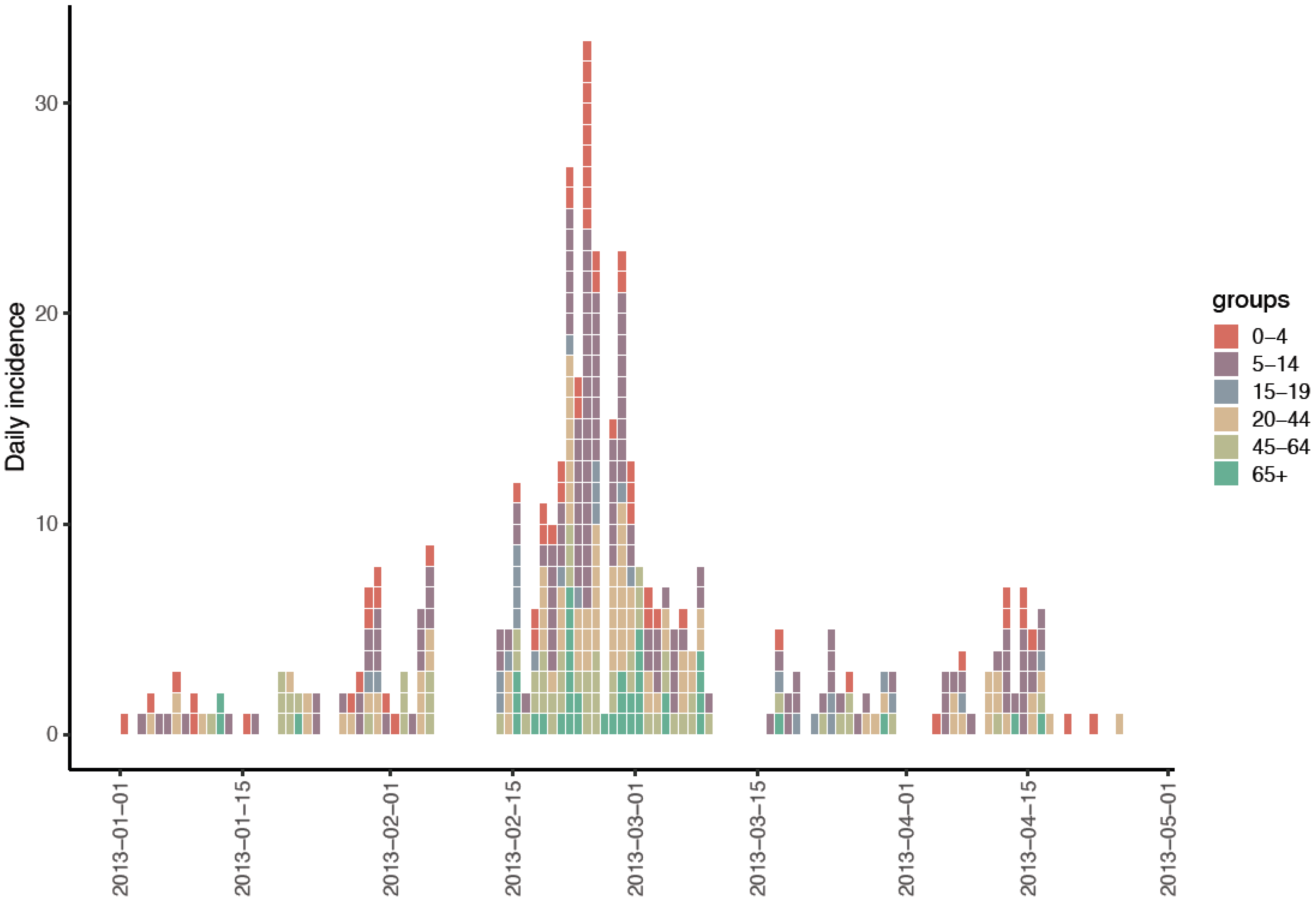
3.1. Study Cohort and ILI Incidence
3.2. Influenza and Rhinoviruses Were Most Commonly Detected and Were a Frequently Co-Detected Pair of Viruses
3.3. Whole-Genome Sequencing of RT-PCR-Confirmed Influenza A Viruses
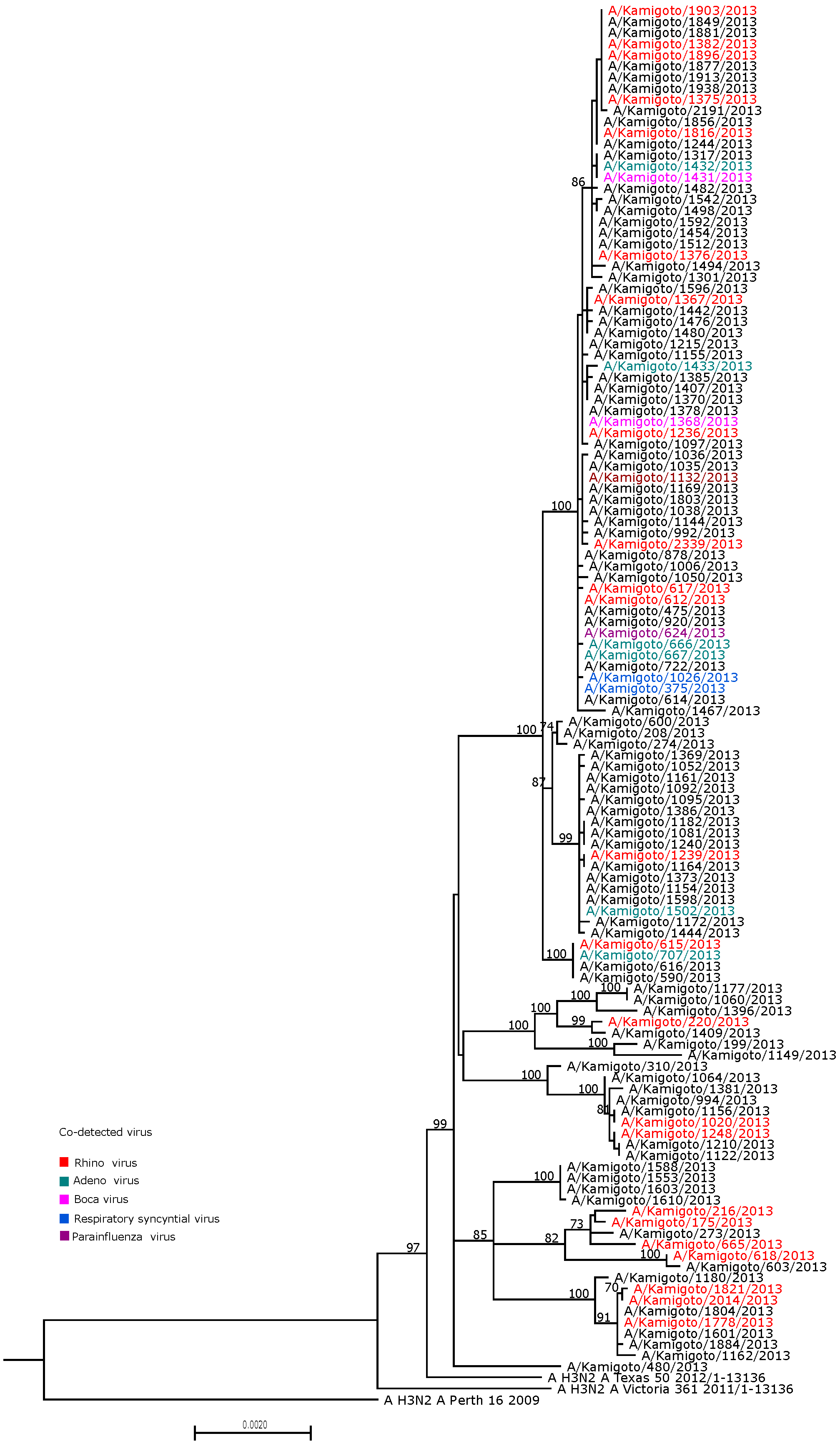
3.4. Identification of Clustering by Viral Co-Detection Status on Phylogeny
3.5. Intra-Host Diversity in Influenza-Only Cases Versus Virus Co-Detected Cases
3.6. Factors Associated with the Number of iSNVs Found in A/H3N2 WGS Samples
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Krammer, F.; Smith, G.J.D.; Fouchier, R.A.M.; Peiris, M.; Kedzierska, K.; Doherty, P.C.; Palese, P.; Shaw, M.L.; Treanor, J.; Webster, R.G.; et al. Influenza. Nat. Rev. Dis. Primers 2018, 4, 3. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- World Health Organization. FluNet Summary. 2022. Available online: https://www.who.int/tools/flunet/flunet-summary (accessed on 13 April 2023).
