
Recent Advances on Targeting Proteases for Antiviral Development

 , and
, and
Abstract
1. Introduction
1.1. Virus Replication Cycle
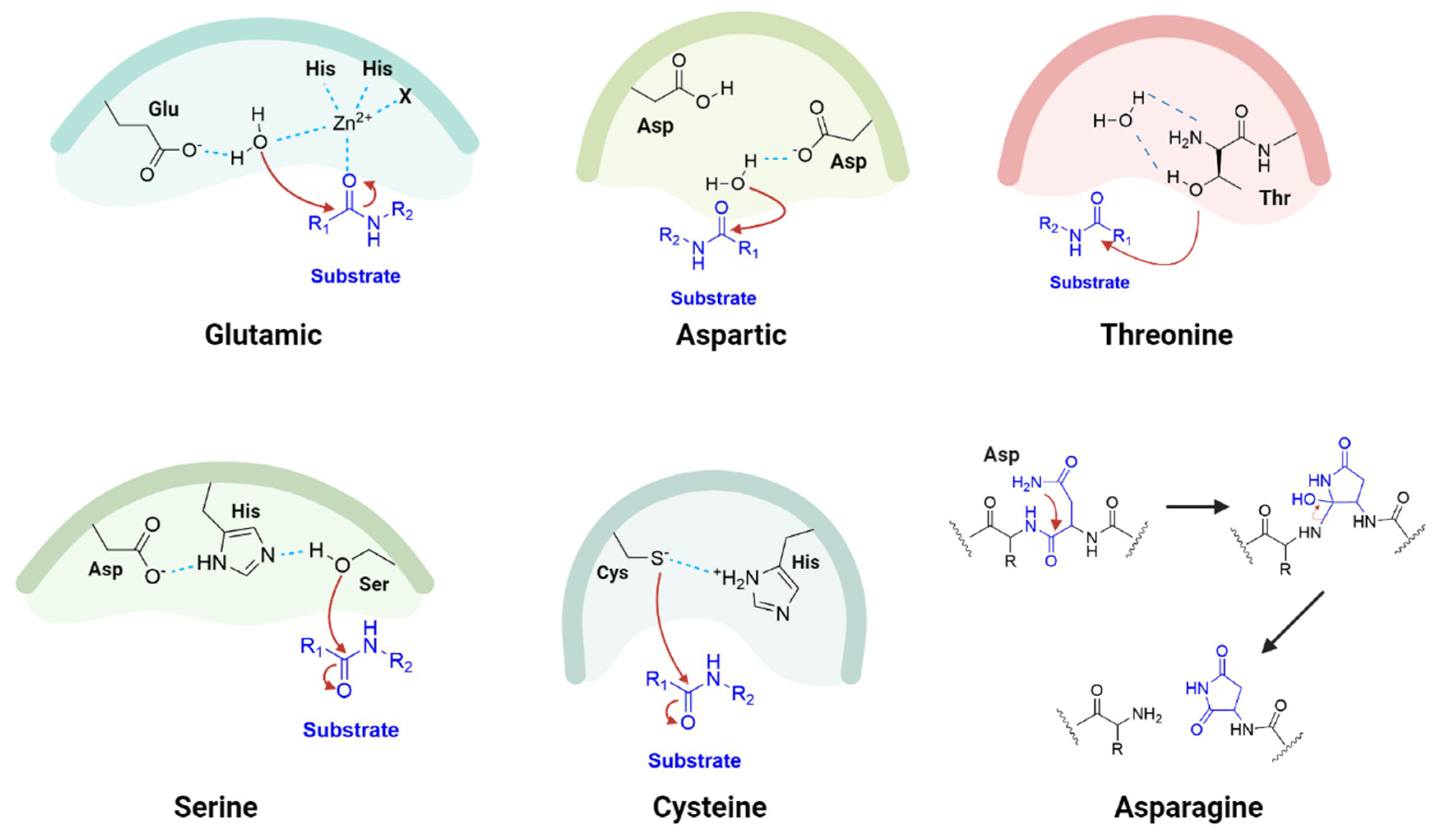
1.2. Proteases as Targets for Antivirals
2. Inhibition of Viral Proteases by Targeting the Active Site
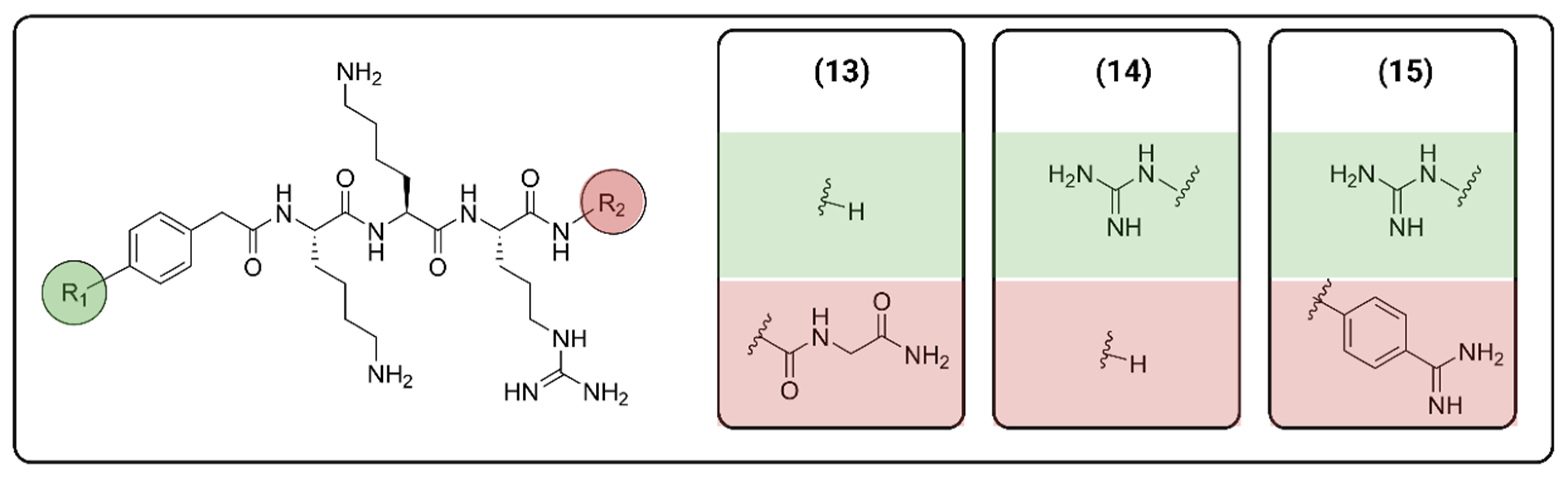
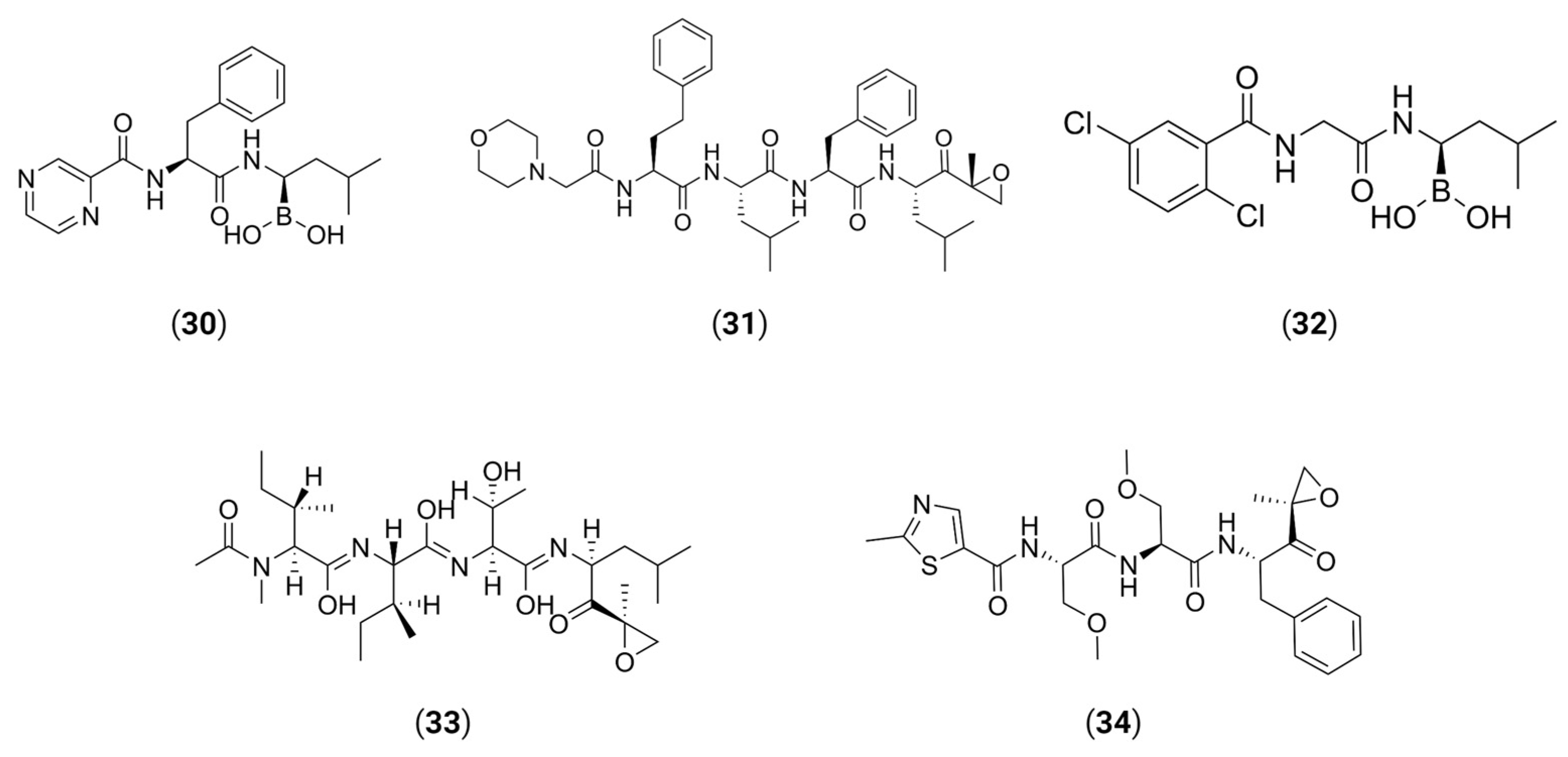
2.1. Peptide-Based Molecules
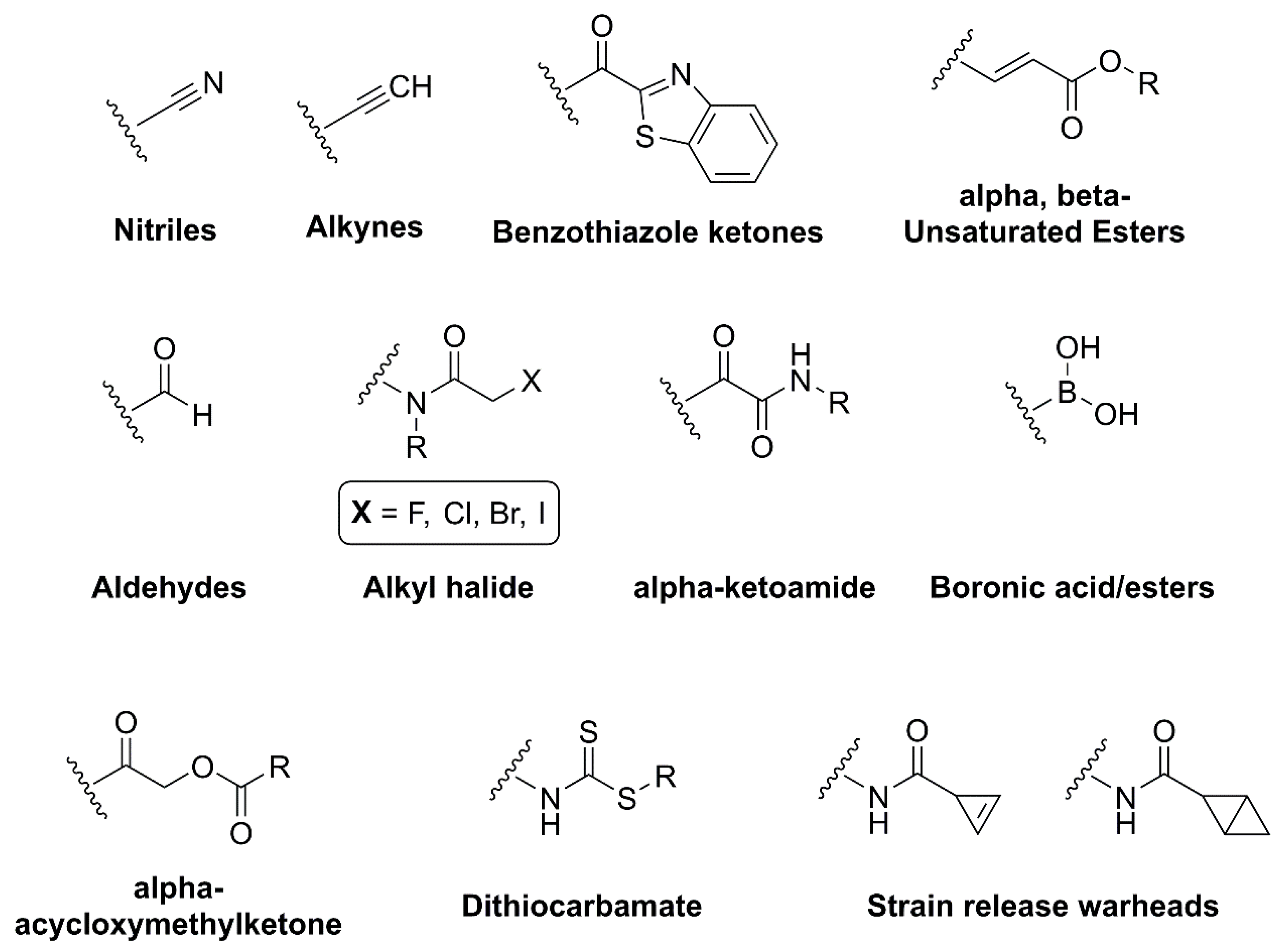
2.2. Covalent Inhibitors
2.3. In Silico-Designed Inhibitors
3. Inhibition of Viral Proteases through Allosteric Site Modulation
4. Host Proteases as Potential Drug Targets for Antiviral Therapies
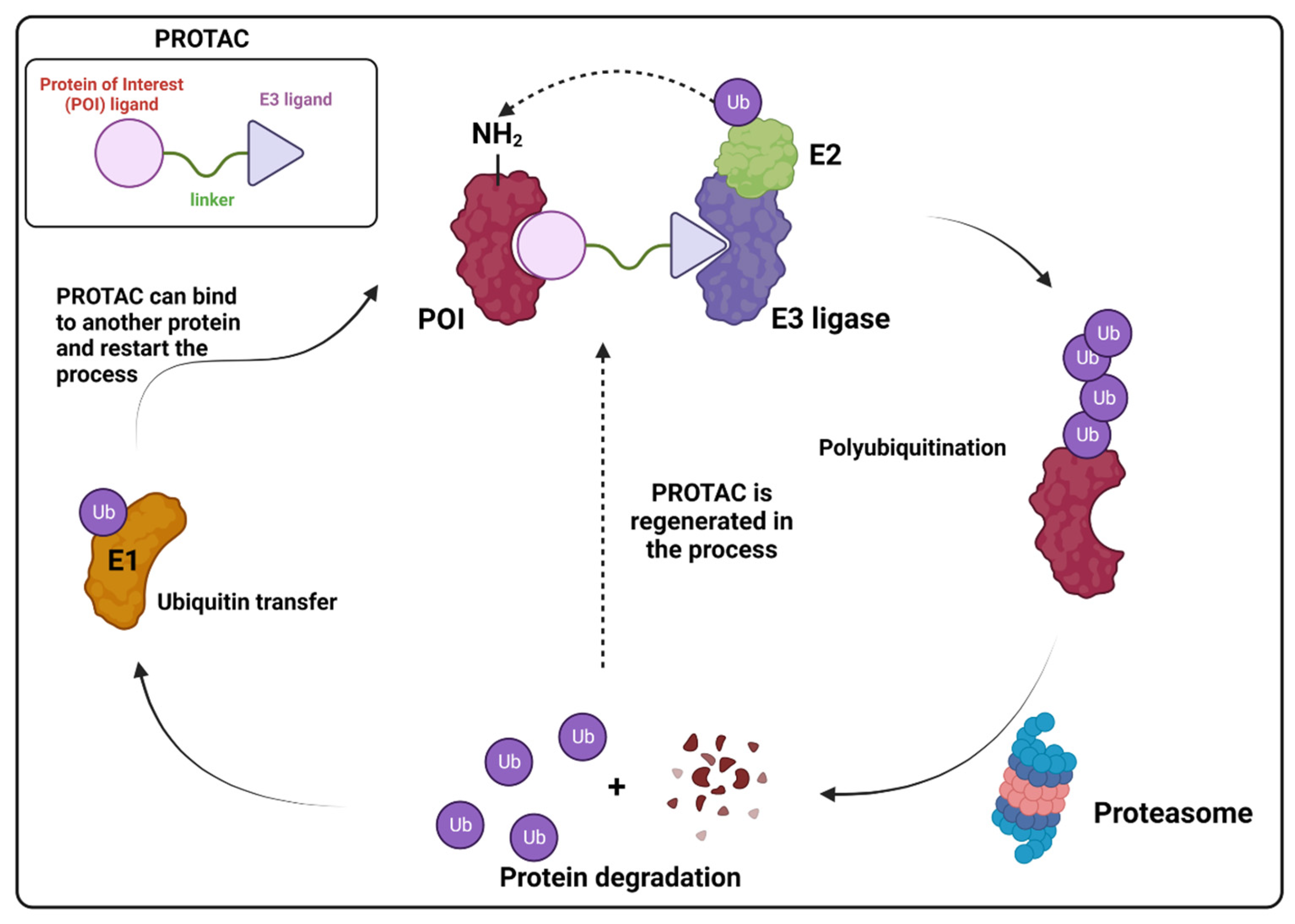
5. Targeted Protein Degradation Strategies
6. Natural Compounds as a Source of Novel Antiviral Drugs
7. Combination Therapies and Synergistic Approaches
8. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Piret, J.; Boivin, G. Pandemics Throughout History. Front. Microbiol. 2021, 11, 631736. [Google Scholar] [CrossRef]
- Baker, R.E.; Mahmud, A.S.; Miller, I.F.; Rajeev, M.; Rasambainarivo, F.; Rice, B.L.; Takahashi, S.; Tatem, A.J.; Wagner, C.E.; Wang, L.-F.; et al. Infectious disease in an era of global change. Nat. Rev. Microbiol. 2022, 20, 193–205. [Google Scholar] [CrossRef]
- Kaiwan, O.; Sethi, Y.; Khehra, N.; Padda, I.; Chopra, H.; Chandran, D.; Dhama, K.; Chakraborty, C.; Islam, A.; Kaka, N. Emerging and re-emerging viral diseases, predisposing risk factors, and implications of international travel: A call for action for increasing vigilance and imposing restrictions under the current threats of recently emerging multiple Omicron subvariants. Int. J. Surg. 2023, 109, 589–591. [Google Scholar] [CrossRef]
- Kumar, M.; Kuroda, K.; Dhangar, K.; Mazumder, P.; Sonne, C.; Rinklebe, J.; Kitajima, M. Potential Emergence of Antiviral-Resistant Pandemic Viruses via Environmental Drug Exposure of Animal Reservoirs. Environ. Sci. Technol. 2020, 54, 8503–8505. [Google Scholar] [CrossRef]
- Debroas, D.; Siguret, C. Viruses as key reservoirs of antibiotic resistance genes in the environment. ISME J. 2019, 13, 2856–2867. [Google Scholar] [CrossRef]
- Strasfeld, L.; Chou, S. Antiviral Drug Resistance: Mechanisms and Clinical Implications. Infect. Dis. Clin. N. Am. 2010, 24, 413–437. [Google Scholar] [CrossRef]
- Tompa, D.R.; Immanuel, A.; Srikanth, S.; Kadhirvel, S. Trends and strategies to combat viral infections: A review on FDA approved antiviral drugs. Int. J. Biol. Macromol. 2021, 172, 524–541. [Google Scholar] [CrossRef]
- Fenner, F.; Bachmann, P.A.; Gibbs, E.P.J.; Murphy, F.; Studdert, M.J.; White, D.O. Structure and Composition of Viruses. Vet. Virol. 1987, 3–19. [Google Scholar] [CrossRef]
