
Beyond Impairment of Virion Infectivity: New Activities of the Anti-HIV Host Cell Factor SERINC5


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction: SERINC Proteins as Antiviral Restriction Factors
2. SERINC Protein Organization and Evolution
3. Expression and Role of SERINC Proteins in HIV Pathogenesis
4. Mechanism of S5 Restriction of HIV-1 Virion Infectivity
5. Mechanism of Nef-Mediated Antagonism of S5 Restriction of HIV-1 Virion Infectivity
6. Phospholipid Scramblase Activity of SERINC Proteins
7. S5 Activities in HIV-1 Infection beyond Reducing Virion Infectivity
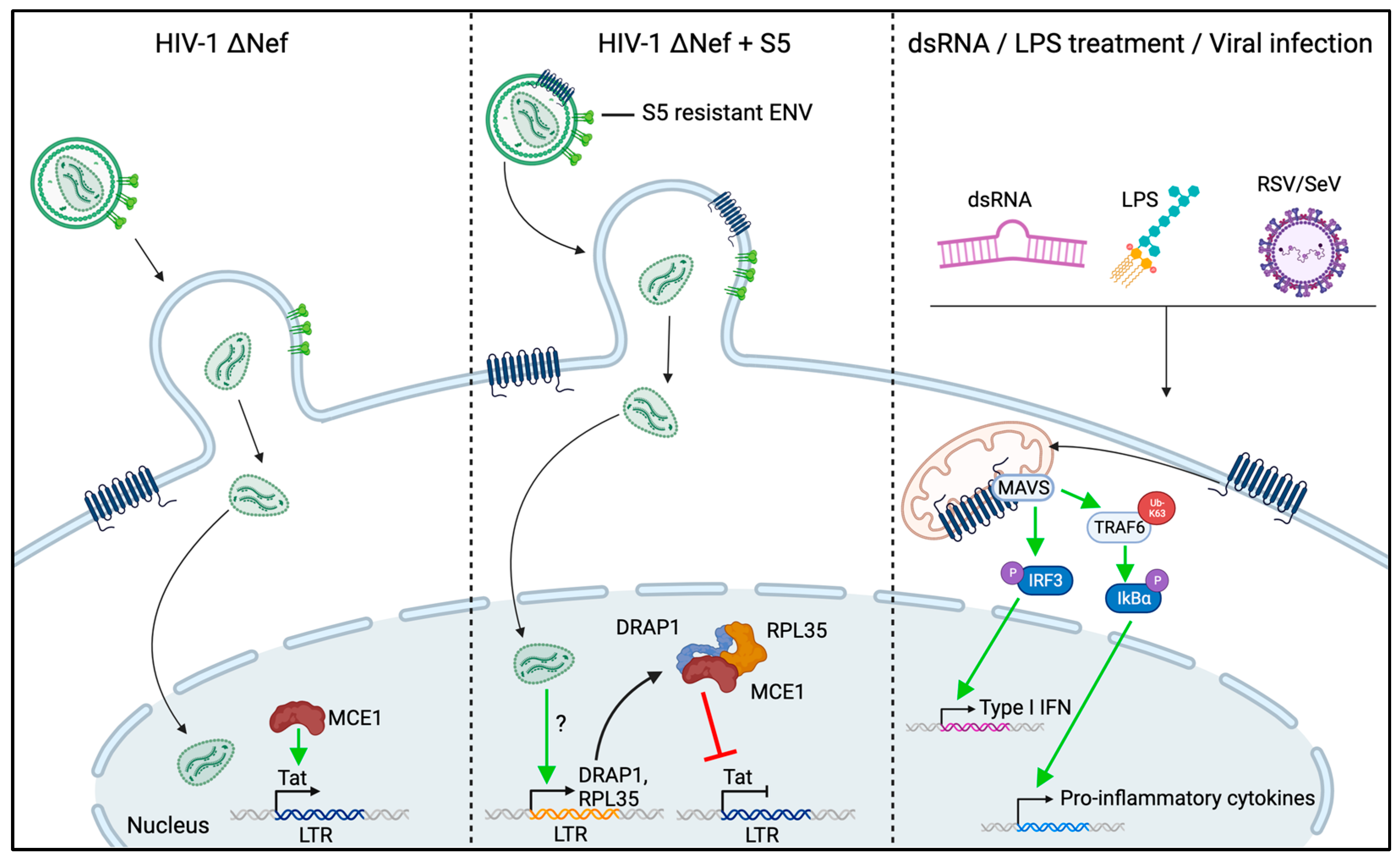
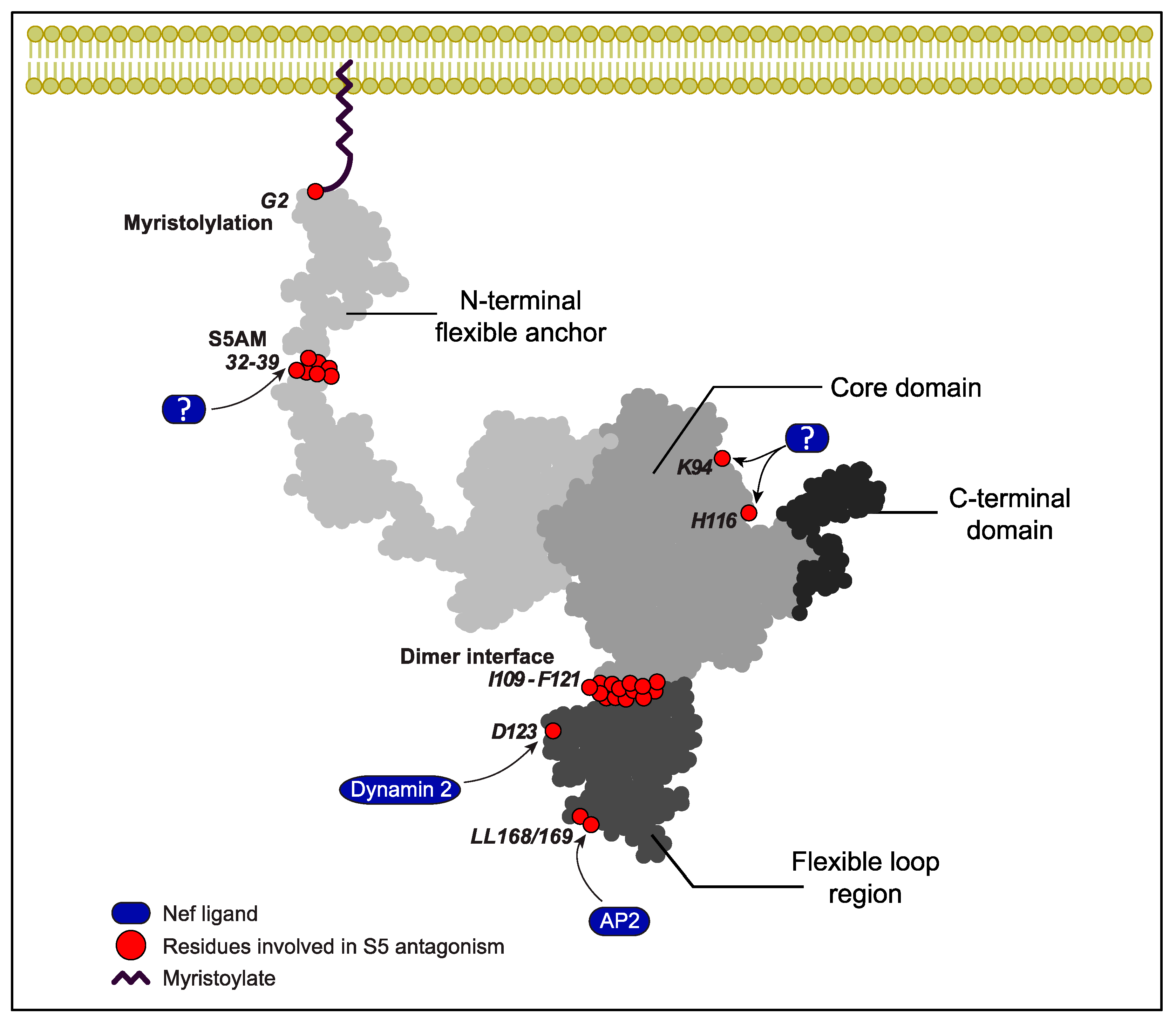
8. Conclusions and Open Questions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pollara, J.; Bonsignori, M.; Moody, M.A.; Pazgier, M.; Haynes, B.F.; Ferrari, G. Epitope specificity of human immunodeficiency virus-1 antibody dependent cellular cytotoxicity [ADCC] responses. Curr. HIV Res. 2013, 11, 378–387. [Google Scholar] [CrossRef]
- Cohen, M.S.; Shaw, G.M.; McMichael, A.J.; Haynes, B.F. Acute HIV-1 Infection. N. Engl. J. Med. 2011, 364, 1943–1954. [Google Scholar] [CrossRef]
- Kaw, S.; Ananth, S.; Tsopoulidis, N.; Morath, K.; Coban, B.M.; Hohenberger, R.; Bulut, O.C.; Klein, F.; Stolp, B.; Fackler, O.T. HIV-1 infection of CD4 T cells impairs antigen-specific B cell function. Embo J. 2020, 39, e105594. [Google Scholar] [CrossRef]
- Sáez-Cirión, A.; Manel, N. Immune Responses to Retroviruses. Annu. Rev. Immunol. 2018, 36, 193–220. [Google Scholar] [CrossRef]
- Altfeld, M.; Gale, M., Jr. Innate immunity against HIV-1 infection. Nat. Immunol. 2015, 16, 554–562. [Google Scholar] [CrossRef]
- Smed-Sörensen, A.; Loré, K. Dendritic cells at the interface of innate and adaptive immunity to HIV-1. Curr. Opin. HIV AIDS 2011, 6, 405–410. [Google Scholar] [CrossRef]
- Sauter, D.; Kirchhoff, F. Evolutionary conflicts and adverse effects of antiviral factors. eLife 2021, 10, e65243. [Google Scholar] [CrossRef]
- Kmiec, D.; Kirchhoff, F. Antiviral factors and their counteraction by HIV-1: Many uncovered and more to be discovered. J. Mol. Cell Biol. 2024. [Google Scholar] [CrossRef]
- Neil, S.J.; Zang, T.; Bieniasz, P.D. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 2008, 451, 425–430. [Google Scholar] [CrossRef]
- Harris, R.S.; Bishop, K.N.; Sheehy, A.M.; Craig, H.M.; Petersen-Mahrt, S.K.; Watt, I.N.; Neuberger, M.S.; Malim, M.H. DNA deamination mediates innate immunity to retroviral infection. Cell 2003, 113, 803–809. [Google Scholar] [CrossRef]
- Chougui, G.; Munir-Matloob, S.; Matkovic, R.; Martin, M.M.; Morel, M.; Lahouassa, H.; Leduc, M.; Ramirez, B.C.; Etienne, L.; Margottin-Goguet, F. HIV-2/SIV viral protein X counteracts HUSH repressor complex. Nat. Microbiol. 2018, 3, 891–897. [Google Scholar] [CrossRef]
- Yurkovetskiy, L.; Guney, M.H.; Kim, K.; Goh, S.L.; McCauley, S.; Dauphin, A.; Diehl, W.E.; Luban, J. Primate immunodeficiency virus proteins Vpx and Vpr counteract transcriptional repression of proviruses by the HUSH complex. Nat. Microbiol. 2018, 3, 1354–1361. [Google Scholar] [CrossRef]
- Geyer, M.; Fackler, O.T.; Peterlin, B.M. Structure–function relationships in HIV-1 Nef. EMBO Rep. 2001, 2, 580–585. [Google Scholar] [CrossRef]
