
Generation of High-Quality African Swine Fever Virus Complete Genome from Field Samples by Next-Generation Sequencing

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Field Samples
2.2. Genome Sequencing
2.3. Sequencing Data Analysis
2.4. Genome Sequences Alignment and Phylogenetic Analysis
3. Results
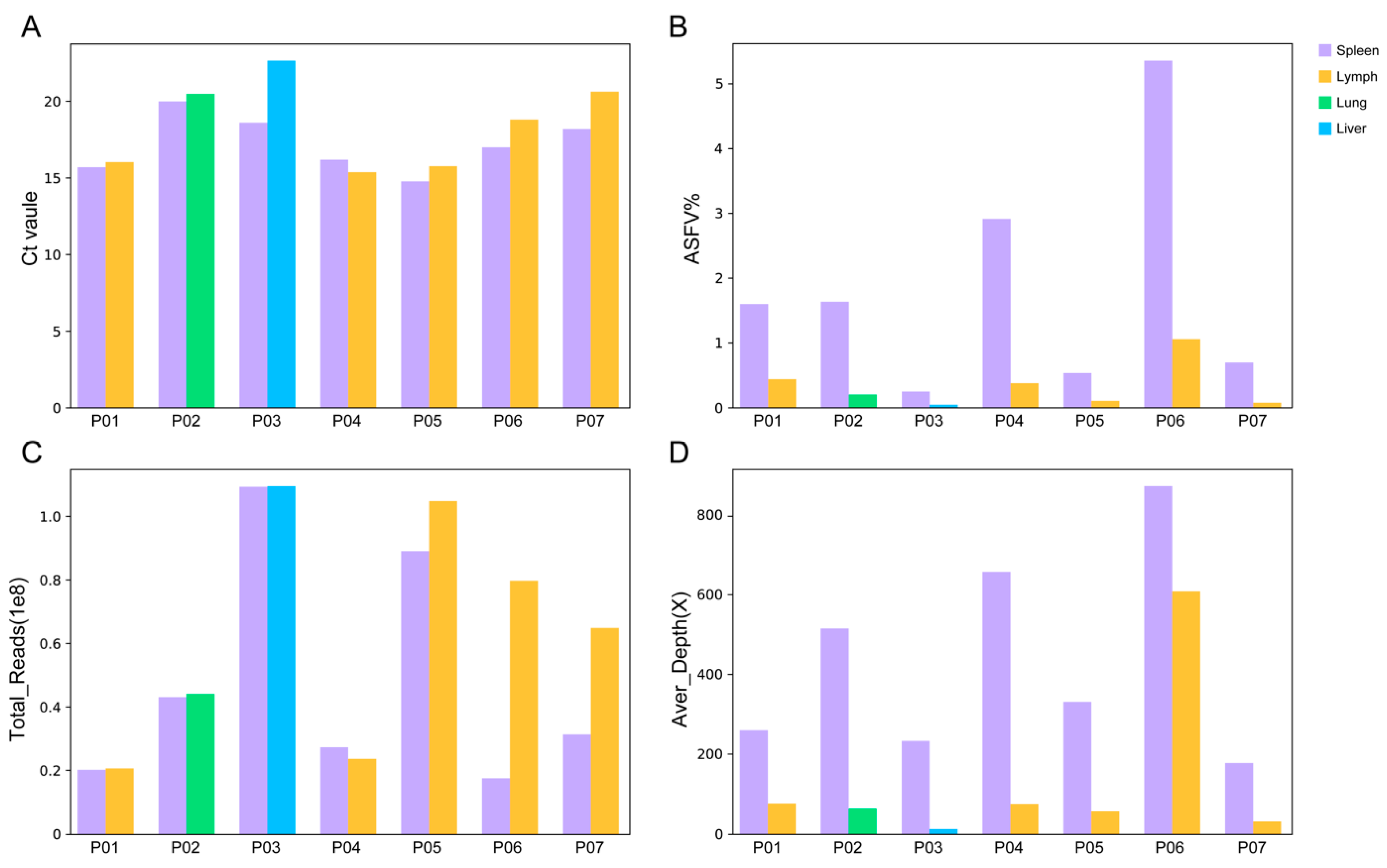
3.1. Performance of mNGS on Different Types of Tissue Samples
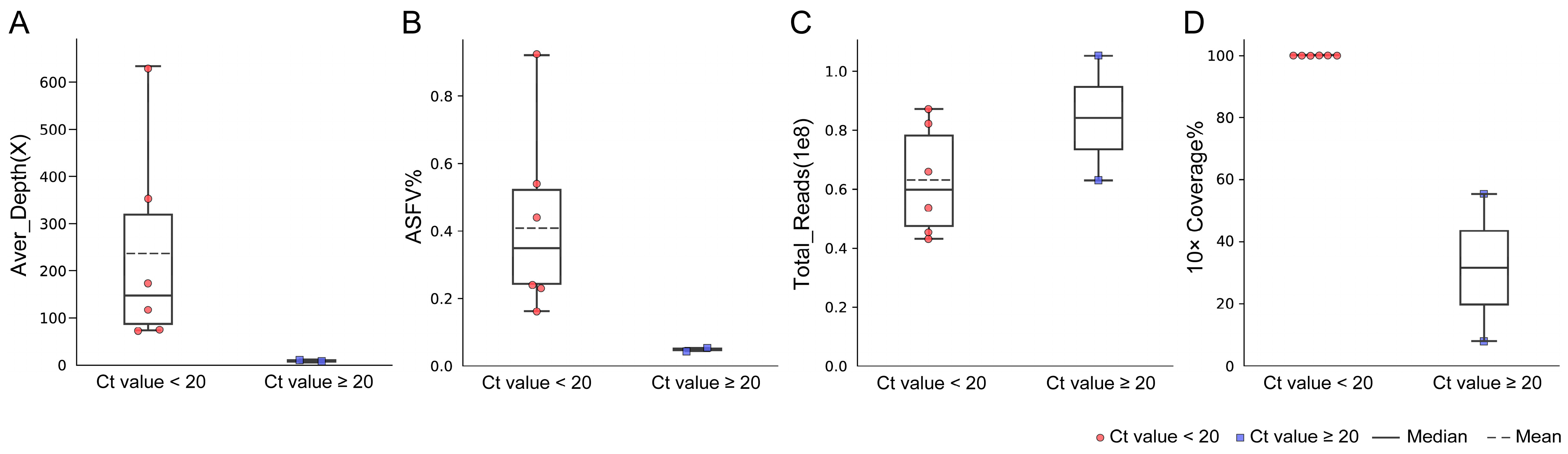
3.2. Performance of mNGS on Spleen Samples with Different Viral Loads
3.3. Performance of mNGS on Blood Samples
3.4. Performance of mNGS for the Detection of SNVs
3.5. Phylogenetic Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Penrith, M.L.; Kivaria, F.M. One hundred years of African swine fever in Africa: Where have we been, where are we now, where are we going? Transbound. Emerg. Dis. 2022, 69, e1179–e1200. [Google Scholar] [CrossRef]
- OIE. African Swine Fever. 2022. Available online: https://www.woah.org/en/disease/african-swine-fever/#ui-id-2 (accessed on 21 December 2023).
