
Optimizing the Conditions for Whole-Genome Sequencing of Avian Reoviruses
 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
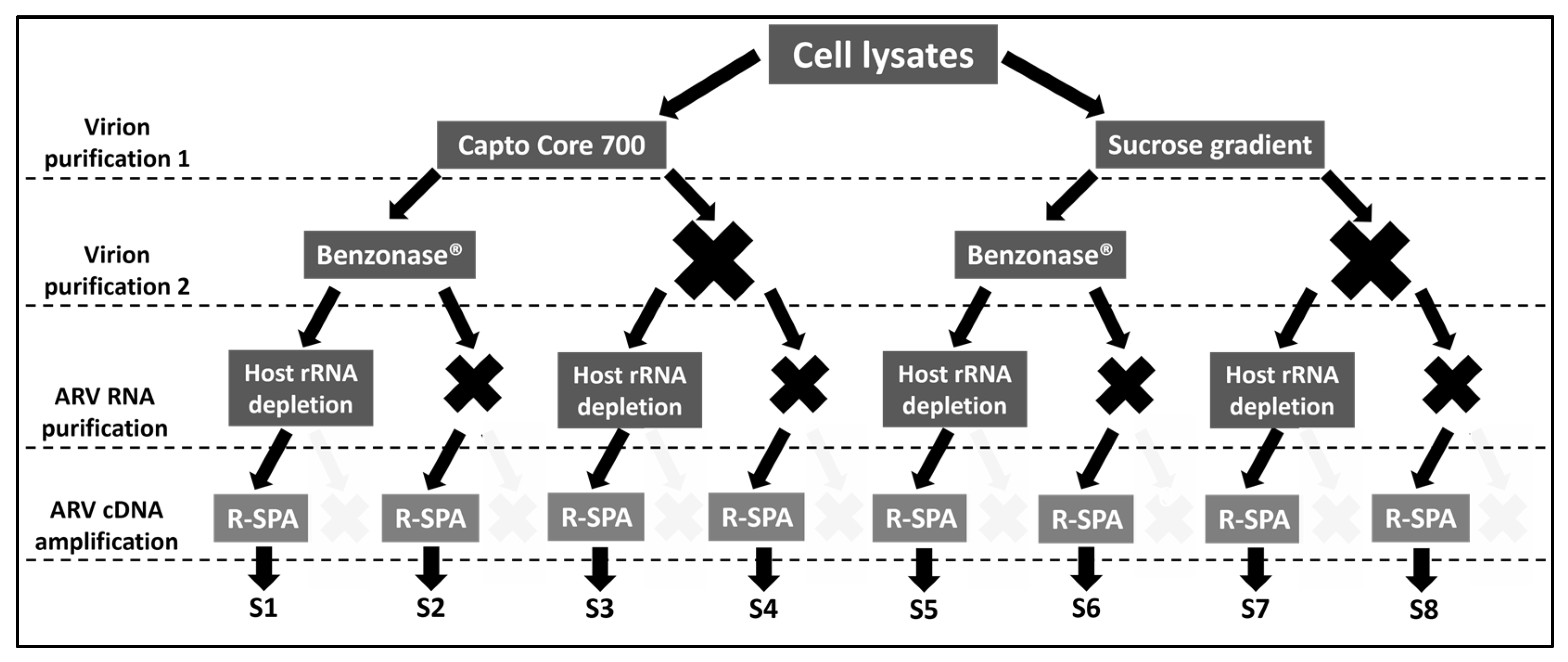
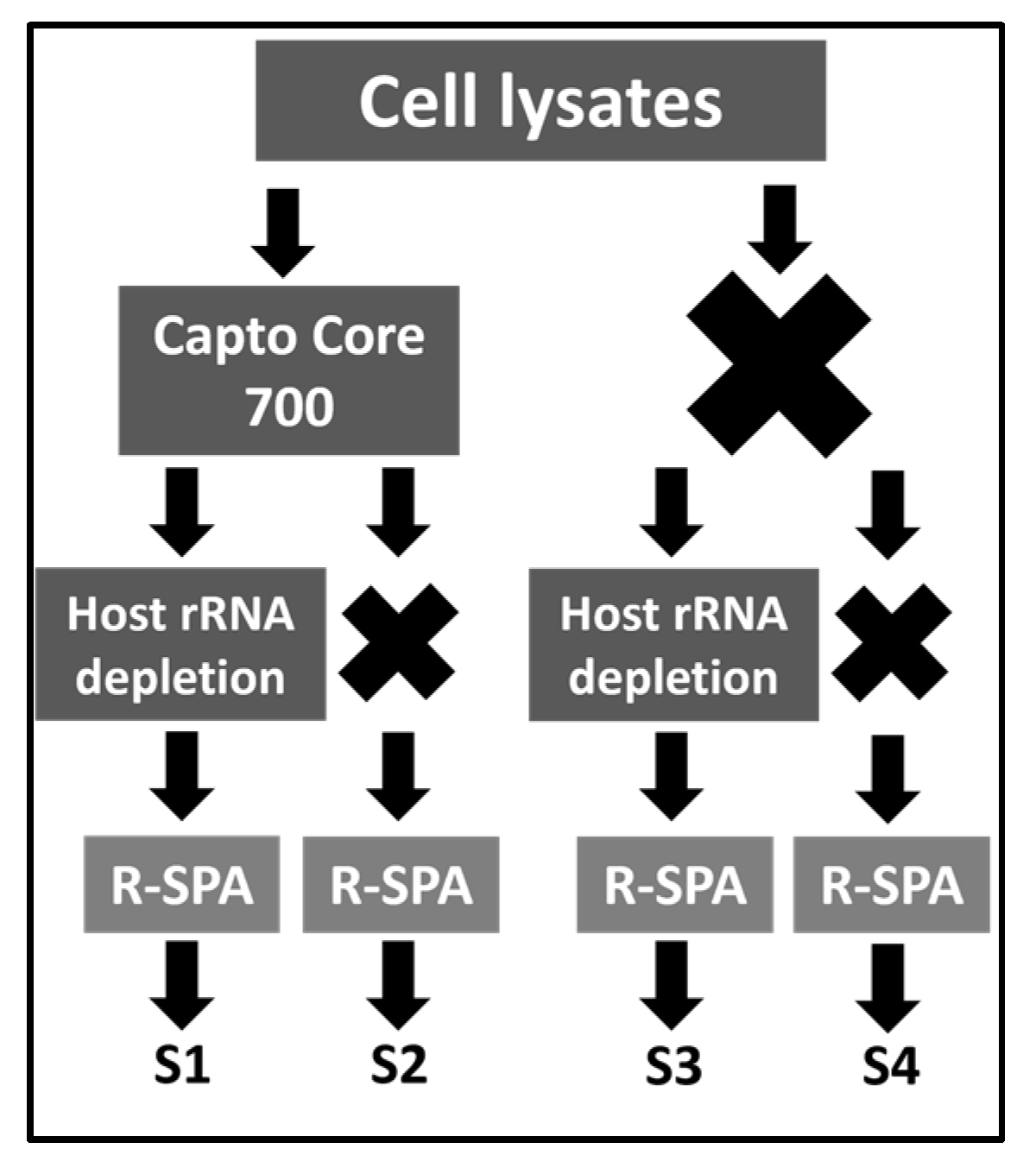
2.1. Experimental Design
2.2. Cell Lines and Culture Conditions
2.3. Purification of ARV Virions Using Capto Core 700 Resin
2.4. Purification of ARV Virions Using a Sucrose Gradient
2.5. Nuclease Treatment
2.6. RNA Extraction and Host/Bacteria rRNA Depletion
2.7. cDNA Production and Amplification using R-SPA
2.8. Whole-Genome Sequencing and Bioinformatic Analysis
2.9. Statistical Analysis
3. Results
3.1. Single Primer Amplification of ARV cDNA (R-SPA) Significantly Increases the Number of ARV-Mapping Reads Recovered after WGS
3.2. Optimization of Quality Reads for ARV Genome Purification
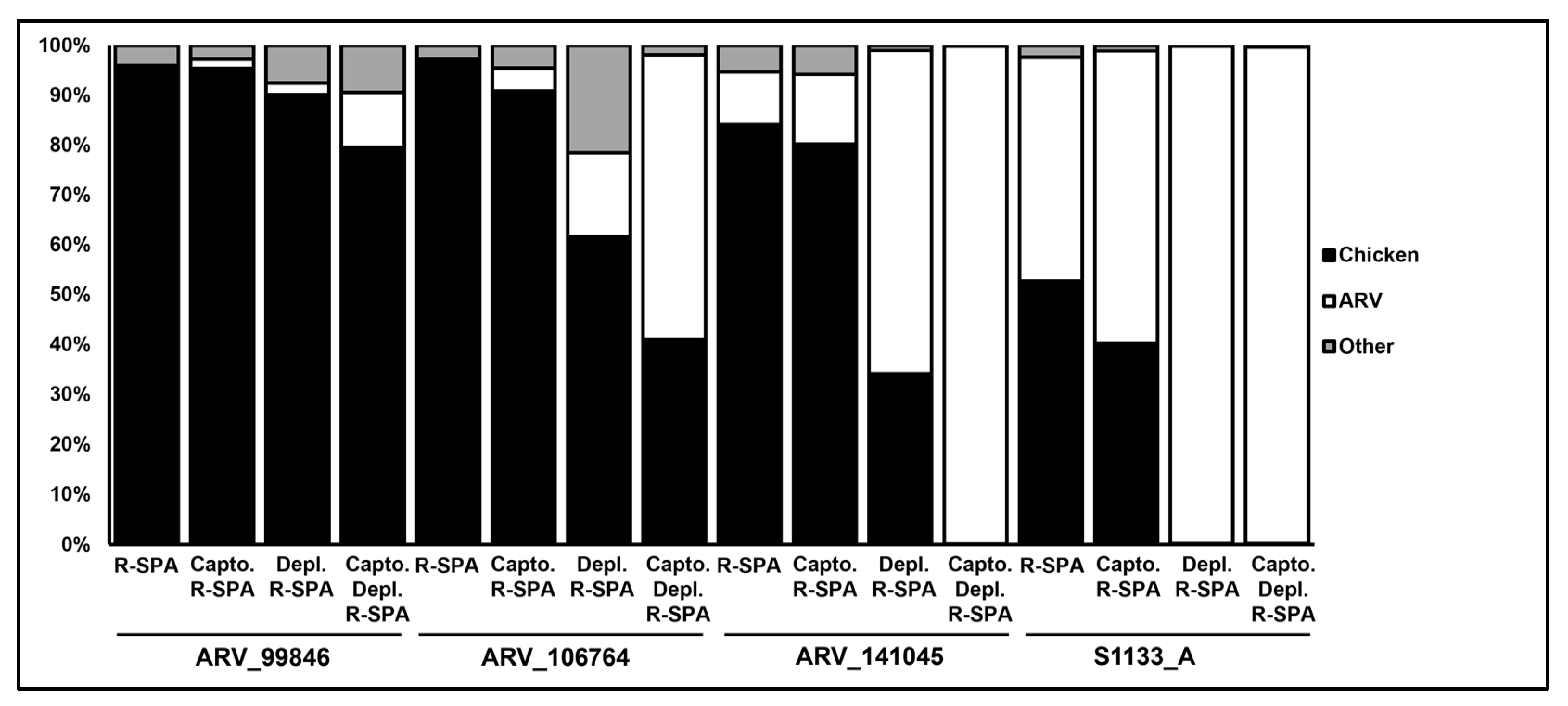
3.3. Short-Read Sequencing Can Detect a Mix of ARVs in a Clinical Isolate
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Benavente, J.; Martinez-Costas, J. Avian reovirus: Structure and biology. Virus Res. 2007, 123, 105–119. [Google Scholar] [CrossRef] [PubMed]
- Egana-Labrin, S.; Hauck, R.; Figueroa, A.; Stoute, S.; Shivaprasad, H.L.; Crispo, M.; Corsiglia, C.; Zhou, H.; Kern, C.; Crossley, B.; et al. Genotypic Characterization of Emerging Avian Reovirus Genetic Variants in California. Sci. Rep. 2019, 9, 9351. [Google Scholar] [CrossRef]
- Lu, H.; Tang, Y.; Dunn, P.A.; Wallner-Pendleton, E.A.; Lin, L.; Knoll, E.A. Isolation and molecular characterization of newly emerging avian reovirus variants and novel strains in Pennsylvania, USA, 2011–2014. Sci. Rep. 2015, 5, 14727. [Google Scholar] [CrossRef]
