
Herpes Simplex Virus Type 2 Blocks IFN-β Production through the Viral UL24 N-Terminal Domain-Mediated Inhibition of IRF-3 Phosphorylation


 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Viruses, Cell Lines, and Abs
2.2. Plasmid Constructions
2.3. UL24 Knockdown
2.4. Cell Viability Assay
2.5. Luciferase Reporter Assay
2.6. RNA Extraction, Reverse Transcription, and Real-Time PCR
2.7. ELISA for IFN-β
2.8. Western Blot Analysis
2.9. Flow Cytometry Analysis (FCA)
2.10. Statistical Analysis
3. Results
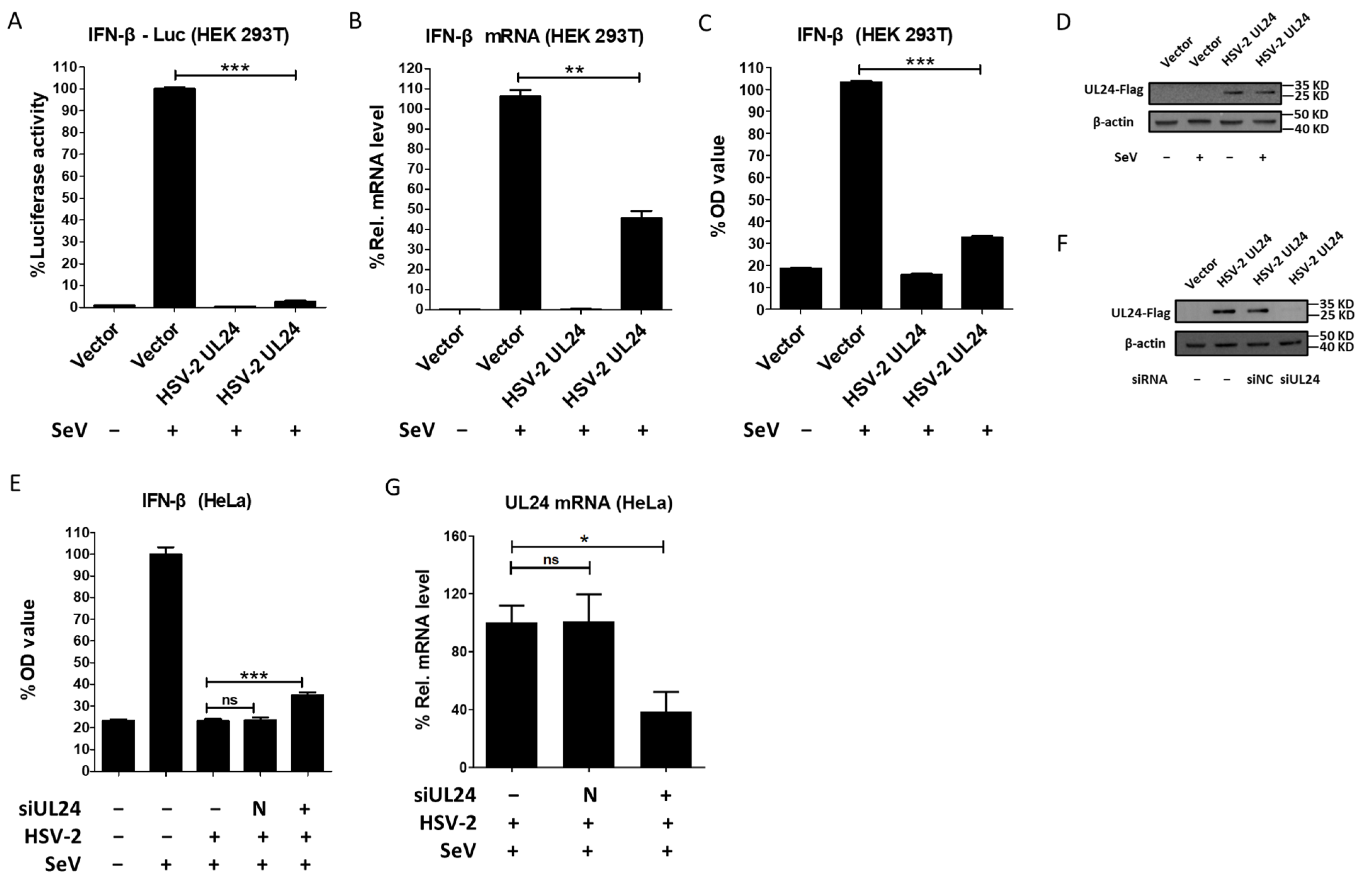
3.1. HSV-2 UL24 Inhibits IFN-β Production
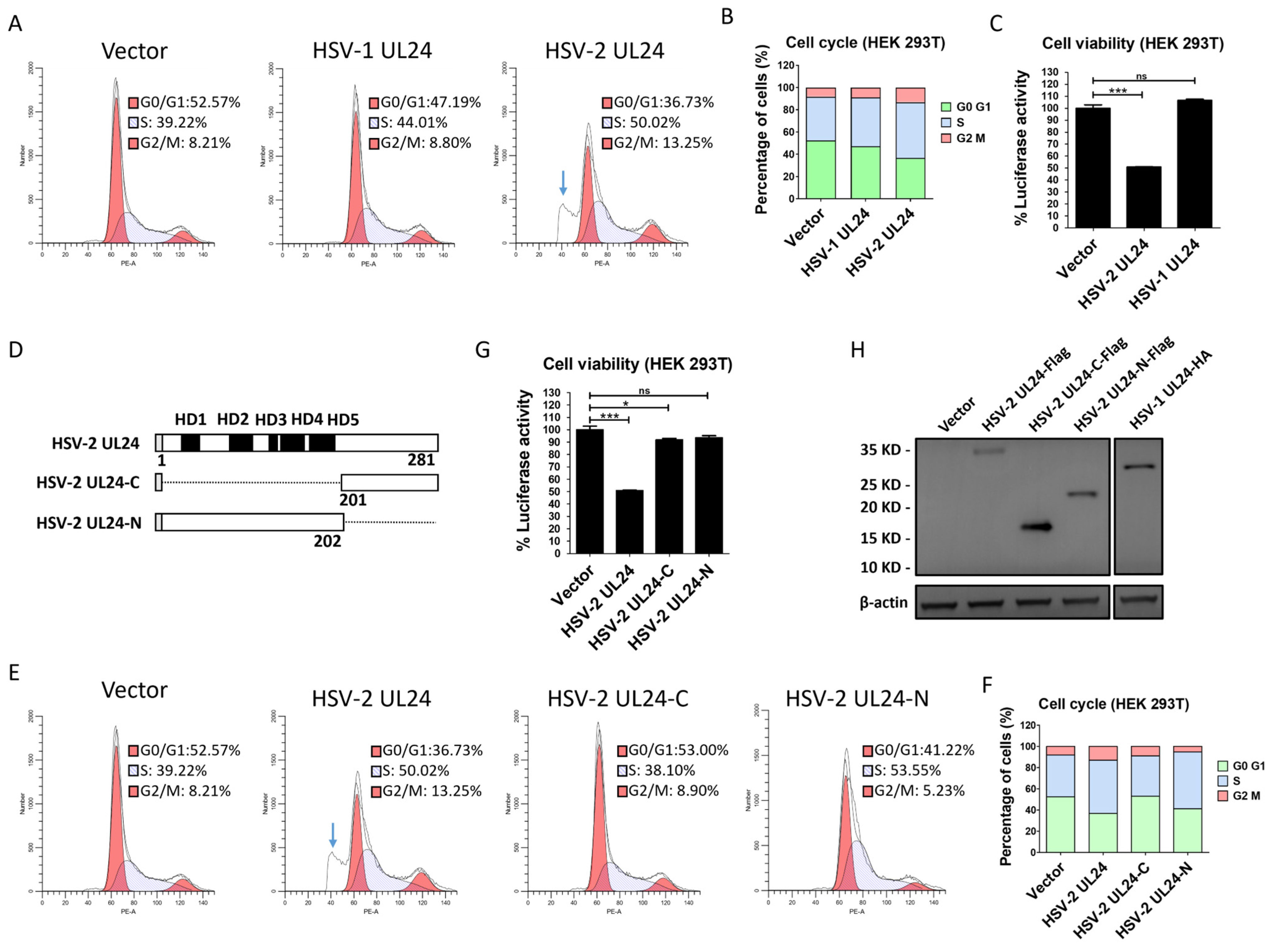
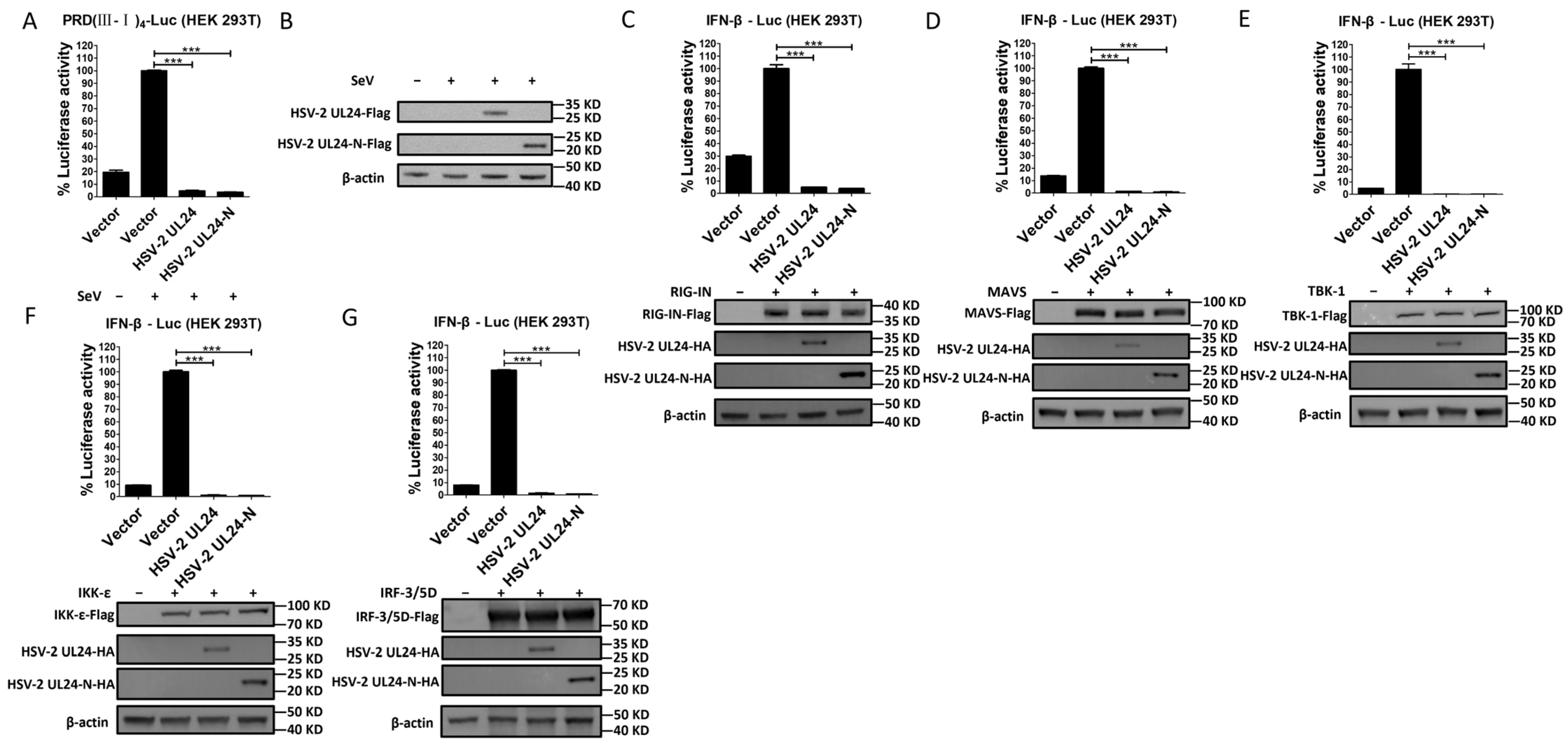
3.2. The N-Terminal 1–202 AA Domain of HSV-2 UL24 Is the Main Functional Region Responsible for HSV-2 UL24–Mediated Inhibition of IFN-β Production
3.3. The N-Terminal 1–202 AA Domain of HSV-2 UL24 Interferes with the IRF-3-Mediated Signaling Pathway
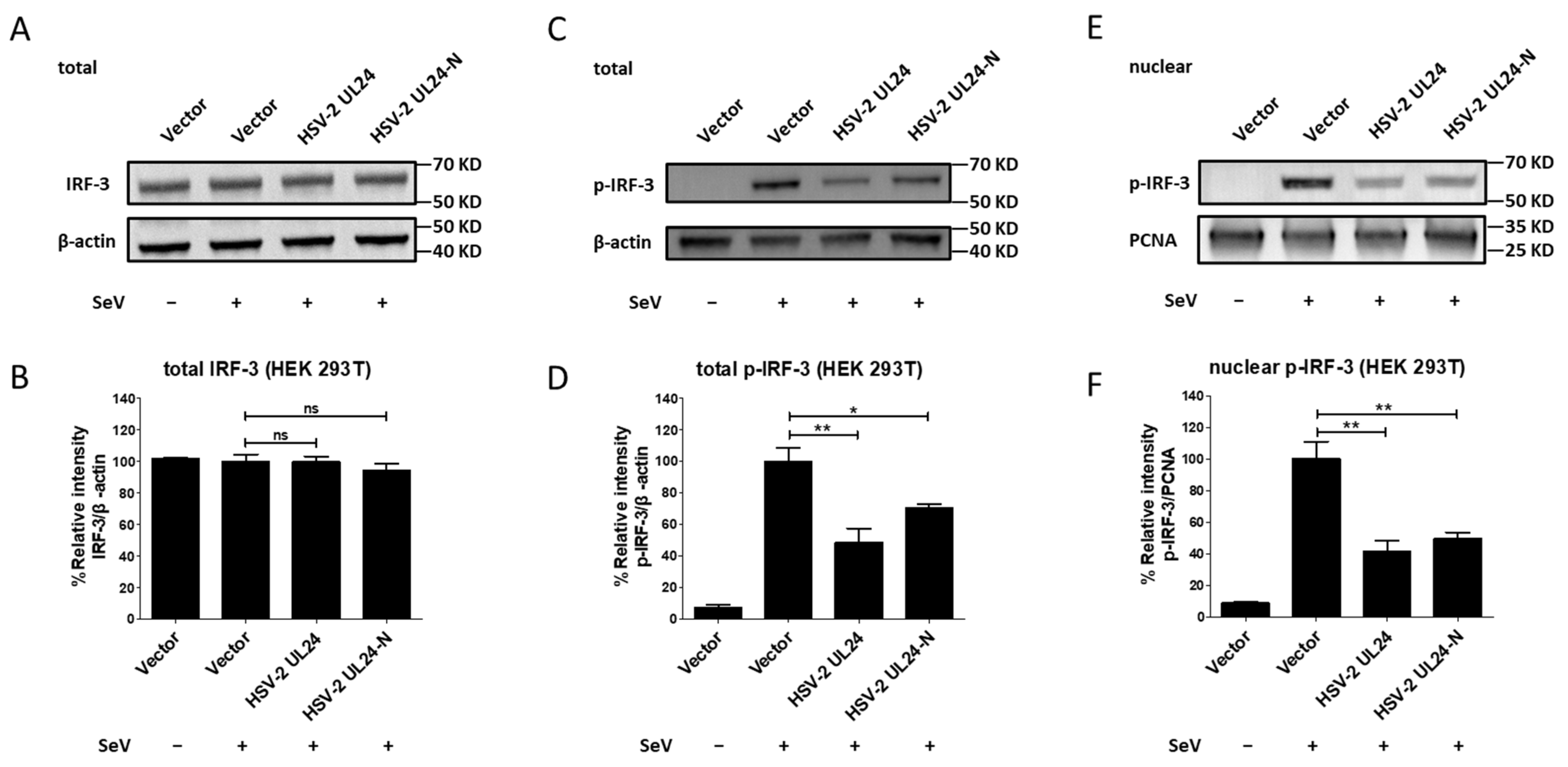
3.4. The N-Terminal 1–202 AA Domain of HSV-2 UL24 Inhibits the Phosphorylation of IRF-3
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Carty, M.; Guy, C.; Bowie, A.G. Detection of Viral Infections by Innate Immunity. Biochem. Pharmacol. 2021, 183, 114316. [Google Scholar] [CrossRef] [PubMed]
- Jondle, C.N.; Tarakanova, V.L. Innate Immunity and Alpha/Gammaherpesviruses: First Impressions Last a Lifetime. Curr. Opin. Virol. 2020, 44, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen Recognition and Innate Immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R. Recognition of Microorganisms and Activation of the Immune Response. Nature 2007, 449, 819–826. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. Innate Immunity to Virus Infection. Immunol. Rev. 2009, 227, 75–86. [Google Scholar] [CrossRef]
- Cai, X.; Chiu, Y.H.; Chen, Z.J. The Cgas-Cgamp-Sting Pathway of Cytosolic DNA Sensing and Signaling. Mol. Cell 2014, 54, 289–296. [Google Scholar] [CrossRef]