- Buffalo, C.Z.; Iwamoto, Y.; Hurley, J.H.; Ren, X. How HIV Nef Proteins Hijack Membrane Traffic To Promote Infection. J. Virol. 2019, 93. [Google Scholar] [CrossRef]
- Rosa, A.; Chande, A.; Ziglio, S.; De Sanctis, V.; Bertorelli, R.; Goh, S.L.; McCauley, S.M.; Nowosielska, A.; Antonarakis, S.E.; Luban, J.; et al. HIV-1 Nef promotes infection by excluding SERINC5 from virion incorporation. Nature 2015, 526, 212–217. [Google Scholar] [CrossRef]
- Usami, Y.; Wu, Y.; Gottlinger, H.G. SERINC3 and SERINC5 restrict HIV-1 infectivity and are counteracted by Nef. Nature 2015, 526, 218–223. [Google Scholar] [CrossRef]
- Fackler, O.T. Spotlight on HIV-1 Nef: SERINC3 and SERINC5 Identified as Restriction Factors Antagonized by the Pathogenesis Factor. Viruses 2015, 7, 6730–6738. [Google Scholar] [CrossRef]
- Firrito, C.; Bertelli, C.; Vanzo, T.; Chande, A.; Pizzato, M. SERINC5 as a New Restriction Factor for Human Immunodeficiency Virus and Murine Leukemia Virus. Annu. Rev. Virol. 2018, 5, 323–340. [Google Scholar] [CrossRef]
- Heigele, A.; Kmiec, D.; Regensburger, K.; Langer, S.; Peiffer, L.; Sturzel, C.M.; Sauter, D.; Peeters, M.; Pizzato, M.; Learn, G.H.; et al. The Potency of Nef-Mediated SERINC5 Antagonism Correlates with the Prevalence of Primate Lentiviruses in the Wild. Cell Host Microbe 2016, 20, 381–391. [Google Scholar] [CrossRef]
- Ahi, Y.S.; Zhang, S.; Thappeta, Y.; Denman, A.; Feizpour, A.; Gummuluru, S.; Reinhard, B.; Muriaux, D.; Fivash, M.J.; Rein, A. Functional Interplay between Murine Leukemia Virus Glycogag, Serinc5, and Surface Glycoprotein Governs Virus Entry, with Opposite Effects on Gammaretroviral and Ebolavirus Glycoproteins. mBio 2016, 7, e01985-16. [Google Scholar] [CrossRef]
- Chande, A.; Cuccurullo, E.C.; Rosa, A.; Ziglio, S.; Carpenter, S.; Pizzato, M. S2 from equine infectious anemia virus is an infectivity factor which counteracts the retroviral inhibitors SERINC5 and SERINC3. Proc. Natl. Acad. Sci. USA 2016, 113, 13197–13202. [Google Scholar] [CrossRef]
- Cano-Ortiz, L.; Gu, Q.; de Sousa-Pereira, P.; Zhang, Z.; Chiapella, C.; Twizerimana, A.P.; Lin, C.; Franco, A.C.; VandeWoude, S.; Luedde, T.; et al. Feline Leukemia Virus-B Envelope Together With its GlycoGag and Human Immunodeficiency Virus-1 Nef Mediate Resistance to Feline SERINC5. J. Mol. Biol. 2022, 434, 167421. [Google Scholar] [CrossRef]
- Zhao, F.; Xu, F.; Liu, X.; Hu, Y.; Wei, L.; Fan, Z.; Wang, L.; Huang, Y.; Mei, S.; Guo, L.; et al. SERINC5 restricts influenza virus infectivity. PLoS Pathog. 2022, 18, e1010907. [Google Scholar] [CrossRef]
- Lai, K.K.; Munro, J.B.; Shi, G.; Majdoul, S.; Compton, A.A.; Rein, A. Restriction of Influenza A Virus by SERINC5. mBio 2022, 13, e02923-22. [Google Scholar] [CrossRef]
- Li, W.; Zhang, Z.; Zhang, L.; Li, H.; Fan, S.; Zhu, E.; Fan, J.; Li, Z.; Chen, W.; Yi, L.; et al. Antiviral Role of Serine Incorporator 5 (SERINC5) Proteins in Classical Swine Fever Virus Infection. Front. Microbiol. 2020, 11, 580233. [Google Scholar] [CrossRef]