- Liu, S.; Luo, Y.; Wang, Y.; Li, S.; Zhao, Z.; Bi, Y.; Sun, J.; Peng, R.; Song, H.; Zhu, D.; et al. Cryo-EM Structure of the African Swine Fever Virus. Cell Host Microbe 2019, 26, 836–843.e3. [Google Scholar] [CrossRef]
- Andres, G.; Charro, D.; Matamoros, T.; Dillard, R.S.; Abrescia, N.G.A. The cryo-EM structure of African swine fever virus unravels a unique architecture comprising two icosahedral protein capsids and two lipoprotein membranes. J. Biol. Chem. 2020, 295, 1–12. [Google Scholar] [CrossRef]
- Wang, N.; Zhao, D.; Wang, J.; Zhang, Y.; Wang, M.; Gao, Y.; Li, F.; Wang, J.; Bu, Z.; Rao, Z.; et al. Architecture of African swine fever virus and implications for viral assembly. Science 2019, 366, 640–644. [Google Scholar] [CrossRef]
- Chapman, D.A.; Tcherepanov, V.; Upton, C.; Dixon, L.K. Comparison of the genome sequences of non-pathogenic and pathogenic African swine fever virus isolates. J. Gen. Virol. 2008, 89 Pt 2, 397–408. [Google Scholar] [CrossRef]
- Chapman, D.A.; Darby, A.C.; Da Silva, M.; Upton, C.; Radford, A.D.; Dixon, L.K. Genomic analysis of highly virulent Georgia 2007/1 isolate of African swine fever virus. Emerg. Infect. Dis. 2011, 17, 599–605. [Google Scholar] [CrossRef]
- Achenbach, J.E.; Gallardo, C.; Nieto-Pelegrin, E.; Rivera-Arroyo, B.; Degefa-Negi, T.; Arias, M.; Jenberie, S.; Mulisa, D.D.; Gizaw, D.; Gelaye, E.; et al. Identification of a New Genotype of African Swine Fever Virus in Domestic Pigs from Ethiopia. Transbound. Emerg. Dis. 2017, 64, 1393–1404. [Google Scholar] [CrossRef]
- Boshoff, C.I.; Bastos, A.D.; Gerber, L.J.; Vosloo, W. Genetic characterisation of African swine fever viruses from outbreaks in southern Africa (1973–1999). Vet. Microbiol. 2007, 121, 45–55. [Google Scholar] [CrossRef]
- Quembo, C.J.; Jori, F.; Vosloo, W.; Heath, L. Genetic characterization of African swine fever virus isolates from soft ticks at the wildlife/domestic interface in Mozambique and identification of a novel genotype. Transbound. Emerg. Dis. 2018, 65, 420–431. [Google Scholar] [CrossRef]
- Wohnke, E.; Fuchs, W.; Hartmann, L.; Blohm, U.; Blome, S.; Mettenleiter, T.C.; Karger, A. Comparison of the Proteomes of Porcine Macrophages and a Stable Porcine Cell Line after Infection with African Swine Fever Virus. Viruses 2021, 13, 2198. [Google Scholar] [CrossRef]
- Ge, S.; Li, J.; Fan, X.; Liu, F.; Li, L.; Wang, Q.; Ren, W.; Bao, J.; Liu, C.; Wang, H.; et al. Molecular Characterization of African Swine Fever Virus, China, 2018. Emerg. Infect. Dis. 2018, 24, 2131–2133. [Google Scholar] [CrossRef]
- Rowlands, R.J.; Michaud, V.; Heath, L.; Hutchings, G.; Oura, C.; Vosloo, W.; Dwarka, R.; Onashvili, T.; Albina, E.; Dixon, L.K. African swine fever virus isolate, Georgia, 2007. Emerg. Infect. Dis. 2008, 14, 1870–1874. [Google Scholar] [CrossRef]
- Ankhanbaatar, U.; Sainnokhoi, T.; Khanui, B.; Ulziibat, G.; Jargalsaikhan, T.; Purevtseren, D.; Settypalli, T.B.K.; Flannery, J.; Dundon, W.G.; Basan, G.; et al. African swine fever virus genotype II in Mongolia, 2019. Transbound. Emerg. Dis. 2021, 68, 2787–2794. [Google Scholar] [CrossRef]