- Chen, Q.; Sun, L.; Chen, Z.J. Regulation and Function of the Cgas-Sting Pathway of Cytosolic DNA Sensing. Nat. Immunol. 2016, 17, 1142–1149. [Google Scholar] [CrossRef]
- Li, S.; Cao, L.; Zhang, Z.; Kuang, M.; Chen, L.; Zhao, Y.; Luo, Y.; Yin, Z.; You, F. Cytosolic and Nuclear Recognition of Virus and Viral Evasion. Mol. Biomed. 2021, 2, 30. [Google Scholar] [CrossRef]
- Rehwinkel, J.; Gack, M.U. RIG-I-like Receptors: Their Regulation and Roles in RNA Sensing. Nat. Rev. Immunol. 2020, 20, 537–551. [Google Scholar] [CrossRef]
- Ramos, H.J.; Gale, M., Jr. Rig-I like Receptors and Their Signaling Crosstalk in the Regulation of Antiviral Immunity. Curr. Opin. Virol. 2011, 1, 167–176. [Google Scholar] [CrossRef]
- Kell, A.M.; Gale, M., Jr. RIG-I in RNA Virus Recognition. Virology 2015, 479–480, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Omarova, S.; Cannon, A.; Weiss, W.; Bruccoleri, A.; Puccio, J. Genital Herpes Simplex Virus—An Updated Review. Adv. Pediatr. 2022, 69, 149–162. [Google Scholar] [CrossRef] [PubMed]
- Wald, A.; Corey, L.; Cone, R.; Hobson, A.; Davis, G.; Zeh, J. Frequent Genital Herpes Simplex Virus 2 Shedding in Immunocompetent Women. Effect of Acyclovir Treatment. J. Clin. Investig. 1997, 99, 1092–1097. [Google Scholar] [CrossRef] [PubMed]
- Van Wagoner, N.; Qushair, F.; Johnston, C. Genital Herpes Infection: Progress and Problems. Infect. Dis. Clin. N. Am. 2023, 37, 351–367. [Google Scholar] [CrossRef]
- Gupta, R.; Warren, T.; Wald, A. Genital Herpes. Lancet 2007, 370, 2127–2137. [Google Scholar] [CrossRef]
- Tronstein, E.; Johnston, C.; Huang, M.L.; Selke, S.; Magaret, A.; Warren, T.; Corey, L.; Wald, A. Genital Shedding of Herpes Simplex Virus among Symptomatic and Asymptomatic Persons with Hsv-2 Infection. Jama 2011, 305, 1441–1449. [Google Scholar] [CrossRef]
- Johnston, C.; Corey, L. Current Concepts for Genital Herpes Simplex Virus Infection: Diagnostics and Pathogenesis of Genital Tract Shedding. Clin. Microbiol. Rev. 2016, 29, 149–161. [Google Scholar] [CrossRef]