- Louten, J. Virus Structure and Classification. Essent. Hum. Virol. 2016, 19–29. [Google Scholar] [CrossRef]
- Lucas, W. Viral Capsids and Envelopes: Structure and Function. In Encyclopedia of Life Sciences; Wiley: Hoboken, NJ, USA, 2010; ISBN 978-0-470-01617-6. [Google Scholar]
- Soderstrom, K. Viral Replication. In xPharm: The Comprehensive Pharmacology Reference; Enna, S.J., Bylund, D.B., Eds.; Elsevier: New York, NY, USA, 2007; pp. 1–5. ISBN 978-0-08-055232-3. [Google Scholar]
- Dimitrov, D.S. Virus entry: Molecular mechanisms and biomedical applications. Nat. Rev. Microbiol. 2004, 2, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Bond, J.S. Proteases: History, discovery, and roles in health and disease. J. Biol. Chem. 2019, 294, 1643–1651. [Google Scholar] [CrossRef]
- Schechter, I.; Berger, A. On the size of the active site in proteases. I. Papain. Biochem. Biophys. Res. Commun. 1967, 27, 157–162. [Google Scholar] [CrossRef]
- Sojka, D.; Šnebergerová, P.; Robbertse, L. Protease Inhibition—An Established Strategy to Combat Infectious Diseases. Int. J. Mol. Sci. 2021, 22, 5762. [Google Scholar] [CrossRef]
- López-Otín, C.; Bond, J.S. Proteases: Multifunctional Enzymes in Life and Disease. J. Biol. Chem. 2008, 283, 30433–30437. [Google Scholar] [CrossRef]
- Rawlings, N.D.; Barrett, A.J.; Bateman, A. Asparagine Peptide Lyases. J. Biol. Chem. 2011, 286, 38321–38328. [Google Scholar] [CrossRef]
- Yost, S.A.; Marcotrigiano, J. Viral precursor polyproteins: Keys of regulation from replication to maturation. Curr. Opin. Virol. 2013, 3, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Zephyr, J.; Kurt Yilmaz, N.; Schiffer, C.A. Viral proteases: Structure, mechanism and inhibition. Enzymes 2021, 50, 301–333. [Google Scholar] [CrossRef] [PubMed]
- Drag, M.; Salvesen, G.S. Emerging principles in protease-based drug discovery. Nat. Rev. Drug Discov. 2010, 9, 690–701. [Google Scholar] [CrossRef] [PubMed]
- Farady, C.J.; Craik, C.S. Mechanisms Of Macromolecular Protease Inhibitors. ChemBioChem 2010, 11, 2341–2346. [Google Scholar] [CrossRef] [PubMed]
- Jimmidi, R. Synthesis and Applications of Peptides and Peptidomimetics in Drug Discovery. Eur. J. Org. Chem. 2023, 26, e202300028. [Google Scholar] [CrossRef]
- El-Faham, A.; de la Torre, B.G.; Albericio, F. Latest Advances on Synthesis, Purification, and Characterization of Peptides and Their Applications. Appl. Sci. 2021, 11, 5593. [Google Scholar] [CrossRef]
- Al Musaimi, O.; Al Shaer, D.; Albericio, F.; de la Torre, B.G. 2022 FDA TIDES (Peptides and Oligonucleotides) Harvest. Pharmaceuticals 2023, 16, 336. [Google Scholar] [CrossRef] [PubMed]
- de la Torre, B.G.; Albericio, F. The Pharmaceutical Industry in 2022: An Analysis of FDA Drug Approvals from the Perspective of Molecules. Molecules 2023, 28, 1038. [Google Scholar] [CrossRef] [PubMed]
