

Genomic Surveillance Reveals the Rapid Expansion of the XBB Lineage among Circulating SARS-CoV-2 Omicron Lineages in Southeastern Wisconsin, USA
Abstract
:1. Introduction
2. Materials and Methods
2.1. SARS-CoV-2-Positive Clinical Specimens
2.2. Clear Dx Sequencing Platform
2.3. MinION Sequencing Platform
2.4. MiSeq Sequencing Platform
2.5. Nextclade Assignment and Phylogenetic Analysis of Sequences
3. Results and Discussion
3.1. SARS-CoV-2-Positive Clinical Specimens and Associated Metadata
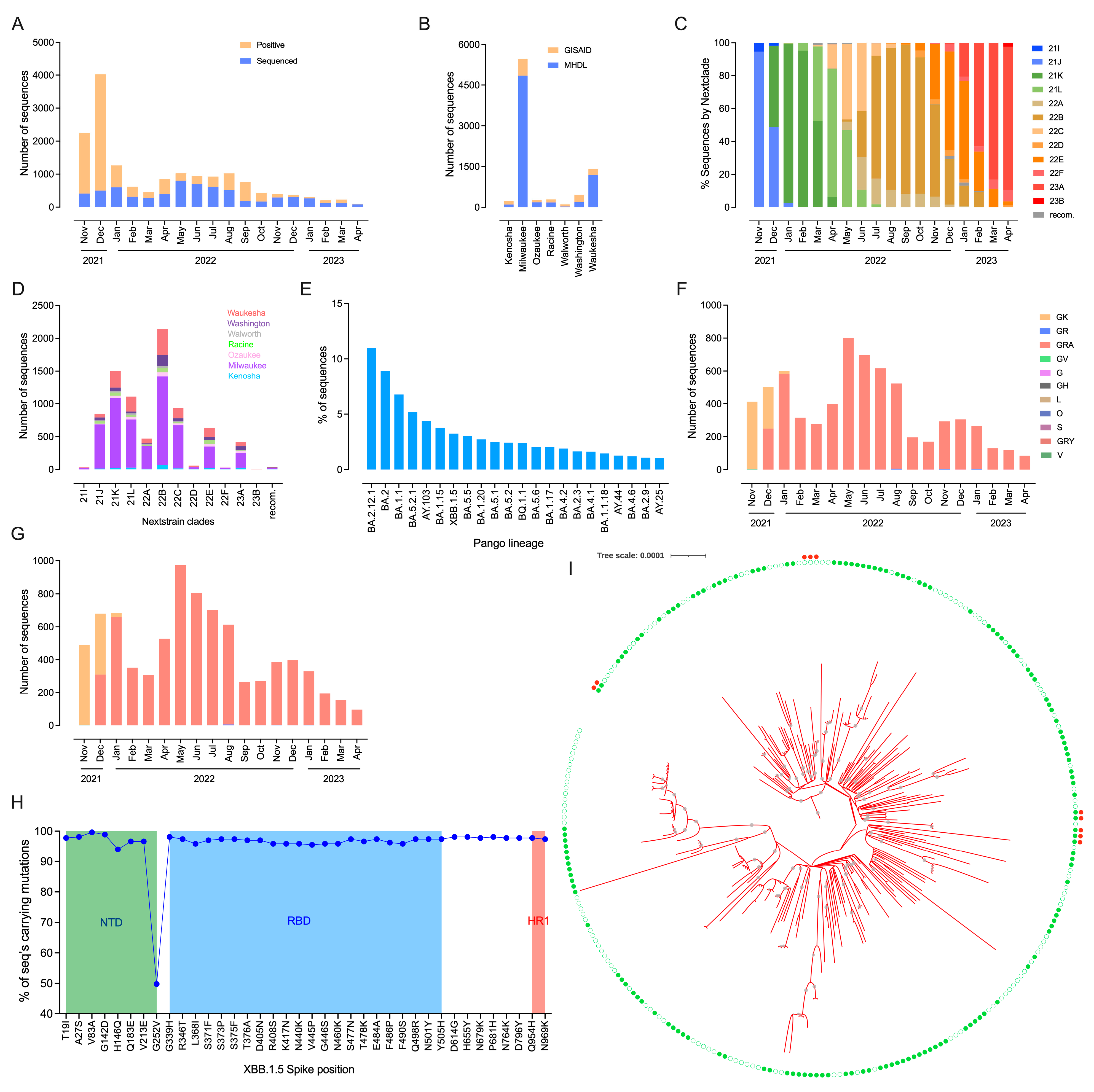
3.2. Identification of Circulating SARS-CoV-2 Lineages Reveals Rapid Spread of the Virus in Most Densely Populated Milwaukee County
3.3. Mutational Profile and Transmission Dynamics of Rapidly Expanded XBB.1.5 Lineage in Southeastern Wisconsin Population
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical Features of Patients Infected with 2019 Novel Coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, D.; Chakravarty, N.; Jeyachandran, A.V.; Jeyakarunakaran, A.; Sinha, S.; Mishra, R.; Arumugaswami, A.; Ramaiah, A. In Silico Genome Analysis Reveals the Evolution and Potential Impact of SARS-CoV-2 Omicron Structural Changes on Host Immune Evasion and Antiviral Therapeutics. Viruses 2022, 14, 2461. [Google Scholar] [CrossRef]
- Ramaiah, A.; Khubbar, M.; Bauer, A.; Scott, S.; Lentz, J.; Akinyemi, K.; Skillman, A.; Weiner, J.; Balakrishnan, N.; Bhattacharyya, S. Genomic surveillance identifies SARS-CoV-2 transmission patterns in local university populations, Wisconsin, USA, 2020-2022. Microb. Genom. 2023, 9, mgen00970. [Google Scholar] [CrossRef]
- Cherian, S.; Potdar, V.; Jadhav, S.; Yadav, P.; Gupta, N.; Das, M.; Rakshit, P.; Singh, S.; Abraham, P.; Panda, S.; et al. SARS-CoV-2 Spike Mutations, L452R, T478K, E484Q and P681R, in the Second Wave of COVID-19 in Maharashtra, India. Microorganisms 2021, 9, 1542. [Google Scholar] [CrossRef]
- Leung, K.; Shum, M.H.; Leung, G.M.; Lam, T.T.; Wu, J.T. Early Transmissibility Assessment of the N501Y Mutant Strains of SARS-CoV-2 in the United Kingdom, October to November 2020. Eurosurveillance 2021, 26, 2002106. [Google Scholar] [CrossRef] [PubMed]
- Yadav, P.D.; Sapkal, G.N.; Abraham, P.; Ella, R.; Deshpande, G.; Patil, D.Y.; Nyayanit, D.A.; Gupta, N.; Sahay, R.R.; Shete, A.M.; et al. Neutralization of Variant under Investigation B.1.617 with Sera of BBV152 Vaccinees. Clin. Infect. Dis. 2021, 74, 366–368. [Google Scholar] [CrossRef]
- Barton, M.I.; MacGowan, S.A.; Kutuzov, M.A.; Dushek, O.; Barton, G.J.; van der Merwe, P.A. Effects of Common Mutations in the SARS-CoV-2 Spike RBD and Its Ligand, the Human ACE2 Receptor on Binding Affinity and Kinetics. eLife 2021, 10, e70658. [Google Scholar] [CrossRef] [PubMed]