- Garland, S.M.; Steben, M. Genital Herpes. Best. Pract. Res. Clin. Obstet. Gynaecol. 2014, 28, 1098–1110. [Google Scholar] [CrossRef]
- Wald, A.; Link, K. Risk of Human Immunodeficiency Virus Infection in Herpes Simplex Virus Type 2-Seropositive Persons: A Meta-Analysis. J. Infect. Dis. 2002, 185, 45–52. [Google Scholar] [CrossRef]
- Schiffer, J.T.; Gottlieb, S.L. Biologic Interactions between Hsv-2 and Hiv-1 and Possible Implications for HSV Vaccine Development. Vaccine 2019, 37, 7363–7371. [Google Scholar] [CrossRef]
- James, C.; Harfouche, M.; Welton, N.J.; Turner, K.M.; Abu-Raddad, L.J.; Gottlieb, S.L.; Looker, K.J. Herpes Simplex Virus: Global Infection Prevalence and Incidence Estimates, 2016. Bull. World Health Organ. 2020, 98, 315–329. [Google Scholar] [CrossRef] [PubMed]
- Stanfield, B.A.; Kousoulas, K.G.; Fernandez, A.; Gershburg, E. Rational Design of Live-Attenuated Vaccines against Herpes Simplex Viruses. Viruses 2021, 13, 1637. [Google Scholar] [CrossRef] [PubMed]
- Sadler, A.J.; Williams, B.R. Interferon-inducible antiviral effectors. Nat. Rev. Immunol. 2008, 8, 559–568. [Google Scholar] [CrossRef]
- Samuel, C.E. Antiviral actions of interferons. Clin. Microbiol. Rev. 2001, 14, 778–809. [Google Scholar] [CrossRef] [PubMed]
- Peng, T.; Zhu, J.; Klock, A.; Phasouk, K.; Huang, M.L.; Koelle, D.M.; Wald, A.; Corey, L. Evasion of the Mucosal Innate Immune System by Herpes Simplex Virus Type 2. J. Virol. 2009, 83, 12559–12568. [Google Scholar] [CrossRef]
- Wang, S.; Wang, K.; Lin, R.; Zheng, C. Herpes Simplex Virus 1 Serine/Threonine Kinase Us3 Hyperphosphorylates Irf3 and Inhibits Beta Interferon Production. J. Virol. 2013, 87, 12814–12827. [Google Scholar] [CrossRef]
- Wang, K.; Ni, L.; Wang, S.; Zheng, C. Herpes Simplex Virus 1 Protein Kinase Us3 Hyperphosphorylates P65/Rela and Dampens Nf-Κb Activation. J. Virol. 2014, 88, 7941–7951. [Google Scholar] [CrossRef]