- Timilsina, U.; Umthong, S.; Ivey, E.B.; Waxman, B.; Stavrou, S. SARS-CoV-2 ORF7a potently inhibits the antiviral effect of the host factor SERINC5. Nat. Commun. 2022, 13, 2935. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, H.; Zhang, J.; Yang, J.; Bai, L.; Zheng, B.; Zheng, T.; Wang, Y.; Li, J.; Zhang, W. SERINC5 Inhibits the Secretion of Complete and Genome-Free Hepatitis B Virions Through Interfering With the Glycosylation of the HBV Envelope. Front. Microbiol. 2020, 11, 697. [Google Scholar] [CrossRef]
- Timilsina, U.; Stavrou, S. SERINC5: One antiviral factor to bind them all. PLoS Pathog. 2023, 19, e1011076. [Google Scholar] [CrossRef]
- Inuzuka, M.; Hayakawa, M.; Ingi, T. Serinc, an activity-regulated protein family, incorporates serine into membrane lipid synthesis. J. Biol. Chem. 2005, 280, 35776–35783. [Google Scholar] [CrossRef]
- Pye, V.E.; Rosa, A.; Bertelli, C.; Struwe, W.B.; Maslen, S.L.; Corey, R.; Liko, I.; Hassall, M.; Mattiuzzo, G.; Ballandras-Colas, A.; et al. A bipartite structural organization defines the SERINC family of HIV-1 restriction factors. Nat. Struct. Mol. Biol. 2020, 27, 78–83. [Google Scholar] [CrossRef]
- Cano-Ortiz, L.; Luedde, T.; Münk, C. HIV-1 restriction by SERINC5. Med. Microbiol. Immunol. 2023, 212, 133–140. [Google Scholar] [CrossRef]
- Schulte, B.; Selyutina, A.; Opp, S.; Herschhorn, A.; Sodroski, J.G.; Pizzato, M.; Diaz-Griffero, F. Localization to detergent-resistant membranes and HIV-1 core entry inhibition correlate with HIV-1 restriction by SERINC5. Virology 2018, 515, 52–65. [Google Scholar] [CrossRef]
- Ramdas, P.; Bhardwaj, V.; Singh, A.; Vijay, N.; Chande, A. Coelacanth SERINC2 Inhibits HIV-1 Infectivity and Is Counteracted by Envelope Glycoprotein from Foamy Virus. J. Virol. 2021, 95, e0022921. [Google Scholar] [CrossRef]
- de Sousa-Pereira, P.; Abrantes, J.; Bauernfried, S.; Pierini, V.; Esteves, P.J.; Keppler, O.T.; Pizzato, M.; Hornung, V.; Fackler, O.T.; Baldauf, H.M. The antiviral activity of rodent and lagomorph SERINC3 and SERINC5 is counteracted by known viral antagonists. J. Gen. Virol. 2019, 100, 278–288. [Google Scholar] [CrossRef]
- Murrell, B.; Vollbrecht, T.; Guatelli, J.; Wertheim, J.O. The Evolutionary Histories of Antiretroviral Proteins SERINC3 and SERINC5 Do Not Support an Evolutionary Arms Race in Primates. J. Virol. 2016, 90, 8085–8089. [Google Scholar] [CrossRef]
- Timilsina, U.; Umthong, S.; Lynch, B.; Stablewski, A.; Stavrou, S. SERINC5 Potently Restricts Retrovirus Infection In Vivo. mBio 2020, 11, e00588-20. [Google Scholar] [CrossRef]
- Janaka, S.K.; Palumbo, A.V.; Tavakoli-Tameh, A.; Evans, D.T. Selective Disruption of SERINC5 Antagonism by Nef Impairs SIV Replication in Primary CD4(+) T Cells. J. Virol. 2021, 95, e01911-20. [Google Scholar] [CrossRef]
- Kruize, Z.; van Nuenen, A.C.; van Wijk, S.W.; Girigorie, A.F.; van Dort, K.A.; Booiman, T.; Kootstra, N.A. Nef Obtained from Individuals with HIV-1 Vary in Their Ability to Antagonize SERINC3- and SERINC5-Mediated HIV-1 Restriction. Viruses 2021, 13, 423. [Google Scholar] [CrossRef]
- Toyoda, M.; Kamori, D.; Tan, T.S.; Goebuchi, K.; Ohashi, J.; Carlson, J.; Kawana-Tachikawa, A.; Gatanaga, H.; Oka, S.; Pizzato, M.; et al. Impaired ability of Nef to counteract SERINC5 is associated with reduced plasma viremia in HIV-infected individuals. Sci. Rep. 2020, 10, 19416. [Google Scholar] [CrossRef]