- Cho, K.H.; Kim, D.Y.; Jang, M.K.; Hong, S.K.; Ryu, J.H.; Kang, H.E.; Park, J.Y. Genetic Characterization of African Swine Fever Virus from Pig Farms in South Korea during Outbreaks in 2019–2021. Viruses 2022, 14, 2621. [Google Scholar] [CrossRef]
- Sun, E.; Huang, L.; Zhang, X.; Zhang, J.; Shen, D.; Zhang, Z.; Wang, Z.; Huo, H.; Wang, W.; Huangfu, H.; et al. Genotype I African swine fever viruses emerged in domestic pigs in China and caused chronic infection. Emerg. Microbes Infect. 2021, 10, 2183–2193. [Google Scholar] [CrossRef]
- Zhao, D.; Sun, E.; Huang, L.; Ding, L.; Zhu, Y.; Zhang, J.; Shen, D.; Zhang, X.; Zhang, Z.; Ren, T.; et al. Highly lethal genotype I and II recombinant African swine fever viruses detected in pigs. Nat. Commun. 2023, 14, 3096. [Google Scholar] [CrossRef]
- Bao, J.; Wang, Q.; Lin, P.; Liu, C.; Li, L.; Wu, X.; Chi, T.; Xu, T.; Ge, S.; Liu, Y.; et al. Genome comparison of African swine fever virus China/2018/AnhuiXCGQ strain and related European p72 Genotype II strains. Transbound. Emerg. Dis. 2019, 66, 1167–1176. [Google Scholar] [CrossRef]
- Hakizimana, J.N.; Yona, C.; Makange, M.R.; Kasisi, E.A.; Netherton, C.L.; Nauwynck, H.; Misinzo, G. Complete genome analysis of African swine fever virus genotypes II, IX and XV from domestic pigs in Tanzania. Sci. Rep. 2023, 13, 5318. [Google Scholar] [CrossRef]
- Okwasiimire, R.; Flint, J.F.; Kayaga, E.B.; Lakin, S.; Pierce, J.; Barrette, R.W.; Faburay, B.; Ndoboli, D.; Ekakoro, J.E.; Wampande, E.M.; et al. Whole Genome Sequencing Shows that African Swine Fever Virus Genotype IX Is Still Circulating in Domestic Pigs in All Regions of Uganda. Pathogens 2023, 12, 912. [Google Scholar] [CrossRef]
- Torresi, C.; Fiori, M.; Bertolotti, L.; Floris, M.; Colitti, B.; Giammarioli, M.; Dei Giudici, S.; Oggiano, A.; Malmberg, M.; De Mia, G.M.; et al. The evolution of African swine fever virus in Sardinia (1978–2014) as revealed by whole-genome sequencing and comparative analysis. Transbound. Emerg. Dis. 2020, 67, 1971–1980. [Google Scholar] [CrossRef]
- Bao, Y.J.; Qiu, J.; Luo, Y.; Rodriguez, F.; Qiu, H.J. The genetic variation landscape of African swine fever virus reveals frequent positive selection and adaptive flexibility. Transbound. Emerg. Dis. 2021, 68, 2703–2721. [Google Scholar] [CrossRef]
- Fiori, M.S.; Sanna, D.; Scarpa, F.; Floris, M.; Di Nardo, A.; Ferretti, L.; Loi, F.; Cappai, S.; Sechi, A.M.; Angioi, P.P.; et al. A Deeper Insight into Evolutionary Patterns and Phylogenetic History of ASFV Epidemics in Sardinia (Italy) through Extensive Genomic Sequencing. Viruses 2021, 13, 1994. [Google Scholar] [CrossRef]
- Shen, Z.J.; Jia, H.; Xie, C.D.; Shagainar, J.; Feng, Z.; Zhang, X.; Li, K.; Zhou, R. Bayesian Phylodynamic Analysis Reveals the Dispersal Patterns of African Swine Fever Virus. Viruses 2022, 14, 889. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, Q.; Zhu, Z.; Wang, S.; Tu, S.; Zhang, Y.; Zou, Y.; Liu, Y.; Liu, C.; Ren, W.; et al. Tracing the Origin of Genotype II African Swine Fever Virus in China by Genomic Epidemiology Analysis. Transbound. Emerg. Dis. 2023, 2023, 4820809. [Google Scholar] [CrossRef]