- Mase, M.; Gotou, M.; Inoue, D.; Masuda, T.; Watanabe, S.; Iseki, H. Genetic Analysis of Avian Reovirus Isolated from Chickens in Japan. Avian Dis. 2021, 65, 346–350. [Google Scholar] [CrossRef]
- Palomino-Tapia, V.; Mitevski, D.; Inglis, T.; van der Meer, F.; Abdul-Careem, M.F. Molecular characterization of emerging avian reovirus variants isolated from viral arthritis cases in Western Canada 2012-2017 based on partial sigma (σ)C gene. Virology 2018, 522, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Zou, Z.; Song, S.; Liu, H.; Gong, X.; Li, B.; Liu, P.; Wang, Q.; Liu, F.; Luan, D.; et al. Epidemiological Analysis of Avian Reovirus in China and Research on the Immune Protection of Different Genotype Strains from 2019 to 2020. Vaccines 2023, 11, 485. [Google Scholar] [CrossRef]
- Spandidos, D.A.; Graham, A.F. Physical and chemical characterization of an avian reovirus. J. Virol. 1976, 19, 968–976. [Google Scholar] [CrossRef]
- Zanaty, A.; Mosaad, Z.; Elfeil, W.M.K.; Badr, M.; Palya, V.; Shahein, M.A.; Rady, M.; Hess, M. Isolation and Genotypic Characterization of New Emerging Avian Reovirus Genetic Variants in Egypt. Poultry 2023, 2, 174–186. [Google Scholar] [CrossRef]
- De Carli, S.; Wolf, J.M.; Graf, T.; Lehmann, F.K.M.; Fonseca, A.S.K.; Canal, C.W.; Lunge, V.R.; Ikuta, N. Genotypic characterization and molecular evolution of avian reovirus in poultry flocks from Brazil. Avian Pathol. 2020, 49, 611–620. [Google Scholar] [CrossRef] [PubMed]
- Ayalew, L.E.; Ahmed, K.A.; Mekuria, Z.H.; Lockerbie, B.; Popowich, S.; Tikoo, S.K.; Ojkic, D.; Gomis, S. The dynamics of molecular evolution of emerging avian reoviruses through accumulation of point mutations and genetic re-assortment. Virus Evol. 2020, 6, veaa025. [Google Scholar] [CrossRef]