- Roberts, N.A.; Martin, J.A.; Kinchington, D.; Broadhurst, A.V.; Craig, J.C.; Duncan, I.B.; Galpin, S.A.; Handa, B.K.; Kay, J.; Kröhn, A.; et al. Rational Design of Peptide-Based HIV Proteinase Inhibitors. Science 1990, 248, 358–361. [Google Scholar] [CrossRef] [PubMed]
- Kosalaraksa, P.; Ananworanich, J.; Puthanakit, T.; Pinyakorn, S.; Lumbiganon, P.; Chuanjaroen, T.; Chobkarjing, U.; Phanuphak, P.; Pancharoen, C.; Bunupuradah, T.; et al. Long-term Lopinavir/Ritonavir Monotherapy in HIV-infected Children. Pediatr. Infect. Dis. J. 2013, 32, 350. [Google Scholar] [CrossRef] [PubMed]
- Pokorná, J.; Machala, L.; Řezáčová, P.; Konvalinka, J. Current and Novel Inhibitors of HIV Protease. Viruses 2009, 1, 1209–1239. [Google Scholar] [CrossRef]
- Weber, I.T.; Wang, Y.-F.; Harrison, R.W. HIV Protease: Historical Perspective and Current Research. Viruses 2021, 13, 839. [Google Scholar] [CrossRef]
- Yang, J.; Qi, J.-L.; Wang, X.-X.; Li, X.-H.; Jin, R.; Liu, B.-Y.; Liu, H.-X.; Rao, H.-Y. The burden of hepatitis C virus in the world, China, India, and the United States from 1990 to 2019. Front. Public Health 2023, 11, 1041201. [Google Scholar] [CrossRef]
- McCauley, J.A.; Rudd, M.T. Hepatitis C virus NS3/4a protease inhibitors. Curr. Opin. Pharmacol. 2016, 30, 84–92. [Google Scholar] [CrossRef]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Shrivastava, S.; Hassanali, M.; Stothard, P.; Chang, Z.; Woolsey, J. DrugBank: A comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006, 34, D668–D672. [Google Scholar] [CrossRef]
- Wang, L.; Wang, N.; Zhang, W.; Cheng, X.; Yan, Z.; Shao, G.; Wang, X.; Wang, R.; Fu, C. Therapeutic peptides: Current applications and future directions. Signal Transduct. Target. Ther. 2022, 7, 48. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef] [PubMed]
- Gutnik, D.; Evseev, P.; Miroshnikov, K.; Shneider, M. Using AlphaFold Predictions in Viral Research. Curr. Issues Mol. Biol. 2023, 45, 3705–3732. [Google Scholar] [CrossRef]
- Owen, D.R.; Allerton, C.M.N.; Anderson, A.S.; Aschenbrenner, L.; Avery, M.; Berritt, S.; Boras, B.; Cardin, R.D.; Carlo, A.; Coffman, K.J.; et al. An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19. Science 2021, 374, 1586–1593. [Google Scholar] [CrossRef]
- Akinosoglou, K.; Schinas, G.; Gogos, C. Oral Antiviral Treatment for COVID-19: A Comprehensive Review on Nirmatrelvir/Ritonavir. Viruses 2022, 14, 2540. [Google Scholar] [CrossRef]
- FDA Approves First Oral Antiviral for Treatment of COVID-19 in Adults. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-oral-antiviral-treatment-covid-19-adults (accessed on 11 December 2023).