- Jin, S.W.; Alsahafi, N.; Kuang, X.T.; Swann, S.A.; Toyoda, M.; Gottlinger, H.; Walker, B.D.; Ueno, T.; Finzi, A.; Brumme, Z.L.; et al. Natural HIV-1 Nef Polymorphisms Impair SERINC5 Downregulation Activity. Cell Rep. 2019, 29, 1449–1457.e5. [Google Scholar] [CrossRef]
- Naicker, D.; Sonela, N.; Jin, S.W.; Mulaudzi, T.; Ojwach, D.; Reddy, T.; Brockman, M.A.; Brumme, Z.L.; Ndung’u, T.; Mann, J.K. HIV-1 subtype C Nef-mediated SERINC5 down-regulation significantly contributes to overall Nef activity. Retrovirology 2023, 20, 3. [Google Scholar] [CrossRef]
- Hernández-López, E.G.; González-Enríquez, G.V.; Torres-Mendoza, B.M.; Cárdenas-Bedoya, J.; Escoto-Delgadillo, M.; Vázquez-Valls, E.; Pérez-Ríos, A.M.; Carbajal-Uribe, D.A.; Rincón-Sánchez, A.R. Downregulation of SERINC5 expression in buffy coats of HIV-1-infected patients with detectable or undetectable viral load. Mol. Biol. Rep. 2021, 48, 4247–4252. [Google Scholar] [CrossRef]
- Wu, Y.; Olety, B.; Weiss, E.R.; Popova, E.; Yamanaka, H.; Gottlinger, H. Potent Enhancement of HIV-1 Replication by Nef in the Absence of SERINC3 and SERINC5. mBio 2019, 10, e01071-19. [Google Scholar] [CrossRef]
- Ramirez, P.W.; Vollbrecht, T.; Acosta, F.M.; Suarez, M.; Angerstein, A.O.; Wallace, J.; RM, O.C.; Guatelli, J. Nef enhances HIV-1 replication and infectivity independently of SERINC5 in CEM T cells. Virology 2023, 578, 154–162. [Google Scholar] [CrossRef]
- Kmiec, D.; Akbil, B.; Ananth, S.; Hotter, D.; Sparrer, K.M.J.; Sturzel, C.M.; Trautz, B.; Ayouba, A.; Peeters, M.; Yao, Z.; et al. SIVcol Nef counteracts SERINC5 by promoting its proteasomal degradation but does not efficiently enhance HIV-1 replication in human CD4+ T cells and lymphoid tissue. PLoS Pathog. 2018, 14, e1007269. [Google Scholar] [CrossRef]
- Passos, V.; Zillinger, T.; Casartelli, N.; Wachs, A.S.; Xu, S.; Malassa, A.; Steppich, K.; Schilling, H.; Franz, S.; Todt, D.; et al. Characterization of Endogenous SERINC5 Protein as Anti-HIV-1 Factor. J. Virol. 2019, 93. [Google Scholar] [CrossRef]
- Zutz, A.; Scholz, C.; Schneider, S.; Pierini, V.; Munchhoff, M.; Sutter, K.; Wittmann, G.; Dittmer, U.; Draenert, R.; Bogner, J.R.; et al. SERINC5 Is an Unconventional HIV Restriction Factor That Is Upregulated during Myeloid Cell Differentiation. J. Innate Immun. 2020, 12, 399–409. [Google Scholar] [CrossRef]
- Ward, A.E.; Kiessling, V.; Pornillos, O.; White, J.M.; Ganser-Pornillos, B.K.; Tamm, L.K. HIV-cell membrane fusion intermediates are restricted by Serincs as revealed by cryo-electron and TIRF microscopy. J. Biol. Chem. 2020, 295, 15183–15195. [Google Scholar] [CrossRef]
- Sood, C.; Marin, M.; Chande, A.; Pizzato, M.; Melikyan, G.B. SERINC5 protein inhibits HIV-1 fusion pore formation by promoting functional inactivation of envelope glycoproteins. J. Biol. Chem. 2017, 292, 6014–6026. [Google Scholar] [CrossRef]
- Chen, Y.C.; Sood, C.; Marin, M.; Aaron, J.; Gratton, E.; Salaita, K.; Melikyan, G.B. Super-Resolution Fluorescence Imaging Reveals That Serine Incorporator Protein 5 Inhibits Human Immunodeficiency Virus Fusion by Disrupting Envelope Glycoprotein Clusters. ACS Nano 2020, 14, 10929–10943. [Google Scholar] [CrossRef]