- Dovrolis, N.; Kassela, K.; Konstantinidis, K.; Kouvela, A.; Veletza, S.; Karakasiliotis, I. ZWA: Viral genome assembly and characterization hindrances from virus-host chimeric reads; a refining approach. PLoS Comput. Biol. 2021, 17, e1009304. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Yu, H.Y.; Jiang, X.N.; Bao, E.D.; Wang, D.; Lu, H.G. Genetic characterization of a novel pheasant-origin orthoreovirus using Next-Generation Sequencing. PLoS ONE 2022, 17, e0277411. [Google Scholar] [CrossRef] [PubMed]
- Chrzastek, K.; Sellers, H.; Kapczynski, D. A universal, single primer amplification protocol (R-SPA) to perform whole genome sequencing of segmented dsRNA reoviruses. bioRxiv 2021. [Google Scholar] [CrossRef]
- James, K.T.; Cooney, B.; Agopsowicz, K.; Trevors, M.A.; Mohamed, A.; Stoltz, D.; Hitt, M.; Shmulevitz, M. Novel High-throughput Approach for Purification of Infectious Virions. Sci. Rep. 2016, 6, 36826. [Google Scholar] [CrossRef]
- Parris, D.J.; Kariithi, H.; Suarez, D.L. Non-target RNA depletion strategy to improve sensitivity of next-generation sequencing for the detection of RNA viruses in poultry. J. Vet. Diagn. Investig. 2022, 34, 638–645. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing, S. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Gangiredla, J.; Rand, H.; Benisatto, D.; Payne, J.; Strittmatter, C.; Sanders, J.; Wolfgang, W.J.; Libuit, K.; Herrick, J.B.; Prarat, M.; et al. GalaxyTrakr: A distributed analysis tool for public health whole genome sequence data accessible to non-bioinformaticians. BMC Genom. 