- Tegally, H.; Wilkinson, E.; Giovanetti, M.; Iranzadeh, A.; Fonseca, V.; Giandhari, J.; Doolabh, D.; Pillay, S.; San, E.J.; Msomi, N.; et al. Detection of a SARS-CoV-2 variant of concern in South Africa. Nature 2021, 592, 438–443. [Google Scholar] [CrossRef]
- Mlcochova, P.; Kemp, S.A.; Dhar, M.S.; Papa, G.; Meng, B.; Ferreira, I.A.T.M.; Datir, R.; Collier, D.A.; Albecka, A.; Singh, S.; et al. SARS-CoV-2 B.1.617.2 Delta Variant Replication and Immune Evasion. Nature 2021, 599, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.C.; Shirk, P.; Lambrou, A.S.; Hassell, N.; Zheng, X.-Y.; Payne, A.B.; Ali, A.R.; Batra, D.; Caravas, J.; Chau, R.; et al. Genomic surveillance for SARS-CoV-2 variants: Circulation of omicron lineages—United States, January 2022-May 2023. MMWR Morb. Mortal. Wkly. Rep. 2023, 72, 651–656. [Google Scholar] [CrossRef]
- Lambrou, A.S.; Shirk, P.; Steele, M.K.; Paul, P.; Paden, C.R.; Cadwell, B.; Reese, H.E.; Aoki, Y.; Hassell, N.; Zheng, X.Y.; et al. Strain Surveillance and Emerging Variants Bioinformatic Working Group; Strain Surveillance and Emerging Variants NS3 Working Group. Genomic Surveillance for SARS-CoV-2 Variants: Predominance of the Delta (B.1.617.2) and Omicron (B.1.1.529) Variants—United States, June 2021-January 2022. MMWR Morb. Mortal. Wkly. Rep. 2022, 71, 206–211. [Google Scholar]
- Galloway, S.E.; Paul, P.; MacCannell, D.R.; Johansson, M.A.; Brooks, J.T.; MacNeil, A.; Slayton, R.B.; Tong, S.; Silk, B.J.; Armstrong, G.L.; et al. Emergence of SARS-CoV-2 B.1.1.7 Lineage—United States, December 29, 2020-January 12, 2021. MMWR Morb. Mortal. Wkly. Rep. 2021, 70, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Scobie, H.M.; Panaggio, M.; Binder, A.M.; Gallagher, M.E.; Duck, W.M.; Graff, P.; Silk, B.J. Correlations and Timeliness of COVID-19 Surveillance Data Sources and Indicators—United States, October 1, 2020-March 22, 2023. MMWR Morb. Mortal. Wkly. Rep. 2023, 72, 529–535. [Google Scholar] [CrossRef]
- Paul, P.; France, A.M.; Aoki, Y.; Batra, D.; Biggerstaff, M.; Dugan, V.; Galloway, S.; Hall, A.J.; Johansson, M.A.; Kondor, R.J.; et al. Genomic Surveillance for SARS-CoV-2 Variants Circulating in the United States, December 2020-May 2021. MMWR Morb. Mortal. Wkly. Rep. 2021, 70, 846–850. [Google Scholar] [CrossRef] [PubMed]
- Ramaiah, A.; Khubbar, M.; Scott, S.; Bauer, A.; Lentz, J.; Akinyemi, K.; Skillman, A.; Weiner, J.; Balakrishnan, N.; Bhattacharyya, S. Implementation and evaluation of the Clear Dx platform for sequencing SARS-CoV-2 genomes in a public health laboratory. Microbiol. Spectr. 2023, 11, e0495722. [Google Scholar] [CrossRef]