- Zhang, D.; Su, C.; Zheng, C. Herpes Simplex Virus 1 Serine Protease VP24 Blocks the DNA-Sensing Signal Pathway by Abrogating Activation of Interferon Regulatory Factor 3. J. Virol. 2016, 90, 5824–5829. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, S.; Wang, K.; Zheng, C. Herpes simplex virus 1 DNA Polymerase Processivity Factor Ul42 Inhibits Tnf-α-Induced Nf-κB Activation by Interacting with P65/RelA and P50/Nf-Κb1. Med. Microbiol. Immunol. 2013, 202, 313–325. [Google Scholar] [CrossRef]
- Lanfranca, M.P.; Mostafa, H.H.; Davido, D.J. Hsv-1 ICP0: An E3 Ubiquitin Ligase That Counteracts Host Intrinsic and Innate Immunity. Cells 2014, 3, 438–454. [Google Scholar] [CrossRef]
- Pourchet, A.; Fuhrmann, S.R.; Pilones, K.A.; Demaria, S.; Frey, A.B.; Mulvey, M.; Mohr, I. CD8(+) T-Cell Immune Evasion Enables Oncolytic Virus Immunotherapy. EBioMedicine 2016, 5, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Baines, J.D.; Pellett, P.E. Genetic Comparison of Human Alphaherpesvirus Genomes. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Arvin, A., Campadelli-Fiume, G., Mocarski, E., Moore, P.S., Roizman, B., Whitley, R., Yamanishi, K., Eds.; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Zhang, M.; Liu, Y.; Wang, P.; Guan, X.; He, S.; Luo, S.; Li, C.; Hu, K.; Jin, W.; Du, T.; et al. Hsv-2 Immediate-Early Protein Us1 Inhibits Ifn-Β Production by Suppressing Association of Irf-3 with Ifn-Β promoter. J. Immunol. 2015, 194, 3102–3115. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Fu, M.; Li, M.; Hu, H.; Gong, S.; Hu, Q. Herpes Simplex Virus Type 2 Inhibits Type I Ifn Signaling Mediated by the Novel E3 Ubiquitin Protein Ligase Activity of Viral Protein Icp22. J. Immunol. 2020, 205, 1281–1292. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.; Zhang, M.; Fu, M.; Luo, S.; Hu, Q. Herpes Simplex Virus Type 2 Immediate Early Protein Icp27 Inhibits Ifn-Β Production in Mucosal Epithelial Cells by Antagonizing Irf3 Activation. Front. Immunol. 2019, 10, 290. [Google Scholar] [CrossRef]