- Forth, J.H.; Calvelage, S.; Fischer, M.; Hellert, J.; Sehl-Ewert, J.; Roszyk, H.; Deutschmann, P.; Reichold, A.; Lange, M.; Thulke, H.H.; et al. African swine fever virus—variants on the rise. Emerg. Microbes Infect. 2023, 12, 2146537. [Google Scholar] [CrossRef]
- Tamas, V.; Righi, C.; Meszaros, I.; D’Errico, F.; Olasz, F.; Casciari, C.; Zadori, Z.; Magyar, T.; Petrini, S.; Feliziani, F. Involvement of the MGF 110-11L Gene in the African Swine Fever Replication and Virulence. Vaccines 2023, 11, 846. [Google Scholar] [CrossRef]
- Teklue, T.; Wang, T.; Luo, Y.; Hu, R.; Sun, Y.; Qiu, H.J. Generation and Evaluation of an African Swine Fever Virus Mutant with Deletion of the CD2v and UK Genes. Vaccines 2020, 8, 763. [Google Scholar] [CrossRef]
- Sun, E.; Zhang, Z.; Wang, Z.; He, X.; Zhang, X.; Wang, L.; Wang, W.; Huang, L.; Xi, F.; Huangfu, H.; et al. Emergence and prevalence of naturally occurring lower virulent African swine fever viruses in domestic pigs in China in 2020. Sci. China Life Sci. 2021, 64, 752–765. [Google Scholar] [CrossRef]
- Sun, Y.; Xu, Z.; Gao, H.; Xu, S.; Liu, J.; Xing, J.; Kuang, Q.; Chen, Y.; Wang, H.; Zhang, G. Detection of a Novel African Swine Fever Virus with Three Large-Fragment Deletions in Genome, China. Microbiol. Spectr. 2022, 10, e0215522. [Google Scholar] [CrossRef]
- Wylezich, C.; Calvelage, S.; Schlottau, K.; Ziegler, U.; Pohlmann, A.; Hoper, D.; Beer, M. Next-generation diagnostics: Virus capture facilitates a sensitive viral diagnosis for epizootic and zoonotic pathogens including SARS-CoV-2. Microbiome 2021, 9, 51. [Google Scholar] [CrossRef]
- Forth, J.H.; Forth, L.F.; King, J.; Groza, O.; Hubner, A.; Olesen, A.S.; Hoper, D.; Dixon, L.K.; Netherton, C.L.; Rasmussen, T.B.; et al. A Deep-Sequencing Workflow for the Fast and Efficient Generation of High-Quality African Swine Fever Virus Whole-Genome Sequences. Viruses 2019, 11, 846. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, Y.; Shi, C.; Huang, Z.; Zhang, Y.; Li, S.; Li, Y.; Ye, J.; Yu, C.; Li, Z.; et al. SOAPnuke: A MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. GigaScience 2018, 7, gix120. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H.; Ruan, J.; Durbin, R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008, 18, 1851–1858. [Google Scholar] [CrossRef]
- Lander, E.S.; Waterman, M.S. Genomic mapping by fingerprinting random clones: A mathematical analysis. Genomics 1988, 2, 231–239. [Google Scholar] [CrossRef]
- Sims, D.; Sudbery, I.; Ilott, N.E.; Heger, A.; Ponting, C.P. Sequencing depth and coverage: Key considerations in genomic analyses. Nat. Rev. Genet. 2014, 15, 121–132. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Bandelt, H.J.; Forster, P.; Rohl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.I.; Nguyen, T.T.H.; Yang, M.S.; Nga, B.T.T.; Bui, V.N.; Le, V.P.; Yi, S.W.; Kim, E.; Hur, T.Y.; Lee, H.S.; et al. Blood parameters and pathological lesions in pigs experimentally infected with Vietnam’s first isolated African swine fever virus. Front. Vet. Sci. 2022, 9, 978398. [Google Scholar] [CrossRef] [PubMed]
- Niederwerder, M.C.; Hefley, T.J. Diagnostic sensitivity of porcine biological samples for detecting African swine fever virus infection after natural consumption in feed and liquid. Transbound. Emerg. Dis. 2022, 69, 2727–2734. [Google Scholar] [CrossRef]