- Tsuzuki, S.; Baguelin, M.; Pebody, R.; van Leeuwen, E. Modelling the optimal target age group for seasonal influenza vaccination in Japan. Vaccine 2019, 38, 752–762. [Google Scholar] [CrossRef]
- Paules, C.; Subbarao, K. Influenza. Lancet 2017, 390, 697–708. [Google Scholar] [CrossRef] [PubMed]
- Lauring, A.S. Within-Host Viral Diversity: A Window into Viral Evolution. Annu. Rev. Virol. 2020, 7, 63–81. [Google Scholar] [CrossRef]
- Xue, K.S.; Moncla, L.H.; Bedford, T.; Bloom, J.D. Within-Host Evolution of Human Influenza Virus. Trends Microbiol. 2018, 26, 781–793. [Google Scholar] [CrossRef]
- Bessiere, P.; Volmer, R. From one to many: The within-host rise of viral variants. PLoS Pathog. 2021, 17, e1009811. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Reed, M.; Mattingly, J.C.; Koelle, K. The impact of host immune status on the within-host and population dynamics of antigenic immune escape. J. R. Soc. Interface 2012, 9, 2603–2613. [Google Scholar] [CrossRef] [PubMed]
- Nelson, C.W.; Hughes, A.L. Within-host nucleotide diversity of virus populations: Insights from next-generation sequencing. Infect. Genet. Evol. 2015, 30, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Debbink, K.; McCrone, J.T.; Petrie, J.G.; Truscon, R.; Johnson, E.; Mantlo, E.K.; Monto, A.S.; Lauring, A.S. Vaccination has minimal impact on the intrahost diversity of H3N2 influenza viruses. PLoS Pathog. 2017, 13, e1006194. [Google Scholar] [CrossRef] [PubMed]
- McCrone, J.T.; Woods, R.J.; Martin, E.T.; Malosh, R.E.; Monto, A.S.; Lauring, A.S. Stochastic processes constrain the within and between host evolution of influenza virus. eLife 2018, 7, e35962. [Google Scholar] [CrossRef]
- Sapoval, N.; de Mesquita, P.J.B.; Liu, Y.; Wang, R.; Liu, T.R.; Garza, J.; Williams, T.; Cadiz, C.; Tan, G.S.; Van Bakel, H.; et al. Intrahost-diversity of influenza A virus in upper and lower respiratory tract derived samples from a college community. medRxiv 2021. [Google Scholar] [CrossRef]
- Knoll, M.; Honce, R.; Meliopoulos, V.; Schultz-Cherry, S.; Ghedin, E.; Gresham, D. Host obesity impacts genetic variation in influenza A viral populations. bioRxiv 2023. [Google Scholar] [CrossRef] [PubMed]
- Destras, G.; Pichon, M.; Simon, B.; Valette, M.; Escuret, V.; Bolze, P.A.; Dubernard, G.; Gaucherand, P.; Lina, B.; Josset, L. Impact of Pregnancy on Intra-Host Genetic Diversity of Influenza A Viruses in Hospitalised Women: A Retrospective Cohort Study. J. Clin. Med. 2019, 8, 1974. [Google Scholar] [CrossRef] [PubMed]