- Bertrand, L.; Leiva-Torres, G.A.; Hyjazie, H.; Pearson, A. Conserved Residues in the UL24 Protein of Herpes Simplex Virus 1 Are Important for Dispersal of the Nucleolar Protein Nucleolin. J. Virol. 2010, 84, 109–118. [Google Scholar] [CrossRef]
- Leiva-Torres, G.A.; Rochette, P.A.; Pearson, A. Differential Importance of Highly Conserved Residues in UL24 for Herpes Simplex Virus 1 Replication in Vivo and Reactivation. J. Gen. Virol. 2010, 91, 1109–1116. [Google Scholar] [CrossRef]
- Carmichael, J.C.; Wills, J.W. Differential Requirements for gE, gI, and UL16 among Herpes Simplex Virus 1 Syncytial Variants Suggest Unique Modes of Dysregulating the Mechanism of Cell-to-Cell Spread. J. Virol. 2019, 93, 10–1128. [Google Scholar] [CrossRef]
- Sanabria-Solano, C.; Gonzalez, C.E.; Richerioux, N.; Bertrand, L.; Dridi, S.; Griffiths, A.; Langelier, Y.; Pearson, A. Regulation of Viral Gene Expression by the Herpes Simplex Virus 1ul24 Protein (Hsv-1UL24 Inhibits Accumulation of Viral Transcripts). Virology 2016, 495, 148–160. [Google Scholar] [CrossRef]
- Xu, H.; Su, C.; Pearson, A.; Mody, C.H.; Zheng, C. Herpes Simplex Virus 1 UL24 Abrogates the DNA Sensing Signal Pathway by Inhibiting Nf-Κb Activation. J. Virol. 2017, 91, 10–1128. [Google Scholar] [CrossRef]
- Nascimento, R.; Dias, J.D.; Parkhouse, R.M. The Conserved UL24 Family of Human Alpha, Beta and Gamma Herpesviruses Induces Cell Cycle Arrest and Inactivation of the cyclinB/cdc2 complex. Arch. Virol. 2009, 154, 1143–1149. [Google Scholar] [CrossRef]