- Pikalo, J.; Deutschmann, P.; Fischer, M.; Roszyk, H.; Beer, M.; Blome, S. African Swine Fever Laboratory Diagnosis-Lessons Learned from Recent Animal Trials. Pathogens 2021, 10, 177. [Google Scholar] [CrossRef] [PubMed]
- Leijonhufvud, G.; Bajalan, A.; Teixeira Soratto, T.A.; Gustafsson, B.; Bogdanovic, G.; Bjerkner, A.; Allander, T.; Ljungman, G.; Andersson, B. Better detection of Torque teno virus in children with leukemia by metagenomic sequencing than by quantitative PCR. J. Med. Virol. 2022, 94, 634–641. [Google Scholar] [CrossRef] [PubMed]
- Pereira De Oliveira, R.; Lucas, P.; Chastagner, A.; De Boisseson, C.; Vial, L.; Le Potier, M.F.; Blanchard, Y. Evaluation of un-methylated DNA enrichment in sequencing of African swine fever virus complete genome. J. Virol. Methods 2020, 285, 113959. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Cheng, T.; Li, D.; Yu, X.; Chen, F.; He, Q. Low-host double MDA workflow for uncultured ASFV positive blood and serum sample sequencing. Front. Vet. Sci. 2022, 9, 936781. [Google Scholar] [CrossRef]
- Ajay, S.S.; Parker, S.C.; Abaan, H.O.; Fajardo, K.V.; Margulies, E.H. Accurate and comprehensive sequencing of personal genomes. Genome Res. 2011, 21, 1498–1505. [Google Scholar] [CrossRef]
- Yuan, J.; Zhou, X.; Xu, G.; Xu, S.; Liu, B. Genetic diversity and population structure of Tongcheng pigs in China using whole-genome SNP chip. Front. Genet. 2022, 13, 910521. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Pig | Type | Ct value | Location | Date |
|---|---|---|---|---|---|
| Group 1 | P01 | Spleen | 15.65 | Liaoning | 16 October 2018 |
| P01 | Lymph node | 15.99 | Liaoning | 16 October 2018 | |
| P02 | Spleen | 19.95 | Liaoning | 16 October 2018 | |
| P02 | Lung | 20.44 | Liaoning | 16 October 2018 | |
| P03 | Spleen | 18.55 | Liaoning | 16 October 20188 | |
| P03 | Liver | 22.60 | Liaoning | 16 October 2018 | |
| P04 | Spleen | 16.14 | Liaoning | 16 October 2018 | |
| P04 | Lymph node | 15.33 | Liaoning | 16 October 2018 | |
| P05 | Spleen | 14.73 | Liaoning | 16 October 2018 | |
| P05 | Lymph node | 15.72 | Liaoning | 16 October 2018 | |
| P06 | Spleen | 16.95 | Liaoning | 16 October 2018 | |
| P06 | Lymph node | 18.76 | Liaoning | 16 October 2018 | |
| P07 | Spleen | 18.14 | Liaoning | 16 October 2018 | |
| P07 | Lymph node | 20.58 | Liaoning | 16 October 2018 | |
| Group 2 | P08 | Spleen | 20.04 | Liaoning | 16 October 2018 |
| P09 | Spleen | 20.28 | Liaoning | 16 October 2018 | |
| P10 | Spleen | 20.30 | Liaoning | 16 October 2018 | |
| P11 | Spleen | 20.50 | Liaoning | 16 October 2018 | |
| P12 | Spleen | 20.53 | Liaoning | 16 October 2018 | |
| P13 | Spleen | 20.60 | Liaoning | 16 October 2018 | |
| P14 | Spleen | 21.10 | Liaoning | 16 October 2018 | |
| P15 | Spleen | 21.57 | Liaoning | 16 October 2018 | |
| P16 | Spleen | 22.00 | Liaoning | 16 October 2018 | |
| P17 | Spleen | 22.19 | Liaoning | 16 October 2018 | |
| P18 | Spleen | 22.48 | Liaoning | 16 October 2018 | |
| P19 | Spleen | 24.28 | Liaoning | 16 October 2018 | |