- Choudhry, H.; Alzahrani, F.A.; Hassan, M.A.; Alghamdi, A.; Abdulaal, W.H.; Bakhrebah, M.A.; Zamzami, M.A.; Helmi, N.; Bokhari, F.F.; Zeyadi, M.; et al. Zika Virus Targeting by Screening Inhibitors against NS2B/NS3 Protease. BioMed Res. Int. 2019, 2019, e3947245. [Google Scholar] [CrossRef] [PubMed]
- Faye, O.; Faye, O.; Dupressoir, A.; Weidmann, M.; Ndiaye, M.; Alpha Sall, A. One-step RT-PCR for detection of Zika virus. J. Clin. Virol. 2008, 43, 96–101. [Google Scholar] [CrossRef]
- Lowe, R.; Barcellos, C.; Brasil, P.; Cruz, O.G.; Honório, N.A.; Kuper, H.; Carvalho, M.S. The Zika Virus Epidemic in Brazil: From Discovery to Future Implications. Int. J. Environ. Res. Public Health 2018, 15, 96. [Google Scholar] [CrossRef] [PubMed]
- Musso, D.; Ko, A.I.; Baud, D. Zika Virus Infection—After the Pandemic. N. Engl. J. Med. 2019, 381, 1444–1457. [Google Scholar] [CrossRef]
- Murtuja, S.; Shilkar, D.; Sarkar, B.; Sinha, B.N.; Jayaprakash, V. A short survey of dengue protease inhibitor development in the past 6 years (2015–2020) with an emphasis on similarities between DENV and SARS-CoV-2 proteases. Bioorg. Med. Chem. 2021, 49, 116415. [Google Scholar] [CrossRef]
- Luo, D.; Xu, T.; Watson, R.P.; Scherer-Becker, D.; Sampath, A.; Jahnke, W.; Yeong, S.S.; Wang, C.H.; Lim, S.P.; Strongin, A.; et al. Insights into RNA unwinding and ATP hydrolysis by the flavivirus NS3 protein. EMBO J. 2008, 27, 3209–3219. [Google Scholar] [CrossRef]
- Bazan, J.F.; Fletterick, R.J. Detection of a trypsin-like serine protease domain in flaviviruses and pestviruses. Virology 1989, 171, 637–639. [Google Scholar] [CrossRef] [PubMed]
- Nitsche, C. Proteases from dengue, West Nile and Zika viruses as drug targets. Biophys. Rev. 2019, 11, 157–165. [Google Scholar] [CrossRef]
- Nitsche, C.; Holloway, S.; Schirmeister, T.; Klein, C.D. Biochemistry and Medicinal Chemistry of the Dengue Virus Protease. Chem. Rev. 2014, 114, 11348–11381. [Google Scholar] [CrossRef] [PubMed]
- Phoo, W.W.; Zhang, Z.; Wirawan, M.; Chew, E.J.C.; Chew, A.B.L.; Kouretova, J.; Steinmetzer, T.; Luo, D. Structures of Zika virus NS2B-NS3 protease in complex with peptidomimetic inhibitors. Antivir. Res. 2018, 160, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Vagner, J.; Qu, H.; Hruby, V.J. Peptidomimetics, a synthetic tool of drug discovery. Curr. Opin. Chem. Biol. 2008, 12, 292–296. [Google Scholar] [CrossRef] [PubMed]
- Behrouz, S.; Kühl, N.; Klein, C.D. N-sulfonyl peptide-hybrids as a new class of dengue virus protease inhibitors. Eur. J. Med. Chem. 2023, 251, 115227. [Google Scholar] [CrossRef] [PubMed]
- de Bont, D.B.A.; Sliedregt-Bol, K.M.; Hofmeyer, L.J.F.; Liskamp, R.M.J. Increased stability of peptidesulfonamide peptidomimetics towards protease catalyzed degradation. Bioorg. Med. Chem. 1999, 7, 1043–1047. [Google Scholar] [CrossRef] [PubMed]
- Love, R.A.; Parge, H.E.; Wickersham, J.A.; Hostomsky, Z.; Habuka, N.; Moomaw, E.W.; Adachi, T.; Hostomska, Z. The Crystal Structure of Hepatitis C Virus NS3 Proteinase Reveals a Trypsin-like Fold and a Structural Zinc Binding Site. Cell 1996, 87, 331–342. [Google Scholar] [CrossRef]
- Venkatraman, S.; Njoroge, F.G.; Girijavallabhan, V.M.; Madison, V.S.; Yao, N.H.; Prongay, A.J.; Butkiewicz, N.; Pichardo, J. Design and Synthesis of Depeptidized Macrocyclic Inhibitors of Hepatitis C NS3-4A Protease Using Structure-Based Drug Design. J. Med. Chem. 2005, 48, 5088–5091. [Google Scholar] [CrossRef]
- de Leuw, P.; Stephan, C. Protease inhibitors for the treatment of hepatitis C virus infection. GMS Infect. Dis. 2017, 5, Doc08. [Google Scholar] [CrossRef]
- Cummings, M.D.; Sekharan, S. Structure-Based Macrocycle Design in Small-Molecule Drug Discovery and Simple Metrics to Identify Opportunities for Macrocyclization of Small-Molecule Ligands. J. Med. Chem. 2019, 62, 6843–6853. [Google Scholar] [CrossRef] [PubMed]
- Vinogradov, A.A.; Yin, Y.; Suga, H. Macrocyclic Peptides as Drug Candidates: Recent Progress and Remaining Challenges. J. Am. Chem. Soc. 2019, 141, 4167–4181. [Google Scholar] [CrossRef]
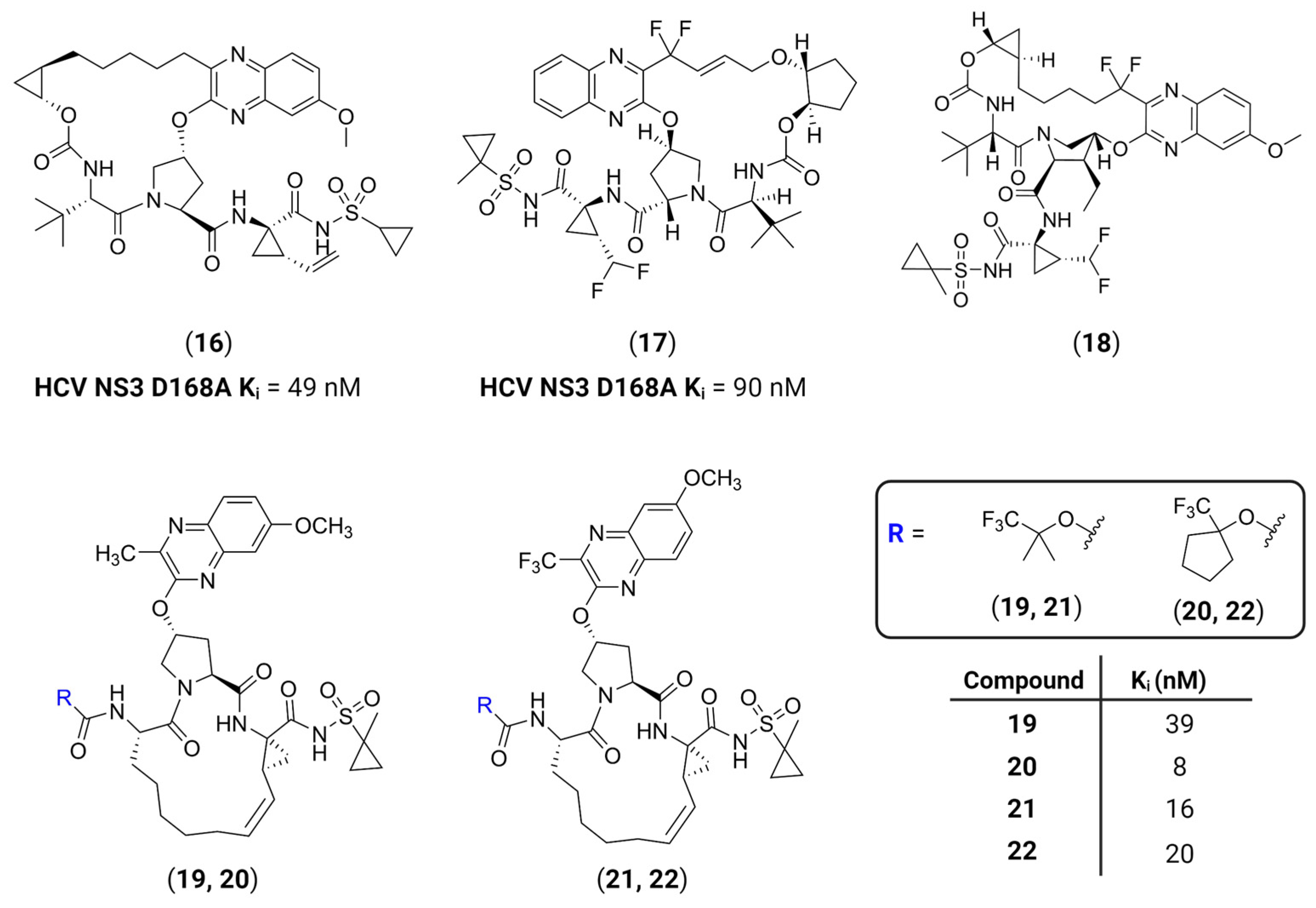
- Nageswara Rao, D.; Zephyr, J.; Henes, M.; Chan, E.T.; Matthew, A.N.; Hedger, A.K.; Conway, H.L.; Saeed, M.; Newton, A.; Petropoulos, C.J.; et al. Discovery of Quinoxaline-Based P1–P3 Macrocyclic NS3/4A Protease Inhibitors with Potent Activity against Drug-Resistant Hepatitis C Virus Variants. J. Med. Chem. 2021, 64, 11972–11989. [Google Scholar] [CrossRef]
- Li Petri, G.; Di Martino, S.; De Rosa, M. Peptidomimetics: An Overview of Recent Medicinal Chemistry Efforts toward the Discovery of Novel Small Molecule Inhibitors. J. Med. Chem. 2022, 65, 7438–7475. [Google Scholar] [CrossRef]
- Sutanto, F.; Konstantinidou, M.; Dömling, A. Covalent inhibitors: A rational approach to drug discovery. RSC Med. Chem. 2020, 11, 876–884. [Google Scholar] [CrossRef]
- Aljoundi, A.; Bjij, I.; El Rashedy, A.; Soliman, M.E.S. Covalent Versus Non-covalent Enzyme Inhibition: Which Route Should We Take? A Justification of the Good and Bad from Molecular Modelling Perspective. Protein J. 2020, 39, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Bonatto, V.; Lameiro, R.F.; Rocho, F.R.; Lameira, J.; Leitão, A.; Montanari, C.A. Nitriles: An attractive approach to the development of covalent inhibitors. RSC Med. Chem. 2023, 14, 201–217. [Google Scholar] [CrossRef] [PubMed]
- Brogi, S.; Ibba, R.; Rossi, S.; Butini, S.; Calderone, V.; Gemma, S.; Campiani, G. Covalent Reversible Inhibitors of Cysteine Proteases Containing the Nitrile Warhead: Recent Advancement in the Field of Viral and Parasitic Diseases. Molecules 2022, 27, 2561. [Google Scholar] [CrossRef]
- Ngo, C.; Fried, W.; Aliyari, S.; Feng, J.; Qin, C.; Zhang, S.; Yang, H.; Shanaa, J.; Feng, P.; Cheng, G.; et al. Alkyne as a Latent Warhead to Covalently Target SARS-CoV-2 Main Protease. J. Med. Chem. 2023, 66, 12237–12248. [Google Scholar] [CrossRef]
- Dai, W.; Zhang, B.; Su, H.; Li, J.; Zhao, Y.; Xie, X.; Jin, Z.; Liu, F.; Li, C.; Li, Y.; et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 2020, 368, 1331–1335. [Google Scholar] [CrossRef] [PubMed]
- Fairhurst, R.A.; Knoepfel, T.; Buschmann, N.; Leblanc, C.; Mah, R.; Todorov, M.; Nimsgern, P.; Ripoche, S.; Niklaus, M.; Warin, N.; et al. Discovery of Roblitinib (FGF401) as a Reversible-Covalent Inhibitor of the Kinase Activity of Fibroblast Growth Factor Receptor 4. J. Med. Chem. 2020, 63, 12542–12573. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Xi, K.; Ratia, K.; Santarsiero, B.D.; Fu, W.; Harcourt, B.H.; Rota, P.A.; Baker, S.C.; Johnson, M.E.; Mesecar, A.D. Design and Synthesis of Peptidomimetic Severe Acute Respiratory Syndrome Chymotrypsin-like Protease Inhibitors. J. Med. Chem. 2005, 48, 6767–6771. [Google Scholar] [CrossRef] [PubMed]