- Ben Abdeljelil, N.; Rochette, P.A.; Pearson, A. The UL24 Protein of Herpes Simplex Virus 1 Affects the Sub-Cellular Distribution of Viral Glycoproteins Involved in Fusion. Virology 2013, 444, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, J.G.; Chen, S.H.; Cook, W.J.; Kramer, M.F.; Coen, D.M. Importance of the Herpes Simplex Virus UL24 Gene for Productive Ganglionic Infection in Mice. Virology 1998, 242, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Lieber, D.; Bailer, S.M. Determination of HSV-1 Infectivity by Plaque Assay and a Luciferase Reporter Cell Line. Methods Mol. Biol. 2013, 1064, 171–181. [Google Scholar]
- Iwasaki, A. Exploiting Mucosal Immunity for Antiviral Vaccines. Annu. Rev. Immunol. 2016, 34, 575–608. [Google Scholar] [CrossRef]
- Hong-Yan, Z.; Murata, T.; Goshima, F.; Takakuwa, H.; Koshizuka, T.; Yamauchi, Y.; Nishiyama, Y. Identification and Characterization of the UL24 Gene Product of Herpes Simplex Virus Type 2. Virus Genes 2001, 22, 321–327. [Google Scholar] [CrossRef]
- Jacobson, J.G.; Martin, S.L.; Coen, D.M. A Conserved Open Reading Frame That Overlaps the Herpes Simplex Virus Thymidine Kinase Gene is Important for Viral Growth in Cell Culture. J. Virol. 1989, 63, 1839–1843. [Google Scholar] [CrossRef]
- Bertrand, L.; Pearson, A. The conserved N-Terminal Domain of Herpes Simplex Virus 1 Ul24 Protein Is Sufficient to Induce the Spatial Redistribution of Nucleolin. J. Gen. Virol. 2008, 89 Pt 5, 1142–1151. [Google Scholar] [CrossRef]
- Au, W.C.; Moore, P.A.; Lowther, W.; Juang, Y.T.; Pitha, P.M. Identification of a Member of the Interferon Regulatory Factor Family That Binds to the Interferon-Stimulated Response Element and Activates Expression of Interferon-Induced Genes. Proc. Natl. Acad. Sci. USA 1995, 92, 11657–11661. [Google Scholar] [CrossRef]
- Hiscott, J. Triggering the Innate Antiviral Response through IRF-3 Activation. J. Biol. Chem. 2007, 282, 15325–15329. [Google Scholar] [CrossRef]
- Lin, R.; Mamane, Y.; Hiscott, J. Structural and Functional Analysis of Interferon Regulatory Factor 3: Localization of the Transactivation and Autoinhibitory Domains. Mol. Cell. Biol. 1999, 19, 2465–2474. [Google Scholar] [CrossRef]