- Raghunath, G.; Chen, Y.C.; Marin, M.; Wu, H.; Melikyan, G.B. SERINC5-Mediated Restriction of HIV-1 Infectivity Correlates with Resistance to Cholesterol Extraction but Not with Lipid Order of Viral Membrane. Viruses 2022, 14, 1636. [Google Scholar] [CrossRef]
- Beitari, S.; Ding, S.; Pan, Q.; Finzi, A.; Liang, C. Effect of HIV-1 Env on SERINC5 Antagonism. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Kirschman, J.; Marin, M.; Chen, Y.C.; Chen, J.; Herschhorn, A.; Smith, A.B., 3rd; Melikyan, G.B. SERINC5 Restricts HIV-1 Infectivity by Promoting Conformational Changes and Accelerating Functional Inactivation of Env. Viruses 2022, 14, 1388. [Google Scholar] [CrossRef]
- Staropoli, I.; Dufloo, J.; Ducher, A.; Commere, P.H.; Sartori-Rupp, A.; Novault, S.; Bruel, T.; Lorin, V.; Mouquet, H.; Schwartz, O.; et al. Flow Cytometry Analysis of HIV-1 Env Conformations at the Surface of Infected Cells and Virions: Role of Nef, CD4, and SERINC5. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- Featherstone, A.; Aiken, C. SERINC5 Inhibits HIV-1 Infectivity by Altering the Conformation of gp120 on HIV-1 Particles. J. Virol. 2020, 94, e00594-20. [Google Scholar] [CrossRef]
- Zhang, X.; Shi, J.; Qiu, X.; Chai, Q.; Frabutt, D.A.; Schwartz, R.C.; Zheng, Y.H. CD4 Expression and Env Conformation Are Critical for HIV-1 Restriction by SERINC5. J. Virol. 2019, 93, e00544-19. [Google Scholar] [CrossRef]
- Usami, Y.; Gottlinger, H. HIV-1 Nef responsiveness is determined by Env variable regions involved in trimer association and correlates with neutralization sensitivity. Cell Rep. 2013, 5, 802–812. [Google Scholar] [CrossRef]
- Haider, T.; Snetkov, X.; Jolly, C. HIV envelope tail truncation confers resistance to SERINC5 restriction. Proc. Natl. Acad. Sci. USA 2021, 118, e2101450118. [Google Scholar] [CrossRef]
- Nkuwi, E.; Judicate, G.P.; Tan, T.S.; Barabona, G.; Toyoda, M.; Sunguya, B.; Kamori, D.; Ueno, T. Relative resistance of patient-derived envelope sequences to SERINC5-mediated restriction of HIV-1 infectivity. J. Virol. 2023, 97, e0082323. [Google Scholar] [CrossRef]
- Diehl, W.E.; Guney, M.H.; Vanzo, T.; Kyawe, P.P.; White, J.M.; Pizzato, M.; Luban, J. Influence of Different Glycoproteins and of the Virion Core on SERINC5 Antiviral Activity. Viruses 2021, 13, 1279. [Google Scholar] [CrossRef]
- Tan, T.S.; Toyoda, M.; Tokunaga, K.; Ueno, T. Aromatic Side Chain at Position 412 of SERINC5 Exerts Restriction Activity toward HIV-1 and Other Retroviruses. J. Virol. 2021, 95, e00634-21. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, I.; Li, S.; Li, R.; Chai, Q.; Zhang, L.; Wang, B.; Yu, C.; Zheng, Y.H. The retroviral accessory proteins S2, Nef, and glycoMA use similar mechanisms for antagonizing the host restriction factor SERINC5. J. Biol. Chem. 2019, 294, 7013–7024. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Ahmad, I.; Shi, J.; Wang, B.; Yu, C.; Zhang, L.; Zheng, Y.H. Murine Leukemia Virus Glycosylated Gag Reduces Murine SERINC5 Protein Expression at Steady-State Levels via the Endosome/Lysosome Pathway to Counteract SERINC5 Antiretroviral Activity. J. Virol. 2019, 93, e01651-18. [Google Scholar] [CrossRef] [PubMed]
- Pereira, E.A.; daSilva, L.L. HIV-1 Nef: Taking Control of Protein Trafficking. Traffic 2016, 17, 976–996. [Google Scholar] [CrossRef] [PubMed]