2021, 22, 114. [Google Scholar] [CrossRef]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Nakagomi, O. Fundamentals of Ultracentrifugal Virus Purification; Coulter, B., Ed.; Beckman Coulter: Nagasaki, Japan, 1998; p. 4. [Google Scholar]
- Taylor, M.K.; Williams, E.P.; Wongsurawat, T.; Jenjaroenpun, P.; Nookaew, I.; Jonsson, C.B. Amplicon-Based, Next-Generation Sequencing Approaches to Characterize Single Nucleotide Polymorphisms of Orthohantavirus Species. Front. Cell Infect. Microbiol. 2020, 10, 565591. [Google Scholar] [CrossRef] [PubMed]
- Ulhuq, F.R.; Barge, M.; Falconer, K.; Wild, J.; Fernandes, G.; Gallagher, A.; McGinley, S.; Sugadol, A.; Tariq, M.; Maloney, D.; et al. Analysis of the ARTIC V4 and V4.1 SARS-CoV-2 primers and their impact on the detection of Omicron BA.1 and BA.2 lineage-defining mutations. Microb. Genom. 2023, 9, mgen000991. [Google Scholar] [CrossRef] [PubMed]
- No, J.S.; Kim, W.K.; Cho, S.; Lee, S.H.; Kim, J.A.; Lee, D.; Song, D.H.; Gu, S.H.; Jeong, S.T.; Wiley, M.R.; et al. Comparison of targeted next-generation sequencing for whole-genome sequencing of Hantaan orthohantavirus in Apodemus agrarius lung tissues. Sci. Rep. 2019, 9, 16631. [Google Scholar] [CrossRef]
- Cytiva. Capto Core 400 and Capto Core 700 Multimodial Chromatography. 2020; p. 7. Available online: https://www.google.com/url?sa=t&rct=j&q=&esrc=s&source=web&cd=&ved=2ahUKEwiDofeZoqqBAxULhP0HHQ96A4gQFnoECB0QAQ&url=https%3A%2F%2Fcdn.cytivalifesciences.com%2Fapi%2Fpublic%2Fcontent%2Fdigi-16188-pdf&usg=AOvVaw35ufiJqO7JurIjpe82jYSK&opi=89978449 (accessed on 1 November 2022).