- Shie, J.-J.; Fang, J.-M.; Kuo, T.-H.; Kuo, C.-J.; Liang, P.-H.; Huang, H.-J.; Wu, Y.-T.; Jan, J.-T.; Cheng, Y.-S.E.; Wong, C.-H. Inhibition of the severe acute respiratory syndrome 3CL protease by peptidomimetic α,β-unsaturated esters. Bioorg. Med. Chem. 2005, 13, 5240–5252. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Fonović, M.; Verhelst, S.H.L.; Blum, G.; Bogyo, M. Evaluation of α,β-unsaturated ketone-based probes for papain-family cysteine proteases. Bioorg. Med. Chem. 2009, 17, 1071–1078. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Douangamath, A.; Fearon, D.; Gehrtz, P.; Krojer, T.; Lukacik, P.; Owen, C.D.; Resnick, E.; Strain-Damerell, C.; Aimon, A.; Ábrányi-Balogh, P.; et al. Crystallographic and electrophilic fragment screening of the SARS-CoV-2 main protease. Nat. Commun. 2020, 11, 5047. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Han, X.; Xiao, X.; Zhou, J. Covalent Warheads Targeting Cysteine Residue: The Promising Approach in Drug Development. Molecules 2022, 27, 7728. [Google Scholar] [CrossRef]
- Gehringer, M.; Laufer, S.A. Emerging and Re-Emerging Warheads for Targeted Covalent Inhibitors: Applications in Medicinal Chemistry and Chemical Biology. J. Med. Chem. 2019, 62, 5673–5724. [Google Scholar] [CrossRef]
- Hu, X.; Lin, C.; Xu, Q.; Zhou, X.; Zeng, P.; McCormick, P.J.; Jiang, H.; Li, J.; Zhang, J. Structural Basis for the Inhibition of Coronaviral Main Proteases by a Benzothiazole-Based Inhibitor. Viruses 2022, 14, 2075. [Google Scholar] [CrossRef]
- Martin, J.S.; MacKenzie, C.J.; Fletcher, D.; Gilbert, I.H. Characterising covalent warhead reactivity. Bioorg. Med. Chem. 2019, 27, 2066–2074. [Google Scholar] [CrossRef] [PubMed]
- Müller, P.; Meta, M.; Meidner, J.L.; Schwickert, M.; Meyr, J.; Schwickert, K.; Kersten, C.; Zimmer, C.; Hammerschmidt, S.J.; Frey, A.; et al. Investigation of the Compatibility between Warheads and Peptidomimetic Sequences of Protease Inhibitors—A Comprehensive Reactivity and Selectivity Study. Int. J. Mol. Sci. 2023, 24, 7226. [Google Scholar] [CrossRef]
- Vankadara, S.; Dawson, M.D.; Fong, J.Y.; Oh, Q.Y.; Ang, Q.A.; Liu, B.; Chang, H.Y.; Koh, J.; Koh, X.; Tan, Q.W.; et al. A Warhead Substitution Study on the Coronavirus Main Protease Inhibitor Nirmatrelvir. ACS Med. Chem. Lett. 2022, 13, 1345–1350. [Google Scholar] [CrossRef]
- Singh, J.; Petter, R.C.; Baillie, T.A.; Whitty, A. The resurgence of covalent drugs. Nat. Rev. Drug Discov. 2011, 10, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Péczka, N.; Orgován, Z.; Ábrányi-Balogh, P.; Keserű, G.M. Electrophilic warheads in covalent drug discovery: An overview. Expert Opin. Drug Discov. 2022, 17, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Eaton, J.K.; Furst, L.; Ruberto, R.A.; Moosmayer, D.; Hilpmann, A.; Ryan, M.J.; Zimmermann, K.; Cai, L.L.; Niehues, M.; Badock, V.; et al. Selective covalent targeting of GPX4 using masked nitrile-oxide electrophiles. Nat. Chem. Biol. 2020, 16, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Eaton, J.K.; Ruberto, R.A.; Kramm, A.; Viswanathan, V.S.; Schreiber, S.L. Diacylfuroxans Are Masked Nitrile Oxides That Inhibit GPX4 Covalently. J. Am. Chem. Soc. 2019, 141, 20407–20415. [Google Scholar] [CrossRef]
- Li, L.; Chenna, B.C.; Yang, K.S.; Cole, T.R.; Goodall, Z.T.; Giardini, M.; Moghadamchargari, Z.; Hernandez, E.A.; Gomez, J.; Calvet, C.M.; et al. Self-Masked Aldehyde Inhibitors: A Novel Strategy for Inhibiting Cysteine Proteases. J. Med. Chem. 2021, 64, 11267–11287. [Google Scholar] [CrossRef]
- Mehta, N.V.; Degani, M.S. The expanding repertoire of covalent warheads for drug discovery. Drug Discov. Today 2023, 28, 103799. [Google Scholar] [CrossRef]
- Sabe, V.T.; Ntombela, T.; Jhamba, L.A.; Maguire, G.E.M.; Govender, T.; Naicker, T.; Kruger, H.G. Current trends in computer aided drug design and a highlight of drugs discovered via computational techniques: A review. Eur. J. Med. Chem. 2021, 224, 113705. [Google Scholar] [CrossRef]
- Lei, J.; Hansen, G.; Nitsche, C.; Klein, C.D.; Zhang, L.; Hilgenfeld, R. Crystal structure of Zika virus NS2B-NS3 protease in complex with a boronate inhibitor. Science 2016, 353, 503–505. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, Z.; Phoo, W.W.; Loh, Y.R.; Wang, W.; Liu, S.; Chen, M.W.; Hung, A.W.; Keller, T.H.; Luo, D.; et al. Structural Dynamics of Zika Virus NS2B-NS3 Protease Binding to Dipeptide Inhibitors. Structure 2017, 25, 1242–1250.e3. [Google Scholar] [CrossRef]
- Phoo, W.W.; Li, Y.; Zhang, Z.; Lee, M.Y.; Loh, Y.R.; Tan, Y.B.; Ng, E.Y.; Lescar, J.; Kang, C.; Luo, D. Structure of the NS2B-NS3 protease from Zika virus after self-cleavage. Nat. Commun. 2016, 7, 13410. [Google Scholar] [CrossRef]
- Shin, H.J.; Kim, M.-H.; Lee, J.-Y.; Hwang, I.; Yoon, G.Y.; Kim, H.S.; Kwon, Y.-C.; Ahn, D.-G.; Kim, K.-D.; Kim, B.-T.; et al. Structure-Based Virtual Screening: Identification of a Novel NS2B-NS3 Protease Inhibitor with Potent Antiviral Activity against Zika and Dengue Viruses. Microorganisms 2021, 9, 545. [Google Scholar] [CrossRef]
- Hossain, S.; Shovon, T.I.; Hasan, R.; Hakim, F.T.; Hasan, M.M.; Esha, S.A.; Tasnim, S.; Nazir, S.; Akhter, F.; Ali, M.A.; et al. Therapeutic Potential of Antiviral Peptides against the NS2B/NS3 Protease of Zika Virus. ACS Omega 2023, 8, 35207–35218. [Google Scholar] [CrossRef]
- Xiong, Y.; Cheng, F.; Zhang, J.; Su, H.; Hu, H.; Zou, Y.; Li, M.; Xu, Y. Structure-based design of a novel inhibitor of the ZIKA virus NS2B/NS3 protease. Bioorg. Chem. 2022, 128, 106109. [Google Scholar] [CrossRef] [PubMed]
- Pant, S.; Bhattacharya, G.; Jena, N.R. Structures and dynamics of peptide and peptidomimetic inhibitors bound to the NS2B-NS3 protease of the ZIKA virus. J. Biomol. Struct. Dyn. 2023, 41, 3076–3088. [Google Scholar] [CrossRef]
- Paul, D.; Sanap, G.; Shenoy, S.; Kalyane, D.; Kalia, K.; Tekade, R.K. Artificial intelligence in drug discovery and development. Drug Discov. Today 2021, 26, 80–93. [Google Scholar] [CrossRef]
- Bender, A.; Cortés-Ciriano, I. Artificial intelligence in drug discovery: What is realistic, what are illusions? Part 1: Ways to make an impact, and why we are not there yet. Drug Discov. Today 2021, 26, 511–524. [Google Scholar] [CrossRef] [PubMed]
- Saramago, L.C.; Santana, M.V.; Gomes, B.F.; Dantas, R.F.; Senger, M.R.; Oliveira Borges, P.H.; Ferreira, V.N.D.S.; Dos Santos Rosa, A.; Tucci, A.R.; Dias Miranda, M.; et al. AI-Driven Discovery of SARS-CoV-2 Main Protease Fragment-like Inhibitors with Antiviral Activity In Vitro. J. Chem. Inf. Model. 2023, 63, 2866–2880. [Google Scholar] [CrossRef]