- Haller, C.; Muller, B.; Fritz, J.V.; Lamas-Murua, M.; Stolp, B.; Pujol, F.M.; Keppler, O.T.; Fackler, O.T. HIV-1 Nef and Vpu are functionally redundant broad-spectrum modulators of cell surface receptors, including tetraspanins. J. Virol. 2014, 88, 14241–14257. [Google Scholar] [CrossRef] [PubMed]
- Matheson, N.J.; Sumner, J.; Wals, K.; Rapiteanu, R.; Weekes, M.P.; Vigan, R.; Weinelt, J.; Schindler, M.; Antrobus, R.; Costa, A.S.; et al. Cell Surface Proteomic Map of HIV Infection Reveals Antagonism of Amino Acid Metabolism by Vpu and Nef. Cell Host Microbe 2015, 18, 409–423. [Google Scholar] [CrossRef]
- Xu, S.; Zheng, Z.; Pathak, J.L.; Cheng, H.; Zhou, Z.; Chen, Y.; Wu, Q.; Wang, L.; Zeng, M.; Wu, L. The Emerging Role of the Serine Incorporator Protein Family in Regulating Viral Infection. Front. Cell Dev. Biol. 2022, 10, 856468. [Google Scholar] [CrossRef]
- Trautz, B.; Pierini, V.; Wombacher, R.; Stolp, B.; Chase, A.J.; Pizzato, M.; Fackler, O.T. The Antagonism of HIV-1 Nef to SERINC5 Particle Infectivity Restriction Involves the Counteraction of Virion-Associated Pools of the Restriction Factor. J. Virol. 2016, 90, 10915–10927. [Google Scholar] [CrossRef]
- Welker, R.; Kottler, H.; Kalbitzer, H.R.; Krausslich, H.G. Human immunodeficiency virus type 1 Nef protein is incorporated into virus particles and specifically cleaved by the viral proteinase. Virology 1996, 219, 228–236. [Google Scholar] [CrossRef]
- Schorr, J.; Kellner, R.; Fackler, O.; Freund, J.; Konvalinka, J.; Kienzle, N.; Krausslich, H.G.; Mueller-Lantzsch, N.; Kalbitzer, H.R. Specific cleavage sites of Nef proteins from human immunodeficiency virus types 1 and 2 for the viral proteases. J. Virol. 1996, 70, 9051–9054. [Google Scholar] [CrossRef]
- Firrito, C.; Bertelli, C.; Rosa, A.; Chande, A.; Ananth, S.; van Dijk, H.; Fackler, O.T.; Stoneham, C.; Singh, R.; Guatelli, J.; et al. A Conserved Acidic Residue in the C-Terminal Flexible Loop of HIV-1 Nef Contributes to the Activity of SERINC5 and CD4 Downregulation. Viruses 2023, 15, 652. [Google Scholar] [CrossRef]
- Staudt, R.P.; Smithgall, T.E. Nef homodimers down-regulate SERINC5 by AP-2-mediated endocytosis to promote HIV-1 infectivity. J. Biol. Chem. 2020, 295, 15540–15552. [Google Scholar] [CrossRef]
- Obermaier, B.; Ananth, S.; Tibroni, N.; Pierini, V.; Shytaj, I.L.; Diaz, R.S.; Lusic, M.; Fackler, O.T. Patient-Derived HIV-1 Nef Alleles Reveal Uncoupling of CD4 Downregulation and SERINC5 Antagonism Functions of the Viral Pathogenesis Factor. J. Acquir. Immune Defic. Syndr. (1999) 2020, 85, e23–e26. [Google Scholar] [CrossRef]
- Mumby, M.J.; Johnson, A.L.; Trothen, S.M.; Edgar, C.R.; Gibson, R.; Stathopulos, P.B.; Arts, E.J.; Dikeakos, J.D. An Amino Acid Polymorphism within the HIV-1 Nef Dileucine Motif Functionally Uncouples Cell Surface CD4 and SERINC5 Downregulation. J. Virol. 2021, 95, e00588-21. [Google Scholar] [CrossRef]
- Dai, W.; Usami, Y.; Wu, Y.; Gottlinger, H. A Long Cytoplasmic Loop Governs the Sensitivity of the Anti-viral Host Protein SERINC5 to HIV-1 Nef. Cell Rep. 2018, 22, 869–875. [Google Scholar] [CrossRef]
- Stoneham, C.A.; Ramirez, P.W.; Singh, R.; Suarez, M.; Debray, A.; Lim, C.; Jia, X.; Xiong, Y.; Guatelli, J. A Conserved Acidic-Cluster Motif in SERINC5 Confers Partial Resistance to Antagonism by HIV-1 Nef. J. Virol. 2020, 94, e01554-19. [Google Scholar] [CrossRef]