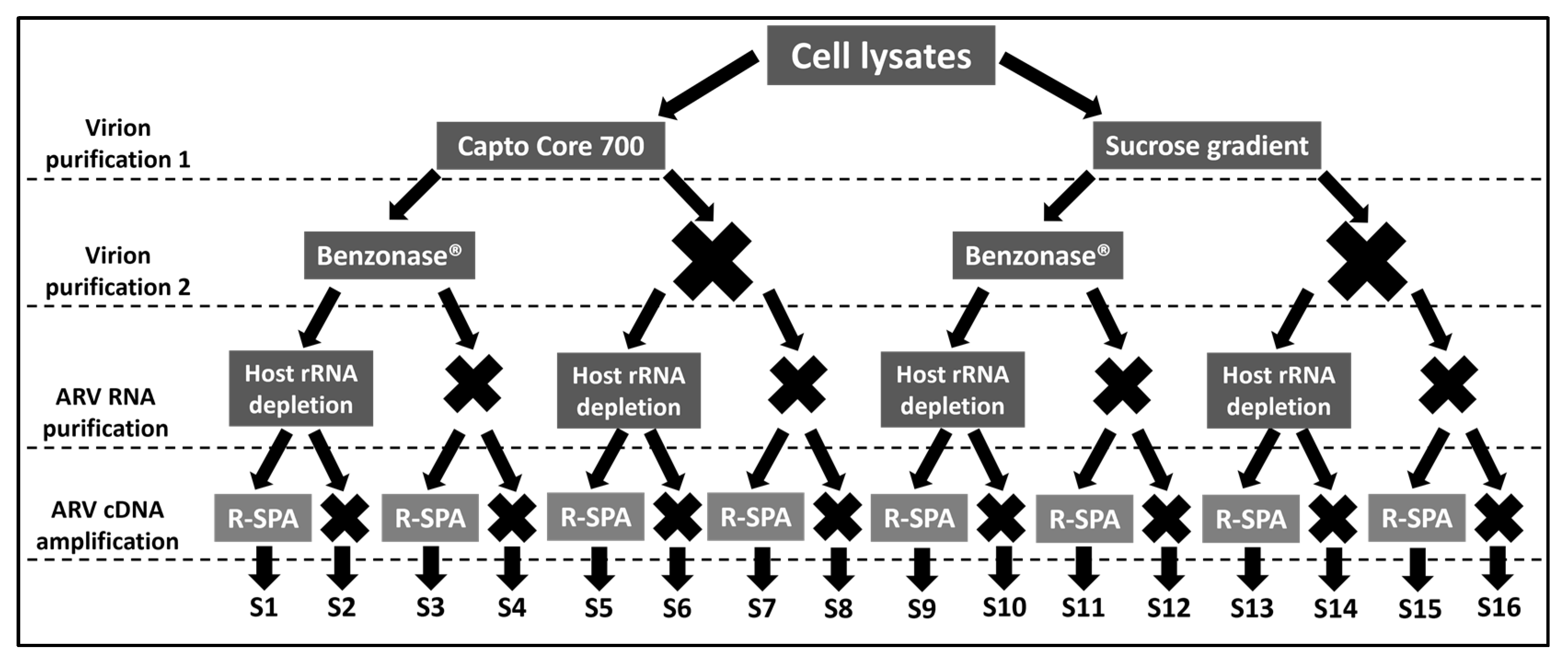
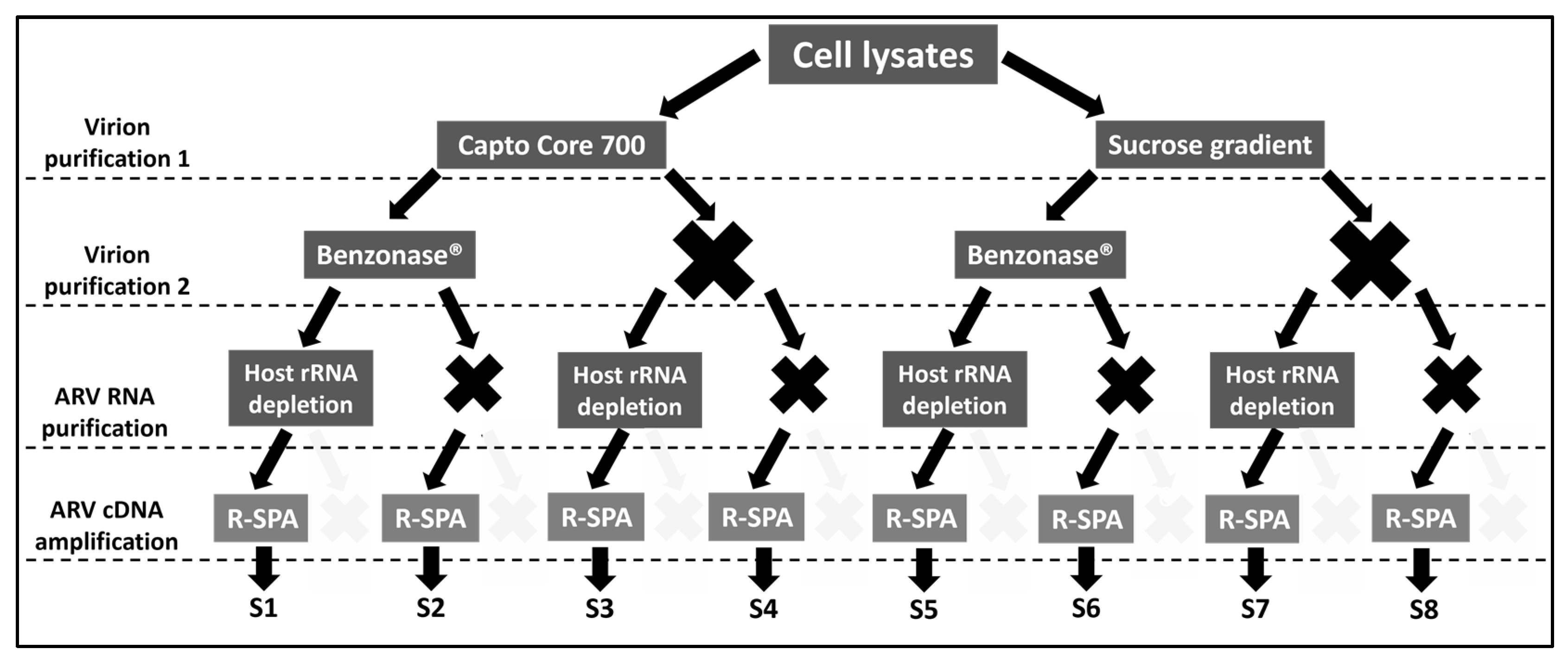
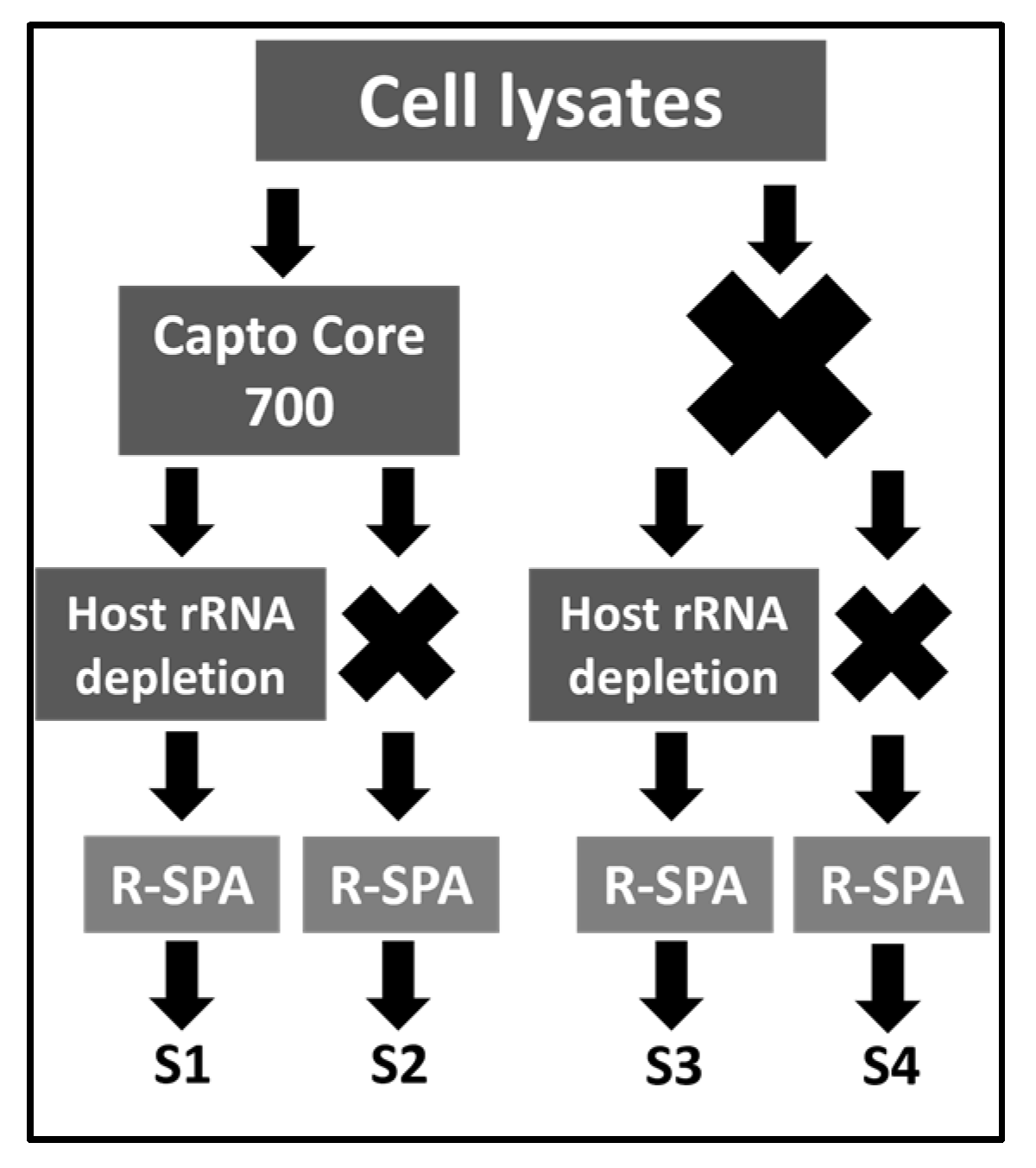



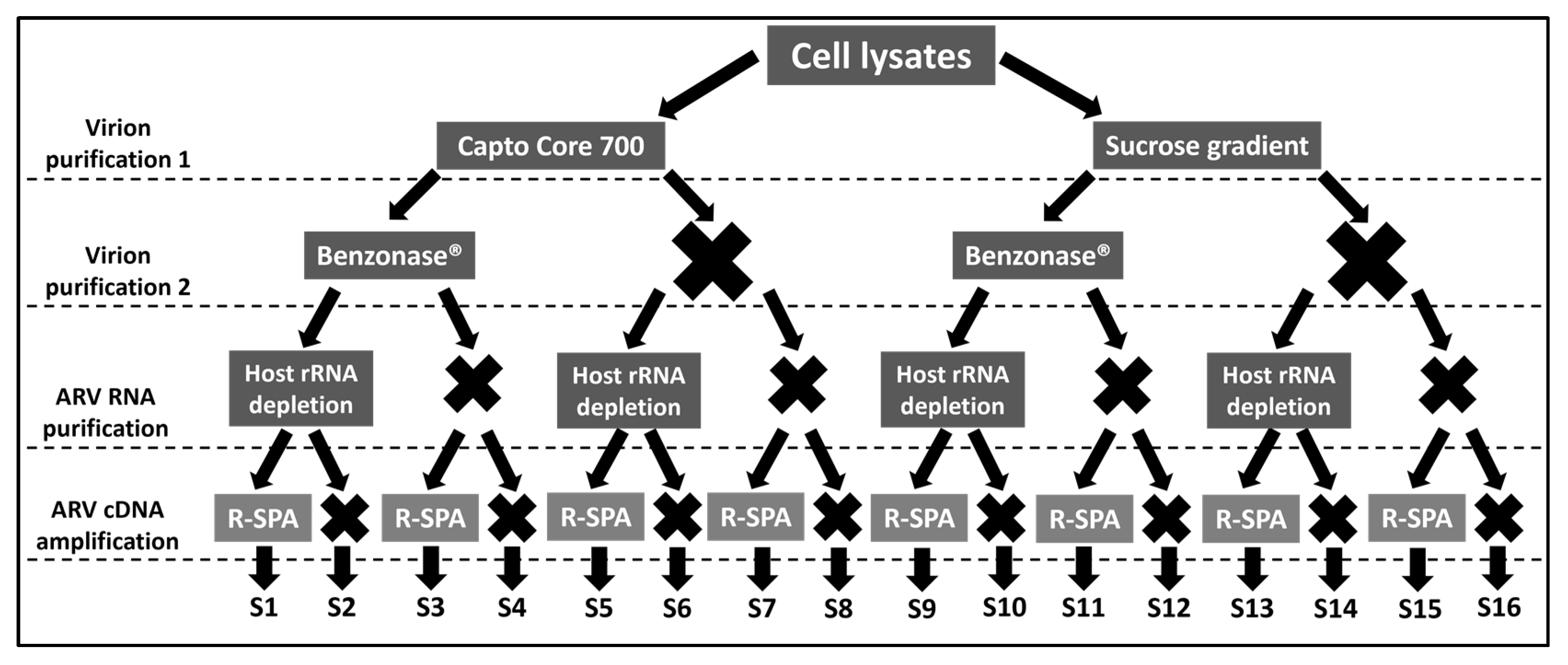{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Virion Purification Method | Benzonase Treatment | Host rRNA Depletion | R-SPA | Filtered Reads | Chicken- Mapping Filtered Reads | ARV-Mapping Filtered Reads | Total Contigs | ARV Contigs | Estimated Genome Coverage | Estimated Genome Length (bp) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| S1 | CaptoCore700 | - | - | - | 66,527 | 99.8% | 0 | 0 | 0 | 0 | 0 |
| S2 | CaptoCore700 | Benzonase | - | - | 204,945 | 54.2% | 45.1% | 32 | 23 | 951 | 24,320 |
| S3 | CaptoCore700 | - | Depletion | - | 44,945 | 63.3% | 39.0% | 19 | 13 | 202 | 20,769 |
| S4 | CaptoCore700 | Benzonase | Depletion | - | 7879 | 43.0% | 52.7% | 59 | 30 | 36 | 26,963 |
| S5 | CaptoCore700 | - | - | R-SPA | 30,665 | 98.3% | 0.1% | 0 | 0 | 0 | 0 |
| S6 | CaptoCore700 | Benzonase | - | R-SPA | 22,516 | 32.8% | 50.7% | 12 | 10 | 157 | 21,566 |
| S7 | CaptoCore700 | - | Depletion | R-SPA | 12,482 | 32.3% | 56.7% | 14 | 11 | 87 | 22,381 |