- Groves, M.J. Genital Herpes: A Review. Am. Fam. Physician 2016, 93, 928–934. [Google Scholar] [PubMed]
- Chan, T.; Barra, N.G.; Lee, A.J.; Ashkar, A.A. Innate and Adaptive Immunity against Herpes Simplex Virus Type 2 in the Genital Mucosa. J. Reprod. Immunol. 2011, 88, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, A.; Medzhitov, R. Control of Adaptive Immunity by the Innate Immune System. Nat. Immunol. 2015, 16, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Sin, W.X.; Li, P.; Yeong, J.P.; Chin, K.C. Activation and Regulation of Interferon-Β in Immune Responses. Immunol. Res. 2012, 53, 25–40. [Google Scholar] [CrossRef]
- Chiu, Y.H.; Macmillan, J.B.; Chen, Z.J. RNA Polymerase III Detects Cytosolic DNA and Induces Type I Interferons through the RIG-I Pathway. Cell 2009, 138, 576–591. [Google Scholar] [CrossRef]
- Lymberopoulos, M.H.; Bourget, A.; Ben Abdeljelil, N.; Pearson, A. Involvement of the UL24 Protein in Herpes Simplex Virus 1-Induced Dispersal of B23 and in Nuclear Egress. Virology 2011, 412, 341–348. [Google Scholar] [CrossRef]
- Gonzalez, C.E.; Ben Abdeljelil, N.; Pearson, A. The Disruption of a Nuclear Export Signal in the C-Terminus of the Herpes Simplex Virus 1 Determinant of Pathogenicity UL24 Protein Leads to a Syncytial Plaque Phenotype. Viruses 2023, 15, 1971. [Google Scholar] [CrossRef]
- Pattabhi, S.; Wilkins, C.R.; Dong, R.; Knoll, M.L.; Posakony, J.; Kaiser, S.; Mire, C.E.; Wang, M.L.; Ireton, R.C.; Geisbert, T.W.; et al. Targeting Innate Immunity for Antiviral Therapy through Small Molecule Agonists of the RLR Pathway. J. Virol. 2015, 90, 2372–2387. [Google Scholar] [CrossRef]
- Christensen, M.H.; Jensen, S.B.; Miettinen, J.J.; Luecke, S.; Prabakaran, T.; Reinert, L.S.; Mettenleiter, T.; Chen, Z.J.; Knipe, D.M.; Sandri-Goldin, R.M.; et al. Hsv-1 Icp27 Targets the Tbk1-Activated Sting Signalsome To inhibit Virus-Induced Type I Ifn Expression. EMBO J. 2016, 35, 1385–1399. [Google Scholar] [CrossRef]