- Nickbakhsh, S.; Mair, C.; Matthews, L.; Reeve, R.; Johnson, P.C.D.; Thorburn, F.; von Wissmann, B.; Reynolds, A.; McMenamin, J.; Gunson, R.N.; et al. Virus-virus interactions impact the population dynamics of influenza and the common cold. Proc. Natl. Acad. Sci. USA 2019, 116, 27142–27150. [Google Scholar] [CrossRef] [PubMed]
- Pinky, L.; Dobrovolny, H.M. Coinfections of the Respiratory Tract: Viral Competition for Resources. PLoS ONE 2016, 11, e0155589. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.; Mihaylova, V.T.; Landry, M.L.; Foxman, E.F. Interference between rhinovirus and influenza A virus: A clinical data analysis and experimental infection study. Lancet Microbe 2020, 1, e254–e262. [Google Scholar] [CrossRef]
- Mifsud, E.J.; Kuba, M.; Barr, I.G. Innate Immune Responses to Influenza Virus Infections in the Upper Respiratory Tract. Viruses 2021, 13, 2090. [Google Scholar] [CrossRef] [PubMed]
- Tao, K.P.; Chong, M.K.; Chan, K.Y.; Pun, J.C.; Tsun, J.G.; Chow, S.M.; Ng, C.S.; Wang, M.H.; Chan, P.K.; Li, A.M.; et al. Suppression of influenza virus infection by rhinovirus interference-at the population, individual and cellular levels. Curr. Res. Microb. Sci. 2022, 3, 100147. [Google Scholar]
- Chan, K.F.; Carolan, L.A.; Korenkov, D.; Druce, J.; McCaw, J.; Reading, P.C.; Barr, I.G.; Laurie, K.L. Investigating Viral Interference Between Influenza A Virus and Human Respiratory Syncytial Virus in a Ferret Model of Infection. J. Infect. Dis. 2018, 218, 406–417. [Google Scholar] [CrossRef] [PubMed]
- Han, S.M.; Robert, A.; Masuda, S.; Yasaka, T.; Kanda, S.; Komori, K.; Saito, N.; Suzuki, M.; Endo, A.; Baguelin, M.; et al. Transmission dynamics of seasonal influenza in a remote island population. Sci. Rep. 2023, 13, 5393. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, L.M.; Suzuki, M.; Yamamoto, T.; Nguyen, H.A.; Nguyen, C.D.; Nguyen, A.T.; Oishi, K.; Vu, T.D.; Le, T.H.; Le, M.Q.; et al. Viral pathogens associated with acute respiratory infections in central vietnamese children. Pediatr. Infect. Dis. J. 2010, 29, 75–77. [Google Scholar] [CrossRef]
- Zhou, B.; Donnelly, M.E.; Scholes, D.T.; St George, K.; Hatta, M.; Kawaoka, Y.; Wentworth, D.E. Single-reaction genomic amplification accelerates sequencing and vaccine production for classical and Swine origin human influenza a viruses. J. Virol. 2009, 83, 10309–10313. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Larsson, A. AliView: A fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 2014, 30, 3276–3278. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; Von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed]
- FigTree v1.4.4. Available online: http://tree.bio.ed.ac.uk/software (accessed on 1 January 2020).