| S8 | CaptoCore700 | Benzonase | Depletion | R-SPA | 17,978 | 30.6% | 59.1% | 20 | 14 | 130 | 23,638 |
| S9 | Sucrose | - | - | - | 1082 | 85.8% | 6.1% | 0 | 0 | 0 | 0 |
| S10 | Sucrose | Benzonase | - | - | 7419 | 61.3% | 4.9% | 76 | 15 | 10 | 10,064 |
| S11 | Sucrose | - | Depletion | - | 1235 | 95.5% | 0.8% | 0 | 0 | 0 | 0 |
| S12 | Sucrose | Benzonase | Depletion | - | 971 | 78.1% | 1.6% | 0 | 0 | 0 | 0 |
| S13 | Sucrose | - | - | R-SPA | 81,379 | 75.6% | 22.6% | 24 | 17 | 671 | 12,081 |
| S14 | Sucrose | Benzonase | - | R-SPA | 35,942 | 22.2% | 77.6% | 13 | 12 | 316 | 22,011 |
| S15 | Sucrose | - | Depletion | R-SPA | 150,048 | 83.6% | 16.2% | 15 | 13 | 435 | 15,486 |
| S16 | Sucrose | Benzonase | Depletion | R-SPA | 71,016 | 65.5% | 34.4% | 19 | 13 | 300 | 18,042 |
| Virion Purification Method | Benzonase Treatment | Host rRNA Depletion | R-SPA | Filtered Reads | % Chicken- Mapping Filtered Reads | % ARV- Mapping Filtered Reads | Total Contigs | ARV Contigs | Average Genome Coverage | Estimated Genome Length (bp) |
|---|---|---|---|---|---|---|---|---|---|---|
| CaptoCore700 | - | - | R-SPA | 19,664 ± 7907 | 98 ± 0.2 | 1.2 ± 0.5 | 5 ± 5 | 5 ± 5 | 3 ± 3 | |