- Moreno, G.K.; Braun, K.M.; Riemersma, K.K.; Martin, M.A.; Halfmann, P.J.; Crooks, C.M.; Prall, T.; Baker, D.; Baczenas, J.J.; Heffron, A.S.; et al. Revealing fine-scale spatiotemporal differences in SARS-CoV-2 introduction and spread. Nat. Commun. 2020, 11, 5558. [Google Scholar] [CrossRef]
- Jung, A.; Droit, L.; Febles, B.; Fronick, C.; Cook, L.; Handley, S.A.; Parikh, B.A.; Wang, D. Tracking the Prevalence and Emergence of SARS CoV2 Variants of Concern Using a Regional Genomic Surveillance Program. medRxiv 2023, 10. Available online: https://www.medrxiv.org/content/10.1101/2023.05.08.23289687v1 (accessed on 15 August 2023).
- Payen, S.H.; Gorzalski, A.; Siao, D.D.; Pandori, M.; Verma, S.C.; Rossetto, C.C. Analysis of SARS-CoV-2 variants from patient specimens in Nevada from October 2020 to August 2021. Infect. Genet. Evol. 2023, 111, 105434. [Google Scholar] [CrossRef]
- Oltean, H.N.; Allen, K.J.; Frisbie, L.; Lunn, S.M.; Torres, L.M.; Manahan, L.; Painter, I.; Russell, D.; Singh, A.; Peterson, J.M.; et al. Sentinel Surveillance System Implementation and Evaluation for SARS-CoV-2 Genomic Data, Washington, USA, 2020-2021. Emerg. Infect. Dis. 2023, 29, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Mediavilla, J.R.; Lozy, T.; Lee, A.; Kim, J.; Kan, V.W.; Titova, E.; Amin, A.; Zody, M.C.; Corvelo, A.; Oschwald, D.M.; et al. Molecular and Clinical Epidemiology of SARS-CoV-2 Infection among Vaccinated and Unvaccinated Individuals in a Large Healthcare Organization from New Jersey. Viruses 2023, 15, 1699. [Google Scholar] [CrossRef]
- Baker, J.M.; Nakayama, J.Y.; O’Hegarty, M.; McGowan, A.; Teran, R.A.; Bart, S.M.; Mosack, K.; Roberts, N.; Campos, B.; Paegle, A.; et al. SARS-CoV-2 B.1.1.529 (Omicron) Variant Transmission Within Households—Four U.S. Jurisdictions, November 2021–February 2022. MMWR Morb. Mortal. Wkly. Rep. 2022, 71, 341–346. [Google Scholar] [CrossRef]
- Paradis, H.; Katrichis, J.; Stevenson, M.; Tomaro, N.; Mukai, R.; Torres, G.; Bhattacharyya, S.; Kowalik, J.; Schlanger, K.; Leidman, E. Notes from the Field: Public Health Efforts to Mitigate COVID-19 Transmission During the April 7, 2020, Election—City of Milwaukee, Wisconsin, March 13-May 5, 2020. MMWR Morb. Mortal. Wkly. Rep. 2020, 69, 1002–1003. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. Interim Guidelines for Collecting and Handling of Clinical Specimens for COVID-19 Testing. Summary of Recent Changes. Key Points. Collecting and Handling Specimens Safely. 2021. Available online: https://www.cdc.gov/coronavirus/2019-nCoV/lab/guidelines-clinical-specimens.html (accessed on 15 April 2023).