- Borges, V.; Pinheiro, M.; Pechirra, P.; Guiomar, R.; Gomes, J.P. INSaFLU: An automated open web-based bioinformatics suite “from-reads” for influenza whole-genome-sequencing-based surveillance. Genome Med. 2018, 10, 46. [Google Scholar] [CrossRef]
- Quick, J.; Grubaugh, N.D.; Pullan, S.T.; Claro, I.M.; Smith, A.D.; Gangavarapu, K.; Oliveira, G.; Robles-Sikisaka, R.; Rogers, T.F.; Beutler, N.A.; et al. Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat. Protoc. 2017, 12, 1261–1276. [Google Scholar] [CrossRef] [PubMed]
- Kurskaya, O.; Ryabichenko, T.; Leonova, N.; Shi, W.; Bi, H.; Sharshov, K.; Kazachkova, E.; Sobolev, I.; Prokopyeva, E.; Kartseva, T.; et al. Viral etiology of acute respiratory infections in hospitalized children in Novosibirsk City, Russia (2013–2017). PLoS ONE 2018, 13, e0200117. [Google Scholar] [CrossRef]
- Nascimento-Carvalho, A.C.; Vilas-Boas, A.L.; Fontoura, M.H.; Vuorinen, T.; Nascimento-Carvalho, C.M.; PNEUMOPAC-Efficacy Study Group. Respiratory viruses among children with non-severe community-acquired pneumonia: A prospective cohort study. J. Clin. Virol. 2018, 105, 77–83. [Google Scholar] [CrossRef]
- Arden, K.E.; McErlean, P.; Nissen, M.D.; Sloots, T.P.; Mackay, I.M. Frequent detection of human rhinoviruses, paramyxoviruses, coronaviruses, and bocavirus during acute respiratory tract infections. J. Med. Virol. 2006, 78, 1232–1240. [Google Scholar] [CrossRef] [PubMed]
- Esper, F.P.; Spahlinger, T.; Zhou, L. Rate and influence of respiratory virus co-infection on pandemic (H1N1) influenza disease. J. Infect. 2011, 63, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Li, Z.; Zhang, S.; Song, S.; Julong, W.; Lin, Y.; Guo, N.; Xing, C.; Xu, A.; Bi, Z.; et al. Viral Etiology of acute respiratory tract infections in hospitalized children and adults in Shandong Province, China. Virol. J. 2015, 12, 168. [Google Scholar] [CrossRef] [PubMed]
- Han, A.X.; Felix Garza, Z.C.; Welkers, M.R.; Vigeveno, R.M.; Tran, N.D.; Le, T.Q.M.; Pham Quang, T.; Dang, D.T.; Tran, T.N.A.; Ha, M.T.; et al. Within-host evolutionary dynamics of seasonal and pandemic human influenza A viruses in young children. eLife 2021, 10, e68917. [Google Scholar] [CrossRef]
- Dinis, J.M.; Florek, N.W.; Fatola, O.O.; Moncla, L.H.; Mutschler, J.P.; Charlier, O.K.; Meece, J.K.; Belongia, E.A.; Friedrich, T.C. Deep Sequencing Reveals Potential Antigenic Variants at Low Frequencies in Influenza A Virus-Infected Humans. J. Virol. 2016, 90, 3355–3365. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | N (%) |
|---|---|
| Number of ILI episodes | 2407 |
| Age group | |
| <6 | 546 (22.7) |
| 6–18 | 707 (29.3) |
| 19–64 | 693 (28.8) |