| CaptoCore700 | Benzonase | - | R-SPA | 37,309 ± 15,084 | 55 ± 18 | 26 ± 20 | 19 ± 5 | 10 ± 1 | 163 ± 5 | |
| CaptoCore700 | - | Depletion | R-SPA | 15,913 ± 5216 | 11 ± 10 | 82 ± 13 | 13 ± 1 | 11 ± 1 | 135 ± 35 | |
| CaptoCore700 | Benzonase | Depletion | R-SPA | 16,820 ± 5457 | 10 ± 10 | 86 ± 14 | 15 ± 3 | 13 ± 2 | 146 ± 37 | |
| Sucrose | - | - | R-SPA | 45,163 ± 20,557 | 55 ± 12 | 40 ± 10 | 17 ± 4 | 14 ± 2 | 352 ± 172 | |
| Sucrose | Benzonase | - | R-SPA | 46,464 ± 23,480 | 39 ± 28 | 57 ± 25 | 53 ± 38 | 30 ± 15 | 158 ± 80 | |
| Sucrose | - | Depletion | R-SPA | 87,287 ± 42,323 | 42 ± 24 | 53 ± 25 | 24 ± 15 | 15 ± 1 | 349 ± 167 | |
| Sucrose | Benzonase | Depletion | R-SPA | 28,528 ± 18,476 | 38 ± 20 | 62 ± 20 | 17 ± 1 | 13 ± 1 | 140 ± 79 |
| ID | Virion Purification Method | Host rRNA Depletion | R-SPA | Total Filtered Reads | % Chicken- Mapping Filtered Reads | % ARV- Mapping Filtered Reads | Total Contigs | ARV Contigs | Average Genome Coverage | Estimated Genome Length (bp) |
|---|---|---|---|---|---|---|---|---|---|---|
| ARV_99846 | - | - | R-SPA | 17,573 | 96 | 0 | 0 | 0 | 0 | 0 |
| CaptoCore700 | - | R-SPA | 13,942 | 95 | 2 | 3 | 3 | 12 | 6637 | |