- Centers for Disease Control and Prevention. CDC 2019-Novel Corona-Virus (2019- NCoV) Real-Time RT-PCR Diagnostic Panel. For Emergency Use Only. Instructions for Use. 2020. Available online: https://www.fda.gov/media/134922/download (accessed on 14 March 2021).
- Centers for Disease Control and Prevention. CDC Influenza SARS-CoV-2 (flu SC2) Multiplex Assay. For Emergency Use Only. Instructions for Use. 2020. Available online: https://www.fda.gov/media/139743/download (accessed on 21 May 2023).
- SARS-CoV-2 Sequencing on Illumina MiSeq Using ARTIC Protocol: Part 2—Illumina DNA Flex Protocol V.1—Joel Sevinsky, StaPH-B Consortium, Coronavirus Method Development Community. Available online: https://www.protocols.io/private/EF7D7A9B84D611EAAB080242AC110005?step=4 (accessed on 22 May 2023).
- Li, H. New strategies to improve minimap2 alignment accuracy. Bioinformatics. 2021, 37, 4572–4574. [Google Scholar] [CrossRef] [PubMed]
- SARS-CoV-2 Sequencing on Illumina MiSeq Using ARTIC Protocol: Part 2—Illumina DNA Prep Protocol V.1. 2021. Available online: https://www.protocols.io/view/sars-cov-2-sequencing-onillumina-miseq-using-arti-n92ld9w1xg5b/v1 (accessed on 22 May 2023).
- Khare, S.; Gurry, C.; Freitas, L.; Schultz, M.B.; Bach, G.; Diallo, A.; Akite, N.; Ho, J.; Lee, R.T.C.; Yeo, W.; et al. GISAID’s Role in Pandemic Response. China CDC Wkly. 2021, 3, 1049–1051. [Google Scholar] [CrossRef]
- Aksamentov, I.; Roemer, C.; Hodcroft, E.B.; Neher, R.A. Nextclade: Clade assignment, mutation calling and quality control for viral genomes. J. Open Source Softw. 2021, 6, 3773. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Stokes, E.K.; Zambrano, L.D.; Anderson, K.N.; Marder, E.P.; Raz, K.M.; El Burai Felix, S.; Tie, Y.; Fullerton, K.E. Coronavirus Disease 2019 Case Surveillance—United States, January 22-May 30, 2020. MMWR Morb. Mortal. Wkly. Rep. 2020, 69, 759–765. [Google Scholar] [CrossRef]
- Gebhard, C.; Regitz-Zagrosek, V.; Neuhauser, H.K.; Morgan, R.; Klein, S.L. Impact of sex and gender on COVID-19 outcomes in Europe. Biol. Sex Differ. 2020, 11, 29. [Google Scholar] [CrossRef]
- Cai, H. Sex difference and smoking predisposition in patients with COVID-19. Lancet Respir. Med. 2020, 8, e20. [Google Scholar] [CrossRef]
- Gargaglioni, L.H.; Marques, D.A. Let’s talk about sex in the context of COVID-19. J. Appl. Physiol. (1985) 2020, 128, 1533–1538. [Google Scholar] [CrossRef]
- Goren, A.; McCoy, J.; Wambier, C.G.; Vano-Galvan, S.; Shapiro, J.; Dhurat, R.; Washenik, K.; Lotti, T. What does androgenetic alopecia have to do with COVID-19? An insight into a potential new therapy. Dermatol. Ther. 2020, 33, e13365. [Google Scholar] [CrossRef]
- Montopoli, M.; Zumerle, S.; Vettor, R.; Rugge, M.; Zorzi, M.; Catapano, C.V.; Carbone, G.M.; Cavalli, A.; Pagano, F.; Ragazzi, E.; et al. Androgen-deprivation therapies for prostate cancer and risk of infection by SARS-CoV-2: A population-based study (N = 4532). Ann. Oncol. 2020, 31, 1040–1045. [Google Scholar] [CrossRef]
- Cai, G.; Bosse, Y.; Xiao, F.; Kheradmand, F.; Amos, C.I. Tobacco smoking increases the lung gene expression of ACE2, the receptor of SARS-CoV-2. Am. J. Respir. Crit. Care Med. 2020, 201, 1557–1559. [Google Scholar] [CrossRef]