| Group 3 | P20 | Blood | 16.84 | Liaoning | 16 October 2018 |
| P21 | Blood | 17.72 | Liaoning | 16 October 2018 | |
| P22 | Blood | 18.04 | Liaoning | 16 October 2018 | |
| P23 | Blood | 18.25 | Liaoning | 16 October 2018 | |
| P24 | Blood | 18.35 | Liaoning | 16 October 2018 | |
| P25 | Blood | 18.97 | Liaoning | 16 October 2018 | |
| P26 | Blood | 21.40 | Liaoning | 16 October 2018 | |
| P27 | Blood | 25.95 | Liaoning | 16 October 2018 |
| Pig | Type | Ct | Total Reads | Swine Reads | Host% | ASFV Reads | ASFV% | Aver Depth (×) | ≥10× Coverage |
|---|---|---|---|---|---|---|---|---|---|
| P01 | Spleen | 15.65 | 19,300,794 | 19,039,879 | 98.65% | 307,725 | 1.59% | 259 | 100% |
| P01 | LN * | 15.99 | 19,753,224 | 19,482,394 | 98.63% | 85,603 | 0.43% | 75 | 99.96% |
| P02 | Spleen | 19.95 | 41,434,017 | 40,796,077 | 98.46% | 675,114 | 1.63% | 514 | 100% |
| P02 | Lung | 20.44 | 42,428,273 | 42,135,799 | 99.31% | 83,171 | 0.20% | 63 | 99.91% |
| P03 | Spleen | 18.55 | 105,357,666 | 104,789,969 | 99.46% | 254,528 | 0.24% | 232 | 99.93% |
| P03 | Liver | 22.60 | 105,531,945 | 104,924,581 | 99.42% | 42,239 | 0.04% | 12 | 56.41% |
| P04 | Spleen | 16.14 | 26,182,449 | 25,587,744 | 97.73% | 761,136 | 2.91% | 656 | 100% |
| P04 | LN | 15.33 | 22,644,180 | 22,397,705 | 98.91% | 84,216 | 0.37% | 74 | 99.99% |
| P05 | Spleen | 14.73 | 85,820,966 | 85,264,227 | 99.35% | 452,122 | 0.53% | 330 | 99.95% |
| P05 | LN | 15.72 | 101,012,071 | 100,567,586 | 99.56% | 100,290 | 0.10% | 56 | 99.77% |
| P06 | Spleen | 16.95 | 16,732,552 | 16,047,605 | 95.91% | 894,899 | 5.35% | 871 | 100% |
| P06 | LN | 18.76 | 76,822,410 | 76,034,726 | 98.97% | 802,947 | 1.05% | 607 | 100% |
| P07 | Spleen | 18.14 | 30,133,820 | 29,862,047 | 99.10% | 207,573 | 0.69% | 176 | 99.98% |
| P07 | LN | 20.58 | 62,476,949 | 62,132,807 | 99.45% | 43,865 | 0.07% | 31 | 99.19% |
| Group | Pig | Type | Ct | Total Reads | Swine Reads | Host% | ASFV Reads | ASFV% | Aver Depth (×) | ≥10× Coverage |
|---|---|---|---|---|---|---|---|---|---|---|
| Group 1 | P01 | Spleen | 15.65 | 19,300,794 | 19,039,879 | 98.65% | 307,725 | 1.59% | 259 | 100% |
| P02 | Spleen | 19.95 | 41,434,017 | 40,796,077 | 98.46% | 675,114 | 1.63% | 514 | 100% | |
| P03 | Spleen | 18.55 | 105,357,666 | 104,789,969 | 99.46% | 254,528 | 0.24% | 232 | 99.93% | |
| P04 | Spleen | 16.14 | 26,182,449 | 25,587,744 | 97.73% | 761,136 | 2.91% | 656 | 100% | |
| P05 | Spleen | 14.73 | 85,820,966 | 85,264,227 | 99.35% | 452,122 | 0.53% | 330 | 99.95% | |
| P06 | Spleen | 16.95 | 16,732,552 | 16,047,605 | 95.91% | 894,899 | 5.35% | 871 | 100% | |
| P07 | Spleen | 18.14 | 30,133,820 | 29,862,047 | 99.10% | 207,573 | 0.69% | 176 | 99.98% | |
| Group 2 | P08 | Spleen | 20.04 | 95,779,150 | 94,301,391 | 98.46% | 123,268 | 0.13% | 106 | 99.98% |
| P09 | Spleen | 20.28 | 91,428,758 | 90,842,633 | 99.36% | 223,638 | 0.24% | 160 | 99.94% | |
| P10 | Spleen | 20.30 | 76,917,858 | 76,437,533 | 99.38% | 202,494 | 0.26% | 149 | 99.85% | |
| P11 | Spleen | 20.50 | 113,506,138 | 112,655,479 | 99.25% | 98,136 | 0.09% | 73 | 99.98% | |
| P12 | Spleen | 20.53 | 39,908,094 | 39,648,842 | 99.35% | 15,519 | 0.04% | 12 | 65.69% | |