| ≥65 | 450 (18.7) |
| Total sampled | 2305 |
| By pathogen identification | |
| Influenza A (IAV) | 538 (23.3) |
| Influenza B (IBV) | 22 (0.9) |
| Human rhinovirus (hRV) | 264 (11.5) |
| Human metapneumovirus (hMPV) | 8 (0.4) |
| Parainfluenza virus (I-IV) (PIV I-IV) | 43 (1.9) |
| Respiratory syncytial virus (RSV) | 20 (0.9) |
| Human coronavirus (OC43, 229E) (hCOV) | 14 (0.6) |
| Adenovirus (AdV) | 44 (1.9) |
| Bocavirus (BoV) | 26 (1.1) |
| By number of viruses detected | |
| Single virus infection | 777 (33.7) |
| No virus detected | 1427 (61.9) |
| Multiple infections | |
| 2 viruses | 96 (4.2) |
| >2 viruses | 5 (0.2) |
| Virus Detected | hRV | PIV | hCoV | hMPV | RSV | IBV | AdV | BoV | IAV |
|---|---|---|---|---|---|---|---|---|---|
| hRV | 264 | 5 | 0 | 1 | 0 | 6 | 8 | 4 | 48 |
| PIV | 43 | 0 | 0 | 0 | 0 | 0 | 0 | 4 | |
| hCoV | 14 | 0 | 0 | 0 | 0 | 1 | 2 | ||
| hMPV | 8 | 0 | 1 | 0 | 0 | 0 | |||
| RSV | 20 | 1 | 0 | 0 | 11 | ||||
| IBV | 22 | 0 | 0 | 4 | |||||
| AdV | 44 | 0 | 7 | ||||||
| BoV | 26 | 3 | |||||||
| IAV | 538 |
| Pair | Observed | Expected | Odd Ratios | p-Value * |
|---|---|---|---|---|
| hRV & PIV | 5 | 5 | 0.90 (0.28–2.31) | 1.000 |
| hRV & hCoV | 0 | 2 | 0.27 (0.00–2.34) | 0.391 |
| hRV & hMPV | 1 | 1 | 0.97 (0.02–7.26) | 1.000 |
| hRV & RSV | 0 | 2 | 0.19 (0.00–2.34) | 0.156 |
| hRV & IBV | 6 | 3 | 2.09 (0.69–5.38) | 0.129 |
| hRV & AdV | 8 | 5 | 1.38 (0.64–2.96) | 0.544 |
| hRV & BoV | 4 | 3 | 1.18 (0.30–3.43) | 0.772 |
| hRV & IAV | 48 | 61 | 0.11 (0.08–0.14) | <0.001 |
| PIV & Adeno | 0 | 1 | 0.59 (0.00–4.85) | 1.000 |
| PIV & IAV | 4 | 10 | 0.31 (0.11–0.86) | 0.014 |
| hCoV & BoV | 1 | 0 | 6.24 (0.14–44.04) | 0.162 |
| hCoV & IAV | 2 | 3 | 0.47 (0.05–2.05) | 0.389 |
| hMPV & IBV | 1 | 0 | 12.92 (0.28–103.77) | 0.086 |
| hMPV & IAV | 0 | 2 | 0.19 (0.00–1.93) | 0.211 |
| RSV & IBV | 1 | 0 | 5.16 (0.12–35.31) | 0.190 |
| RSV & IAV | 11 | 5 | 1.79 (0.85–3.75) | 0.136 |
| IBV & IAV | 4 | 5 | 0.59 (0.20–1.73) | 0.484 |
| AdV & IAV | 7 | 10 | 0.51 (0.23–1.14) | 0.131 |
| BoV & IAV | 3 | 6 | 0.38 (0.11–1.24) | 0.121 |
| Characteristics | Only Influenza (n = 110) | Viral Co-Detection (n = 40) |
|---|---|---|
| Age group | ||
| <6 | 8 | 5 |
| 6–18 | 41 | 12 |
| 19–65 | 52 | 18 |
| ≥65 | 6 | 4 |
| Sex | ||
| Male | 52 | 15 |
| Female | 58 | 25 |
| Vaccination status | ||
| Vaccinated | 80 | 26 |
| Unvaccinated | 29 | 14 |
| Location/Region | ||
| Narao | 2 | 2 |
| Arikawa | 38 | 13 |
| Shinuonome | 23 | 4 |
| Kamigoto | 31 | 13 |
| Wakamatsu | 14 | 51 |
| WGS (passed variant calling criteria) | 99 | 33 |