- Arrigoni, R.; Santacroce, L.; Ballini, A.; Palese, L.L. AI-Aided Search for New HIV-1 Protease Ligands. Biomolecules 2023, 13, 858. [Google Scholar] [CrossRef]
- Tsai, C.-J.; Nussinov, R. A Unified View of “How Allostery Works”. PLoS Comput. Biol. 2014, 10, e1003394. [Google Scholar] [CrossRef]
- Lu, S.; Qiu, Y.; Ni, D.; He, X.; Pu, J.; Zhang, J. Emergence of allosteric drug-resistance mutations: New challenges for allosteric drug discovery. Drug Discov. Today 2020, 25, 177–184. [Google Scholar] [CrossRef]
- Wu, H.; Bock, S.; Snitko, M.; Berger, T.; Weidner, T.; Holloway, S.; Kanitz, M.; Diederich, W.E.; Steuber, H.; Walter, C.; et al. Novel Dengue Virus NS2B/NS3 Protease Inhibitors. Antimicrob. Agents Chemother. 2015, 59, 1100–1109. [Google Scholar] [CrossRef] [PubMed]
- Millies, B.; von Hammerstein, F.; Gellert, A.; Hammerschmidt, S.; Barthels, F.; Göppel, U.; Immerheiser, M.; Elgner, F.; Jung, N.; Basic, M.; et al. Proline-Based Allosteric Inhibitors of Zika and Dengue Virus NS2B/NS3 Proteases. J. Med. Chem. 2019, 62, 11359–11382. [Google Scholar] [CrossRef]
- Gangopadhyay, A.; Saha, A. Exploring allosteric hits of the NS2B-NS3 protease of DENV2 by structure-guided screening. Comput. Biol. Chem. 2023, 104, 107876. [Google Scholar] [CrossRef]
- Batra, R.; Khayat, R.; Tong, L. Molecular mechanism for dimerization to regulate the catalytic activity of human cytomegalovirus protease. Nat. Struct. Mol. Biol. 2001, 8, 810–817. [Google Scholar] [CrossRef] [PubMed]
- Zühlsdorf, M.; Werten, S.; Klupp, B.G.; Palm, G.J.; Mettenleiter, T.C.; Hinrichs, W. Dimerization-Induced Allosteric Changes of the Oxyanion-Hole Loop Activate the Pseudorabies Virus Assemblin pUL26N, a Herpesvirus Serine Protease. PLoS Pathog. 2015, 11, e1005045. [Google Scholar] [CrossRef]
- Kaptan, S.; Girych, M.; Enkavi, G.; Kulig, W.; Sharma, V.; Vuorio, J.; Rog, T.; Vattulainen, I. Maturation of the SARS-CoV-2 virus is regulated by dimerization of its main protease. Comput. Struct. Biotechnol. J. 2022, 20, 3336–3346. [Google Scholar] [CrossRef] [PubMed]
- Shen, A. Allosteric regulation of protease activity by small molecules. Mol. Biosyst. 2010, 6, 1431–1443. [Google Scholar] [CrossRef][Green Version]
- Ghosh, A.K.; Weber, I.T.; Mitsuya, H. Beyond darunavir: Recent development of next generation HIV-1 protease inhibitors to combat drug resistance. Chem. Commun. 2022, 58, 11762–11782. [Google Scholar] [CrossRef]
- Nashed, N.T.; Aniana, A.; Ghirlando, R.; Chiliveri, S.C.; Louis, J.M. Modulation of the monomer-dimer equilibrium and catalytic activity of SARS-CoV-2 main protease by a transition-state analog inhibitor. Commun. Biol. 2022, 5, 160. [Google Scholar] [CrossRef]
- Ferreira, J.C.; Fadl, S.; Rabeh, W.M. Key dimer interface residues impact the catalytic activity of 3CLpro, the main protease of SARS-CoV-2. J. Biol. Chem. 2022, 298, 102023. [Google Scholar] [CrossRef]
- Silvestrini, L.; Belhaj, N.; Comez, L.; Gerelli, Y.; Lauria, A.; Libera, V.; Mariani, P.; Marzullo, P.; Ortore, M.G.; Palumbo Piccionello, A.; et al. The dimer-monomer equilibrium of SARS-CoV-2 main protease is affected by small molecule inhibitors. Sci. Rep. 2021, 11, 9283. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [PubMed]
- Tao, X.; Zhang, L.; Du, L.; Liao, R.; Cai, H.; Lu, K.; Zhao, Z.; Xie, Y.; Wang, P.-H.; Pan, J.-A.; et al. Allosteric inhibition of SARS-CoV-2 3CL protease by colloidal bismuth subcitrate. Chem. Sci. 2021, 12, 14098–14102. [Google Scholar] [CrossRef] [PubMed]
- Samrat, S.K.; Xu, J.; Xie, X.; Gianti, E.; Chen, H.; Zou, J.; Pattis, J.G.; Elokely, K.; Lee, H.; Li, Z.; et al. Allosteric inhibitors of the main protease of SARS-CoV-2. Antivir. Res. 2022, 205, 105381. [Google Scholar] [CrossRef] [PubMed]
- Hulce, K.R.; Jaishankar, P.; Lee, G.M.; Bohn, M.-F.; Connelly, E.J.; Wucherer, K.; Ongpipattanakul, C.; Volk, R.F.; Chuo, S.-W.; Arkin, M.R.; et al. Inhibiting a dynamic viral protease by targeting a non-catalytic cysteine. Cell Chem. Biol. 2022, 29, 785–798.e19. [Google Scholar] [CrossRef] [PubMed]
- Gable, J.E.; Acker, T.M.; Craik, C.S. Current and Potential Treatments for Ubiquitous but Neglected Herpesvirus Infections. Chem. Rev. 2014, 114, 11382–11412. [Google Scholar] [CrossRef]
- Kneller, D.W.; Phillips, G.; Weiss, K.L.; Pant, S.; Zhang, Q.; O’Neill, H.M.; Coates, L.; Kovalevsky, A. Unusual zwitterionic catalytic site of SARS–CoV-2 main protease revealed by neutron crystallography. J. Biol. Chem. 2020, 295, 17365–17373. [Google Scholar] [CrossRef]
- Nussinov, R.; Tsai, C.-J. Allostery in Disease and in Drug Discovery. Cell 2013, 153, 293–305. [Google Scholar] [CrossRef]
- Wold, E.A.; Zhou, J. GPCR Allosteric Modulators: Mechanistic Advantages and Therapeutic Applications. Curr. Top. Med. Chem. 2018, 18, 2002–2006. [Google Scholar] [CrossRef]
- Kenakin, T. Allosteric Theory: Taking Therapeutic Advantage of the Malleable Nature of GPCRs. Curr. Neuropharmacol. 2007, 5, 149–156. [Google Scholar] [CrossRef]
- Böttcher-Friebertshäuser, E.; Klenk, H.; Garten, W. Activation of influenza viruses by proteases from host cells and bacteria in the human airway epithelium. Pathog. Dis. 2013, 69, 87–100. [Google Scholar] [CrossRef]
- Earnest, J.T.; Hantak, M.P.; Park, J.-E.; Gallagher, T. Coronavirus and Influenza Virus Proteolytic Priming Takes Place in Tetraspanin-Enriched Membrane Microdomains. J. Virol. 2015, 89, 6093–6104. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Basso, L.G.M.; Zeraik, A.E.; Felizatti, A.P.; Costa-Filho, A.J. Membranotropic and biological activities of the membrane fusion peptides from SARS-CoV spike glycoprotein: The importance of the complete internal fusion peptide domain. Biochim. Biophys. Acta Biomembr. 2021, 1863, 183697. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Hofmann-Winkler, H.; Pöhlmann, S. Priming Time: How Cellular Proteases Arm Coronavirus Spike Proteins. Act. Viruses Host Proteases 2018, 71–98. [Google Scholar] [CrossRef]
- Fujishima, A.; Imai, Y.; Nomura, T.; Fujisawa, Y.; Yamamoto, Y.; Sugawara, T. The crystal structure of human cathepsin L complexed with E-64. FEBS Lett. 1997, 407, 47–50. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.-M.; Yang, W.-L.; Yang, F.-Y.; Zhang, L.; Huang, W.-J.; Hou, W.; Fan, C.-F.; Jin, R.-H.; Feng, Y.-M.; Wang, Y.-C.; et al. Cathepsin L plays a key role in SARS-CoV-2 infection in humans and humanized mice and is a promising target for new drug development. Signal Transduct. Target. Ther. 2021, 6, 134. [Google Scholar] [CrossRef] [PubMed]