- Chai, Q.; Li, S.; Collins, M.K.; Li, R.; Ahmad, I.; Johnson, S.F.; Frabutt, D.A.; Yang, Z.; Shen, X.; Sun, L.; et al. HIV-1 Nef interacts with the cyclin K/CDK13 complex to antagonize SERINC5 for optimal viral infectivity. Cell Rep. 2021, 36, 109514. [Google Scholar] [CrossRef]
- Li, S.; Li, R.; Ahmad, I.; Liu, X.; Johnson, S.F.; Sun, L.; Zheng, Y.H. Cul3-KLHL20 E3 ubiquitin ligase plays a key role in the arms race between HIV-1 Nef and host SERINC5 restriction. Nat. Commun. 2022, 13, 2242. [Google Scholar] [CrossRef] [PubMed]
- Baur, A.S.; Sass, G.; Laffert, B.; Willbold, D.; Cheng-Mayer, C.; Peterlin, B.M. The N-terminus of Nef from HIV-1/SIV associates with a protein complex containing Lck and a serine kinase. Immunity 1997, 6, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Ananth, S.; Morath, K.; Trautz, B.; Tibroni, N.; Shytaj, I.L.; Obermaier, B.; Stolp, B.; Lusic, M.; Fackler, O.T. Multi-functional roles of the N-terminal region of HIV-1SF2Nef are mediated by three independent protein interaction sites. J. Virol. 2019, 94, e01398-19. [Google Scholar] [CrossRef] [PubMed]
- Brugger, B.; Glass, B.; Haberkant, P.; Leibrecht, I.; Wieland, F.T.; Krausslich, H.G. The HIV lipidome: A raft with an unusual composition. Proc. Natl. Acad. Sci. USA 2006, 103, 2641–2646. [Google Scholar] [CrossRef] [PubMed]
- Trautz, B.; Wiedemann, H.; Luchtenborg, C.; Pierini, V.; Kranich, J.; Glass, B.; Krausslich, H.G.; Brocker, T.; Pizzato, M.; Ruggieri, A.; et al. The host-cell restriction factor SERINC5 restricts HIV-1 infectivity without altering the lipid composition and organization of viral particles. J. Biol. Chem. 2017, 292, 13702–13713. [Google Scholar] [CrossRef] [PubMed]
- Chu, E.P.; Elso, C.M.; Pollock, A.H.; Alsayb, M.A.; Mackin, L.; Thomas, H.E.; Kay, T.W.; Silveira, P.A.; Mansell, A.S.; Gaus, K.; et al. Disruption of Serinc1, which facilitates serine-derived lipid synthesis, fails to alter macrophage function, lymphocyte proliferation or autoimmune disease susceptibility. Mol. Immunol. 2017, 82, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Leonhardt, S.A.; Purdy, M.D.; Grover, J.R.; Yang, Z.; Poulos, S.; McIntire, W.E.; Tatham, E.A.; Erramilli, S.K.; Nosol, K.; Lai, K.K.; et al. Antiviral HIV-1 SERINC restriction factors disrupt virus membrane asymmetry. Nat. Commun. 2023, 14, 4368. [Google Scholar] [CrossRef]
- Ward, A.E.; Sokovikova, D.; Waxham, M.N.; Heberle, F.A.; Levental, I.; Levental, K.R.; Kiessling, V.; White, J.M.; Tamm, L.K. Serinc5 Restricts HIV Membrane Fusion by Altering Lipid Order and Heterogeneity in the Viral Membrane. ACS Infect. Dis. 2023, 9, 773–784. [Google Scholar] [CrossRef]
- Pandori, M.W.; Fitch, N.J.; Craig, H.M.; Richman, D.D.; Spina, C.A.; Guatelli, J.C. Producer-cell modification of human immunodeficiency virus type 1: Nef is a virion protein. J. Virol. 1996, 70, 4283–4290. [Google Scholar] [CrossRef]
- Zeng, C.; Waheed, A.A.; Li, T.; Yu, J.; Zheng, Y.M.; Yount, J.S.; Wen, H.; Freed, E.O.; Liu, S.L. SERINC proteins potentiate antiviral type I IFN production and proinflammatory signaling pathways. Sci. Signal 2021, 14, eabc7611. [Google Scholar] [CrossRef] [PubMed]