| Segment | Number of Minor iSNVs | Mean (SD) Minor iSNVs Frequency of Occurence (SD) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Flu Only (n = 99) | Flu–Other Viruses (n = 33) | Bonferroni-Adjusted p-Value | Flu Only (n = 99) | Flu–Other Viruses (n = 33) | Bonferroni-Adjusted p-Value | |||||
| Total Number | Mean (SD)/Subject | Total Number | Mean (SD)/Subject | Max Freq/Site | Mean (SD)/Site | Max Freq | Mean (SD)/Site | |||
| PB2 | 45 | 1.80 (0.99) | 17 | 1.59 (0.71) | 0.698 | 8 | 0.27 (0.68) | 3 | 0.20 (0.46) | 0.700 |
| PB1 | 38 | 1.40 (0.75) | 11 | 2.00 (1.00) | 0.192 | 3 | 0.18 (0.45) | 2 | 0.16 (0.39) | 0.980 |
| PA | 51 | 1.78 (1.56) | 18 | 1.44 (0.92) | 0.219 | 3 | 0.31 (0.55) | 1 | 0.19 (0.40) | 0.328 |
| HA | 34 | 1.59 (0.93) | 13 | 1.85 (1.57) | 0.164 | 15 | 0.18 (1.03) | 2 | 0.18 (0.40) | 0.328 |
| NP | 41 | 1.95 (1.30) | 9 | 1.44 (1.01) | 0.416 | 19 | 0.27 (1.57) | 3 | 0.10 (0.39) | 0.576 |
| NA | 34 | 1.21 (0.41) | 12 | 1.08 (0.29) | 0.545 | 14 | 0.13 (0.94) | 3 | 0.10 (0.37) | 0.663 |
| M | 19 | 1.05 (0.23) | 9 | 1.22 (0.44) | 0.244 | 11 | 0.07 (0.66) | 3 | 0.08 (0.37) | 0.531 |
| NS | 33 | 1.24 (0.50) | 11 | 1.18 (0.41) | 0.802 | 23 | 0.14 (1.35) | 3 | 0.10 (0.39) | 0.629 |
| Characteristics | N = 132 | IRR 1 (95% Confidence Interval) | Bonferroni-Adjusted p-Value |
|---|---|---|---|
| Age group | |||
| 19–64 | 63 | 1 | |
| <6 | 13 | 0.85 (0.51–1.43) | 1.000 |
| 6–18 | 46 | 1.38 (1.01–1.90) | 0.280 |
| >64 | 10 | 1.80 (1.09–3.06) | 0.173 |
| Vaccination status | |||
| Non-vaccinated | 37 | 1 | |
| Vaccinated | 94 | 0.94 (0.68–1.30) | 1.000 |
| Gender | |||
| Female | 62 | 1 | |
| Male | 70 | 0.98 (0.73–1.30) | 1.000 |
| Days post-symptoms | 0.93 (0.83–1.02) | 0.547 | |
| Viral co-detection | |||
| Only influenza | 99 | 1 | |
| Virus co-detected | 33 | 0.90 (0.65–1.26) | 1.000 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, S.M.; Kubo, Y.; Robert, A.; Baguelin, M.; Ariyoshi, K. Impact of Viral Co-Detection on the Within-Host Viral Diversity of Influenza Patients. Viruses 2025, 17, 152. https://doi.org/10.3390/v17020152
Han SM, Kubo Y, Robert A, Baguelin M, Ariyoshi K. Impact of Viral Co-Detection on the Within-Host Viral Diversity of Influenza Patients. Viruses. 2025; 17(2):152. https://doi.org/10.3390/v17020152
Chicago/Turabian StyleHan, Su Myat, Yoshiano Kubo, Alexis Robert, Marc Baguelin, and Koya Ariyoshi. 2025. "Impact of Viral Co-Detection on the Within-Host Viral Diversity of Influenza Patients" Viruses 17, no. 2: 152. https://doi.org/10.3390/v17020152
APA StyleHan, S. M., Kubo, Y., Robert, A., Baguelin, M., & Ariyoshi, K. (2025). Impact of Viral Co-Detection on the Within-Host Viral Diversity of Influenza Patients. Viruses, 17(2), 152. https://doi.org/10.3390/v17020152