- Majchrzak, M.; Poręba, M. The roles of cellular protease interactions in viral infections and programmed cell death: A lesson learned from the SARS-CoV-2 outbreak and COVID-19 pandemic. Pharmacol. Rep. 2022, 74, 1149–1165. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef]
- Metzdorf, K.; Jacobsen, H.; Greweling-Pils, M.C.; Hoffmann, M.; Lüddecke, T.; Miller, F.; Melcher, L.; Kempf, A.M.; Nehlmeier, I.; Bruder, D.; et al. TMPRSS2 Is Essential for SARS-CoV-2 Beta and Omicron Infection. Viruses 2023, 15, 271. [Google Scholar] [CrossRef]
- Mellott, D.M.; Tseng, C.-T.; Drelich, A.; Fajtová, P.; Chenna, B.C.; Kostomiris, D.H.; Hsu, J.; Zhu, J.; Taylor, Z.W.; Kocurek, K.I.; et al. A Clinical-Stage Cysteine Protease Inhibitor blocks SARS-CoV-2 Infection of Human and Monkey Cells. ACS Chem. Biol. 2021, 16, 642–650. [Google Scholar] [CrossRef]
- Zhu, J.; Li, L.; Drelich, A.; Chenna, B.C.; Mellott, D.M.; Taylor, Z.W.; Tat, V.; Garcia, C.Z.; Katzfuss, A.; Tseng, C.-T.K.; et al. Self-Masked Aldehyde Inhibitors of Human Cathepsin L Are Potent Anti-CoV-2 Agents. Front. Chem. 2022, 10, 867928. [Google Scholar] [CrossRef] [PubMed]
- Rahbar Saadat, Y.; Hosseiniyan Khatibi, S.M.; Zununi Vahed, S.; Ardalan, M. Host Serine Proteases: A Potential Targeted Therapy for COVID-19 and Influenza. Front. Mol. Biosci. 2021, 8, 725528. [Google Scholar] [CrossRef]
- El-Shimy, I.A.; Mohamed, M.M.A.; Hasan, S.S.; Hadi, M.A. Targeting host cell proteases as a potential treatment strategy to limit the spread of SARS-CoV-2 in the respiratory tract. Pharmacol. Res. Perspect. 2020, 9, e00698. [Google Scholar] [CrossRef]
- Izaguirre, G. The Proteolytic Regulation of Virus Cell Entry by Furin and Other Proprotein Convertases. Viruses 2019, 11, 837. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, H.; Li, D.; Zhao, C.; Li, S.; Qin, P.; Li, Y.; Yang, X.; Du, W.; Li, W.; et al. Identification of niclosamide as a novel antiviral agent against porcine epidemic diarrhea virus infection by targeting viral internalization. Virol. Sin. 2023, 38, 296–308. [Google Scholar] [CrossRef] [PubMed]
- Callis, J. The Ubiquitination Machinery of the Ubiquitin System. Arab. Book 2014, 12, e0174. [Google Scholar] [CrossRef]
- Ciechanover, A. The ubiquitin-proteasome pathway: On protein death and cell life. EMBO J. 1998, 17, 7151–7160. [Google Scholar] [CrossRef] [PubMed]
- Stringer, D.K.; Piper, R.C. Terminating protein ubiquitination. Cell Cycle 2011, 10, 3067–3071. [Google Scholar] [CrossRef]
- Madiraju, C.; Novack, J.P.; Reed, J.C.; Matsuzawa, S. K63 ubiquitination in immune signaling. Trends Immunol. 2022, 43, 148–162. [Google Scholar] [CrossRef]
- Murata, S.; Yashiroda, H.; Tanaka, K. Molecular mechanisms of proteasome assembly. Nat. Rev. Mol. Cell Biol. 2009, 10, 104–115. [Google Scholar] [CrossRef]
- Chan, W.C.; Liu, X.; Magin, R.S.; Girardi, N.M.; Ficarro, S.B.; Hu, W.; Tarazona Guzman, M.I.; Starnbach, C.A.; Felix, A.; Adelmant, G.; et al. Accelerating inhibitor discovery for deubiquitinating enzymes. Nat. Commun. 2023, 14, 686. [Google Scholar] [CrossRef]
- Giraldo, M.I.; Xia, H.; Aguilera-Aguirre, L.; Hage, A.; van Tol, S.; Shan, C.; Xie, X.; Sturdevant, G.L.; Robertson, S.J.; McNally, K.L.; et al. Envelope protein ubiquitination drives entry and pathogenesis of Zika virus. Nature 2020, 585, 414–419. [Google Scholar] [CrossRef]
- Wang, Y.; Li, Q.; Hu, D.; Gao, D.; Wang, W.; Wu, K.; Wu, J. USP38 Inhibits Zika Virus Infection by Removing Envelope Protein Ubiquitination. Viruses 2021, 13, 2029. [Google Scholar] [CrossRef]
- Chen, S.; Yun, F.; Yao, Y.; Cao, M.; Zhang, Y.; Wang, J.; Song, X.; Qian, Y. USP38 critically promotes asthmatic pathogenesis by stabilizing JunB protein. J. Exp. Med. 2018, 215, 2850–2867. [Google Scholar] [CrossRef]
- Liu, W.; Zhang, Q.; Fang, Y.; Wang, Y. The deubiquitinase USP38 affects cellular functions through interacting with LSD1. Biol. Res. 2018, 51, 53. [Google Scholar] [CrossRef]
- Chen, D.; Frezza, M.; Schmitt, S.; Kanwar, J.; Dou, Q.P. Bortezomib as the First Proteasome Inhibitor Anticancer Drug: Current Status and Future Perspectives. Curr. Cancer Drug Targets 2011, 11, 239–253. [Google Scholar] [CrossRef] [PubMed]
- Ci, Y.; Yao, B.; Yue, K.; Yang, Y.; Xu, C.; Li, D.; Qin, C.-F.; Shi, L. Bortezomib inhibits ZIKV/DENV by interfering with viral polyprotein cleavage via the ERAD pathway. Cell Chem. Biol. 2023, 30, 527–539.e5. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Hu, M.; Yang, Y.; Du, C.; Zhou, H.; Liu, C.; Chen, Y.; Fan, L.; Ma, H.; Gong, Y.; et al. An overview of PROTACs: A promising drug discovery paradigm. Mol. Biomed. 2022, 3, 46. [Google Scholar] [CrossRef] [PubMed]
- Békés, M.; Langley, D.R.; Crews, C.M. PROTAC targeted protein degraders: The past is prologue. Nat. Rev. Drug Discov. 2022, 21, 181–200. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Sun, Y. PROTACs: A novel strategy for cancer drug discovery and development. MedComm 2023, 4, e290. [Google Scholar] [CrossRef] [PubMed]
- Marinella, M.A. Indomethacin and resveratrol as potential treatment adjuncts for SARS-CoV-2/COVID-19. Int. J. Clin. Pract. 2020, 74, e13535. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Gao, X.; Wu, Z.; Selinger, D.W.; Zhou, Z. Indomethacin has a potent antiviral activity against SARS CoV-2 in vitro and canine coronavirus in vivo. bioRxiv 2020. [Google Scholar] [CrossRef]
- Desantis, J.; Mercorelli, B.; Celegato, M.; Croci, F.; Bazzacco, A.; Baroni, M.; Siragusa, L.; Cruciani, G.; Loregian, A.; Goracci, L. Indomethacin-based PROTACs as pan-coronavirus antiviral agents. Eur. J. Med. Chem. 2021, 226, 113814. [Google Scholar] [CrossRef] [PubMed]
- Alugubelli, Y.R.; Xiao, J.; Khatua, K.; Kumar, S.; Ma, Y.; Ma, X.R.; Vulupala, V.R.; Atla, S.R.; Blankenship, L.; Coleman, D.; et al. Discovery of First-in-Class PROTAC Degraders of SARS-CoV-2 Main Protease 2023. bioRxiv 2023. [Google Scholar] [CrossRef]
- Bond, M.J.; Crews, C.M. Proteolysis targeting chimeras (PROTACs) come of age: Entering the third decade of targeted protein degradation. RSC Chem. Biol. 2021, 2, 725–742. [Google Scholar] [CrossRef]
- Hahn, F.; Hamilton, S.T.; Wangen, C.; Wild, M.; Kicuntod, J.; Brückner, N.; Follett, J.E.L.; Herrmann, L.; Kheimar, A.; Kaufer, B.B.; et al. Development of a PROTAC-Based Targeting Strategy Provides a Mechanistically Unique Mode of Anti-Cytomegalovirus Activity. Int. J. Mol. Sci. 2021, 22, 12858. [Google Scholar] [CrossRef]
- Łukasik, P.; Załuski, M.; Gutowska, I. Cyclin-Dependent Kinases (CDK) and Their Role in Diseases Development–Review. Int. J. Mol. Sci. 2021, 22, 2935. [Google Scholar] [CrossRef]