| P13 | Spleen | 20.60 | 74,872,389 | 74,307,643 | 99.25% | 150,682 | 0.20% | 104 | 99.88% | |
| P14 | Spleen | 21.10 | 38,853,338 | 38,575,461 | 99.28% | 53,950 | 0.14% | 47 | 99.89% | |
| P15 | Spleen | 21.57 | 52,842,857 | 52,245,719 | 98.87% | 20,270 | 0.04% | 15 | 78.61% | |
| P16 | Spleen | 22.00 | 57,136,899 | 56,724,036 | 99.28% | 24,979 | 0.04% | 17 | 80.78% | |
| P17 | Spleen | 22.19 | 57,246,209 | 57,032,875 | 99.63% | 38,330 | 0.07% | 22 | 97.28% | |
| P18 | Spleen | 22.48 | 39,724,759 | 39,441,332 | 99.29% | 16,451 | 0.04% | 13 | 71.31% | |
| P19 | Spleen | 24.28 | 60,321,322 | 59,628,674 | 98.85% | 12,357 | 0.02% | 9 | 33.76% |
| Pig | Type | Ct | Total Reads | Swine Reads | Host% | ASFV Reads | ASFV% | Aver Depth (×) | ≥10× Coverage |
|---|---|---|---|---|---|---|---|---|---|
| P20 | Blood | 16.84 | 43,110,963 | 42,600,180 | 98.82% | 191,101 | 0.44% | 177 | 99.99% |
| P21 | Blood | 17.72 | 53,662,886 | 53,281,393 | 99.29% | 79,969 | 0.15% | 72 | 99.97% |
| P22 | Blood | 18.04 | 45,388,551 | 45,162,234 | 99.50% | 106,663 | 0.23% | 77 | 99.92% |
| P23 | Blood | 18.25 | 87,175,246 | 86,377,934 | 99.09% | 805,588 | 0.92% | 629 | 100% |
| P24 | Blood | 18.35 | 82,171,243 | 81,563,167 | 99.26% | 441,883 | 0.54% | 362 | 99.99% |
| P25 | Blood | 18.97 | 65,916,159 | 65,526,088 | 99.41% | 150,578 | 0.23% | 114 | 99.94% |
| P26 | Blood | 21.40 | 62,907,376 | 62,631,286 | 99.56% | 18,247 | 0.03% | 11 | 54.03% |
| P27 | Blood | 25.95 | 105,264,301 | 104,625,443 | 99.39% | 43,435 | 0.04% | 5 | 8.80% |
| Site | China/LN/2018/1 | China/LN/2018/2 | Type | CDS | CDS Variation | AA Variation |
|---|---|---|---|---|---|---|
| 1382 | poly C10 | poly C8 | poly G/C | 5′ UTR MGF 360-1La | / | / |
| 9042 | C | T | SNV | MGF110-4L | G260A | R87Q |
| 14,225 | poly C13 | poly C11 | poly G/C | MGF 110-14L | G346–, G347– | G116 * (Truncation) |
| 21,794 | - | C | Insert | IGR MGF 300-1L/2R | / | / |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, C.; Wang, Q.; Liu, Y.; Wang, S.; Zhang, Y.; Liu, C.; Hu, Y.; Zheng, D.; Sun, C.; Song, F.; et al. Generation of High-Quality African Swine Fever Virus Complete Genome from Field Samples by Next-Generation Sequencing. Viruses 2024, 16, 312. https://doi.org/10.3390/v16020312
Shi C, Wang Q, Liu Y, Wang S, Zhang Y, Liu C, Hu Y, Zheng D, Sun C, Song F, et al. Generation of High-Quality African Swine Fever Virus Complete Genome from Field Samples by Next-Generation Sequencing. Viruses. 2024; 16(2):312. https://doi.org/10.3390/v16020312
Chicago/Turabian StyleShi, Chuan, Qinghua Wang, Yutian Liu, Shujuan Wang, Yongqiang Zhang, Chunju Liu, Yongxin Hu, Dongxia Zheng, Chengyou Sun, Fangfang Song, and et al. 2024. "Generation of High-Quality African Swine Fever Virus Complete Genome from Field Samples by Next-Generation Sequencing" Viruses 16, no. 2: 312. https://doi.org/10.3390/v16020312
APA StyleShi, C., Wang, Q., Liu, Y., Wang, S., Zhang, Y., Liu, C., Hu, Y., Zheng, D., Sun, C., Song, F., Yu, X., Zhao, Y., Bao, J., & Wang, Z. (2024). Generation of High-Quality African Swine Fever Virus Complete Genome from Field Samples by Next-Generation Sequencing. Viruses, 16(2), 312. https://doi.org/10.3390/v16020312