- Prasad, A.; Remick, J.; Zeichner, S.L. Activation of Human Herpesvirus Replication by Apoptosis. J. Virol. 2013, 87, 10641–10650. [Google Scholar] [CrossRef]
- Ori, D.; Murase, M.; Kawai, T. Cytosolic Nucleic Acid Sensors and Innate Immune Regulation. Int. Rev. Immunol. 2017, 36, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; TenOever, B.R.; Grandvaux, N.; Zhou, G.P.; Lin, R.; Hiscott, J. Triggering the Interferon Antiviral Response through an Ikk-Related Pathway. Science 2003, 300, 1148–1151. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Zhao, W.; Zhao, K.; Zhang, L.; Gao, C. Trim26 Negatively Regulates Interferon-B Production and Antiviral Response through Polyubiquitination and Degradation of Nuclear Irf3. PLoS Pathog. 2015, 11, e1004726. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, B.; Li, Y.; Yang, P.; He, S.; Li, W.; Li, M.; Hu, Q.; Zhang, M. Herpes Simplex Virus Type 2 Blocks IFN-β Production through the Viral UL24 N-Terminal Domain-Mediated Inhibition of IRF-3 Phosphorylation. Viruses 2024, 16, 1601. https://doi.org/10.3390/v16101601
Zhang B, Li Y, Yang P, He S, Li W, Li M, Hu Q, Zhang M. Herpes Simplex Virus Type 2 Blocks IFN-β Production through the Viral UL24 N-Terminal Domain-Mediated Inhibition of IRF-3 Phosphorylation. Viruses. 2024; 16(10):1601. https://doi.org/10.3390/v16101601
Chicago/Turabian StyleZhang, Binman, Yuncheng Li, Ping Yang, Siyu He, Weilin Li, Miaomiao Li, Qinxue Hu, and Mudan Zhang. 2024. "Herpes Simplex Virus Type 2 Blocks IFN-β Production through the Viral UL24 N-Terminal Domain-Mediated Inhibition of IRF-3 Phosphorylation" Viruses 16, no. 10: 1601. https://doi.org/10.3390/v16101601
APA StyleZhang, B., Li, Y., Yang, P., He, S., Li, W., Li, M., Hu, Q., & Zhang, M. (2024). Herpes Simplex Virus Type 2 Blocks IFN-β Production through the Viral UL24 N-Terminal Domain-Mediated Inhibition of IRF-3 Phosphorylation. Viruses, 16(10), 1601. https://doi.org/10.3390/v16101601