- Hume, A.J.; Finkel, J.S.; Kamil, J.P.; Coen, D.M.; Culbertson, M.R.; Kalejta, R.F. Phosphorylation of Retinoblastoma Protein by Viral Protein with Cyclin-Dependent Kinase Function. Science 2008, 320, 797–799. [Google Scholar] [CrossRef]
- Sonntag, E.; Milbradt, J.; Svrlanska, A.; Strojan, H.; Häge, S.; Kraut, A.; Hesse, A.-M.; Amin, B.; Sonnewald, U.; Couté, Y.; et al. Protein kinases responsible for the phosphorylation of the nuclear egress core complex of human cytomegalovirus. J. Gen. Virol. 2017, 98, 2569–2581. [Google Scholar] [CrossRef]
- Hutterer, C.; Eickhoff, J.; Milbradt, J.; Korn, K.; Zeitträger, I.; Bahsi, H.; Wagner, S.; Zischinsky, G.; Wolf, A.; Degenhart, C.; et al. A novel CDK7 inhibitor of the Pyrazolotriazine class exerts broad-spectrum antiviral activity at nanomolar concentrations. Antimicrob. Agents Chemother. 2015, 59, 2062–2071. [Google Scholar] [CrossRef] [PubMed]
- Thomas, E.; Stewart, L.E.; Darley, B.A.; Pham, A.M.; Esteban, I.; Panda, S.S. Plant-Based Natural Products and Extracts: Potential Source to Develop New Antiviral Drug Candidates. Molecules 2021, 26, 6197. [Google Scholar] [CrossRef] [PubMed]
- Choo, M.Z.Y.; Chai, C.L.L. Chapter Two—The polypharmacology of natural products in drug discovery and development. In Annual Reports in Medicinal Chemistry; Altmann, K.-H., Ed.; Natural Products; Academic Press: Cambridge, MA, USA, 2023; Volume 61, pp. 55–100. [Google Scholar]
- Zhao, T.; Li, C.; Wang, S.; Song, X. Green Tea (Camellia sinensis): A Review of Its Phytochemistry, Pharmacology, and Toxicology. Molecules 2022, 27, 3909. [Google Scholar] [CrossRef] [PubMed]
- Coronado, M.A.; Gering, I.; Sevenich, M.; Olivier, D.S.; Mastalipour, M.; Amaral, M.S.; Willbold, D.; Eberle, R.J. The Importance of Epigallocatechin as a Scaffold for Drug Development against Flaviviruses. Pharmaceutics 2023, 15, 803. [Google Scholar] [CrossRef] [PubMed]
- Gil, C.; Ginex, T.; Maestro, I.; Nozal, V.; Barrado-Gil, L.; Cuesta-Geijo, M.Á.; Urquiza, J.; Ramírez, D.; Alonso, C.; Campillo, N.E.; et al. COVID-19: Drug Targets and Potential Treatments. J. Med. Chem. 2020, 63, 12359–12386. [Google Scholar] [CrossRef] [PubMed]
- Barretto, N.; Jukneliene, D.; Ratia, K.; Chen, Z.; Mesecar, A.D.; Baker, S.C. The papain-like protease of severe acute respiratory syndrome coronavirus has deubiquitinating activity. J. Virol. 2005, 79, 15189–15198. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, V.; Brognaro, H.; Prabhu, P.R.; de Souza, E.E.; Günther, S.; Reinke, P.Y.A.; Lane, T.J.; Ginn, H.; Han, H.; Ewert, W.; et al. Antiviral activity of natural phenolic compounds in complex at an allosteric site of SARS-CoV-2 papain-like protease. Commun. Biol. 2022, 5, 1–12. [Google Scholar] [CrossRef]
- Majerová, T.; Konvalinka, J. Viral proteases as therapeutic targets. Mol. Asp. Med. 2022, 88, 101159. [Google Scholar] [CrossRef]
- Shyr, Z.A.; Cheng, Y.-S.; Lo, D.C.; Zheng, W. Drug combination therapy for emerging viral diseases. Drug Discov. Today 2021, 26, 2367–2376. [Google Scholar] [CrossRef]
- Wagoner, J.; Herring, S.; Hsiang, T.-Y.; Ianevski, A.; Biering, S.B.; Xu, S.; Hoffmann, M.; Pöhlmann, S.; Gale, M.; Aittokallio, T.; et al. Combinations of Host- and Virus-Targeting Antiviral Drugs Confer Synergistic Suppression of SARS-CoV-2. Microbiol. Spectr. 2022, 10, e03331-22. [Google Scholar] [CrossRef]
- Wild, M.; Karner, D.; Eickhoff, J.; Wagner, S.; Kicuntod, J.; Chang, W.; Barry, P.; Jonjić, S.; Lenac Roviš, T.; Marschall, M. Combined Treatment with Host-Directed and Anticytomegaloviral Kinase Inhibitors: Mechanisms, Synergisms and Drug Resistance Barriers. Pharmaceutics 2023, 15, 2680. [Google Scholar] [CrossRef]
- Mousavi Maleki, M.S.; Sardari, S.; Ghandehari Alavijeh, A.; Madanchi, H. Recent Patents and FDA-Approved Drugs Based on Antiviral Peptides and Other Peptide-Related Antivirals. Int. J. Pept. Res. Ther. 2022, 29, 5. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Osswald, H.L.; Prato, G. Recent Progress in the Development of HIV-1 Protease Inhibitors for the Treatment of HIV/AIDS. J. Med. Chem. 2016, 59, 5172–5208. [Google Scholar] [CrossRef] [PubMed]
- Mason, S.; Devincenzo, J.P.; Toovey, S.; Wu, J.Z.; Whitley, R.J. Comparison of antiviral resistance across acute and chronic viral infections. Antivir. Res. 2018, 158, 103–112. [Google Scholar] [CrossRef] [PubMed]




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | Structure | Trade Name | Active Ingredient | Year | Indication | Drug Target |
|---|---|---|---|---|---|---|
| 1 |  | Invirase | Saquinavir mesylate | 1995 | Anti-HIV drug | HIV-1 protease |
| 2 |  | Norvir | Ritonavir | 1996 | Anti-HIV drug | HIV-1 protease |
| 3 |  | Crixivan | Indinavir | 1996 | Anti-HIV drug | HIV-1 protease |
| 4 |  | Mavik, Tarka | Trandolapril | 1996 | Hypertension, congestive heart failure | Angiotensin-converting Enzyme |
| 5 |  | Viracept | Nelfinavir | 1997 | anti-HIV drug | HIV-1 protease |
| 6 |  | Galvus, Jalra, Xiliarx | Vildagliptin | 2008 | Antihyperglicemic | Dipeptidyl peptidase-4 (DPP-4) |
| 7 |  | Kombiglyze, Komboglyze, Onglyza, Qtern, Qternmet | Saxagliptin | 2009 | Antihyperglicemic | Dipeptidyl peptidase-4 (DPP-4) |
| 8 |  | Incivek | Telaprevir | 2011 | Antiviral, Human Hepatitis C Virus | NS3/4A serine protease |
| 9 |  | Victrelis | Boceprevir | 2011 | Antiviral, Human Hepatitis C Virus | NS3/4A serine protease |
| 10 |  | Xarelto | Rivaroxaban | 2011 | Deep vein Thrombosis, Pulmonary Embolism | Coagulation factor X |
| 11 |  | Olysio, Galexos | Simeprevir | 2013 | Antiviral, Human Hepatitis C Virus | NS3/4A serine protease |
| 12 |  | Paxlovid | Nirmatrelvir | 2023 | Antiviral, SARS-CoV-2 | 3CLpro cysteine protease (Mpro) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borges, P.H.O.; Ferreira, S.B.; Silva, F.P., Jr. Recent Advances on Targeting Proteases for Antiviral Development. Viruses 2024, 16, 366. https://doi.org/10.3390/v16030366
Borges PHO, Ferreira SB, Silva FP Jr. Recent Advances on Targeting Proteases for Antiviral Development. Viruses. 2024; 16(3):366. https://doi.org/10.3390/v16030366
Chicago/Turabian StyleBorges, Pedro Henrique Oliveira, Sabrina Baptista Ferreira, and Floriano Paes Silva, Jr. 2024. "Recent Advances on Targeting Proteases for Antiviral Development" Viruses 16, no. 3: 366. https://doi.org/10.3390/v16030366
APA StyleBorges, P. H. O., Ferreira, S. B., & Silva, F. P., Jr. (2024). Recent Advances on Targeting Proteases for Antiviral Development. Viruses, 16(3), 366. https://doi.org/10.3390/v16030366