- Walter, L.A.; McGregor, A.J. Sex- and gender-specific observations and implications for COVID-19. West J. Emerg. Med. 2020, 21, 507–509. [Google Scholar] [CrossRef]
- Mukherjee, S.; Pahan, K. Is COVID-19 Gender-sensitive? J. Neuroimmune. Pharmacol. 2021, 16, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Yamasoba, D.; Uriu, K.; Plianchaisuk, A.; Kosugi, Y.; Pan, L.; Zahradnik, J.; Genotype to Phenotype Japan (G2P-Japan) Consortium; Ito, J.; Sato, K. Virological characteristics of the SARS-CoV-2 omicron XBB.1.16 variant. Lancet Infect. Dis. 2023, 23, 655–656. [Google Scholar] [CrossRef]
- Yue, C.; Song, W.; Wang, L.; Jian, F.; Chen, X.; Gao, F.; Shen, Z.; Wang, Y.; Wang, X.; Cao, Y. ACE2 binding and antibody evasion in enhanced transmissibility of XBB.1.5. Lancet Infect Dis. 2023, 23, 278–280. [Google Scholar] [CrossRef]
- Cao, Y.; Jian, F.; Wang, J.; Yu, Y.; Song, W.; Yisimayi, A.; Wang, J.; An, R.; Chen, X.; Zhang, N.; et al. Imprinted SARS-CoV-2 humoral immunity induces convergent Omicron RBD evolution. Nature 2023, 614, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Imai, M.; Ito, M.; Kiso, M.; Yamayoshi, S.; Uraki, R.; Fukushi, S.; Watanabe, S.; Suzuki, T.; Maeda, K.; Sakai-Tagawa, Y.; et al. Efficacy of Antiviral Agents against Omicron Subvariants BQ.1.1 and XBB. N. Engl. J. Med. 2023, 388, 89–91. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Iketani, S.; Li, Z.; Liu, L.; Guo, Y.; Huang, Y.; Bowen, A.D.; Liu, M.; Wang, M.; Yu, J.; et al. Alarming antibody evasion properties of rising SARS-CoV-2 BQ and XBB subvariants. Cell 2023, 186, 279–286.e8. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Features | MHDL Data |
|---|---|
| No. of SARS-CoV-2 specimens sequenced (QC passed) | 6709 |
| Gender | |
| Female | 3377 |
| Male | 2744 |
| Unknown | 588 |
| Age * | |
| Female | 1–103 (38) |
| Male | 1–99 (36) |
| Unknown | 1–95 (39) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramaiah, A.; Khubbar, M.; Akinyemi, K.; Bauer, A.; Carranza, F.; Weiner, J.; Bhattacharyya, S.; Payne, D.; Balakrishnan, N. Genomic Surveillance Reveals the Rapid Expansion of the XBB Lineage among Circulating SARS-CoV-2 Omicron Lineages in Southeastern Wisconsin, USA. Viruses 2023, 15, 1940. https://doi.org/10.3390/v15091940
Ramaiah A, Khubbar M, Akinyemi K, Bauer A, Carranza F, Weiner J, Bhattacharyya S, Payne D, Balakrishnan N. Genomic Surveillance Reveals the Rapid Expansion of the XBB Lineage among Circulating SARS-CoV-2 Omicron Lineages in Southeastern Wisconsin, USA. Viruses. 2023; 15(9):1940. https://doi.org/10.3390/v15091940
Chicago/Turabian StyleRamaiah, Arunachalam, Manjeet Khubbar, Katherine Akinyemi, Amy Bauer, Francisco Carranza, Joshua Weiner, Sanjib Bhattacharyya, David Payne, and Nandhakumar Balakrishnan. 2023. "Genomic Surveillance Reveals the Rapid Expansion of the XBB Lineage among Circulating SARS-CoV-2 Omicron Lineages in Southeastern Wisconsin, USA" Viruses 15, no. 9: 1940. https://doi.org/10.3390/v15091940
APA StyleRamaiah, A., Khubbar, M., Akinyemi, K., Bauer, A., Carranza, F., Weiner, J., Bhattacharyya, S., Payne, D., & Balakrishnan, N. (2023). Genomic Surveillance Reveals the Rapid Expansion of the XBB Lineage among Circulating SARS-CoV-2 Omicron Lineages in Southeastern Wisconsin, USA. Viruses, 15(9), 1940. https://doi.org/10.3390/v15091940