- Pertel, T.; Hausmann, S.; Morger, D.; Zuger, S.; Guerra, J.; Lascano, J.; Reinhard, C.; Santoni, F.A.; Uchil, P.D.; Chatel, L.; et al. TRIM5 is an innate immune sensor for the retrovirus capsid lattice. Nature 2011, 472, 361–365. [Google Scholar] [CrossRef]
- Galao, R.P.; Le Tortorec, A.; Pickering, S.; Kueck, T.; Neil, S.J. Innate sensing of HIV-1 assembly by Tetherin induces NFkappaB-dependent proinflammatory responses. Cell Host Microbe 2012, 12, 633–644. [Google Scholar] [CrossRef]
- Pierini, V.; Gallucci, L.; Stürzel, C.M.; Kirchhoff, F.; Fackler, O.T. SERINC5 Can Enhance Proinflammatory Cytokine Production by Primary Human Myeloid Cells in Response to Challenge with HIV-1 Particles. J. Virol. 2021, 95, e02372-20. [Google Scholar] [CrossRef]
- Shi, Y.; Simpson, S.; Ahmed, S.K.; Chen, Y.; Tavakoli-Tameh, A.; Janaka, S.K.; Evans, D.T.; Serra-Moreno, R. The Antiviral Factor SERINC5 Impairs the Expression of Non-Self-DNA. Viruses 2023, 15, 1961. [Google Scholar] [CrossRef]
- Ramdas, P.; Chande, A. SERINC5 Mediates a Postintegration Block to HIV-1 Gene Expression in Macrophages. mBio 2023, 14, e0016623. [Google Scholar] [CrossRef] [PubMed]
- Chiu, Y.L.; Coronel, E.; Ho, C.K.; Shuman, S.; Rana, T.M. HIV-1 Tat protein interacts with mammalian capping enzyme and stimulates capping of TAR RNA. J. Biol. Chem. 2001, 276, 12959–12966. [Google Scholar] [CrossRef] [PubMed]
- Bracq, L.; Xie, M.; Benichou, S.; Bouchet, J. Mechanisms for Cell-to-Cell Transmission of HIV-1. Front. Immunol. 2018, 9, 260. [Google Scholar] [CrossRef] [PubMed]
- 3D Human Tissue Models for HIV Working Group. 3D human tissue models and microphysiological systems for HIV and related comorbidities. Trends Biotechnol. 2023. [Google Scholar] [CrossRef]
- Calado, M.; Pires, D.; Conceição, C.; Santos-Costa, Q.; Anes, E.; Azevedo-Pereira, J.M. Human immunodeficiency virus transmission-Mechanisms underlying the cell-to-cell spread of human immunodeficiency virus. Rev. Med. Virol. 2023, 33, e2480. [Google Scholar] [CrossRef]
- Imle, A.; Kumberger, P.; Schnellbacher, N.D.; Fehr, J.; Carrillo-Bustamante, P.; Ales, J.; Schmidt, P.; Ritter, C.; Godinez, W.J.; Muller, B.; et al. Experimental and computational analyses reveal that environmental restrictions shape HIV-1 spread in 3D cultures. Nat. Commun. 2019, 10, 2144. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sid Ahmed, S.; Bajak, K.; Fackler, O.T. Beyond Impairment of Virion Infectivity: New Activities of the Anti-HIV Host Cell Factor SERINC5. Viruses 2024, 16, 284. https://doi.org/10.3390/v16020284
Sid Ahmed S, Bajak K, Fackler OT. Beyond Impairment of Virion Infectivity: New Activities of the Anti-HIV Host Cell Factor SERINC5. Viruses. 2024; 16(2):284. https://doi.org/10.3390/v16020284
Chicago/Turabian StyleSid Ahmed, Samy, Kathrin Bajak, and Oliver T. Fackler. 2024. "Beyond Impairment of Virion Infectivity: New Activities of the Anti-HIV Host Cell Factor SERINC5" Viruses 16, no. 2: 284. https://doi.org/10.3390/v16020284
APA StyleSid Ahmed, S., Bajak, K., & Fackler, O. T. (2024). Beyond Impairment of Virion Infectivity: New Activities of the Anti-HIV Host Cell Factor SERINC5. Viruses, 16(2), 284. https://doi.org/10.3390/v16020284