| - | Depletion | R-SPA | 12,056 | 90 | 2 | 18 | 10 | 12 | 8229 | |
| CaptoCore700 | Depletion | R-SPA | 11,045 | 80 | 11 | 36 | 19 | 16 | 23,329 | |
| ARV_106764 | - | - | R-SPA | 5403 | 97 | 0 | 0 | 0 | 0 | 0 |
| CaptoCore700 | - | R-SPA | 14,616 | 91 | 5 | 6 | 5 | 20 | 10,214 | |
| - | Depletion | R-SPA | 7562 | 62 | 17 | 26 | 23 | 13 | 29,403 | |
| CaptoCore700 | Depletion | R-SPA | 34,643 | 41 | 57 | 33 | 23 | 130 | 48,244 | |
| ARV_141045 | - | - | R-SPA | 7743 | 84 | 11 | 11 | 11 | 12 | 17,087 |
| CaptoCore700 | - | R-SPA | 30,754 | 80 | 14 | 12 | 11 | 66 | 21,552 | |
| - | Depletion | R-SPA | 13,036 | 35 | 66 | 11 | 10 | 109 | 23,135 | |
| CaptoCore700 | Depletion | R-SPA | 11,408 | 0 | 100 | 11 | 11 | 115 | 22,870 | |
| S1133 | - | - | R-SPA | 4731 | 53 | 45 | 8 | 8 | 35 | 16,186 |
| CaptoCore700 | - | R-SPA | 5302 | 40 | 59 | 9 | 9 | 47 | 19,113 | |
| - | Depletion | R-SPA | 16,006 | 0 | 100 | 10 | 10 | 152 | 23,499 | |
| CaptoCore700 | Depletion | R-SPA | 4952 | 0 | 100 | 10 | 10 | 50 | 22,681 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Narvaez, S.A.; Harrell, T.L.; Oluwayinka, O.; Sellers, H.S.; Khalid, Z.; Hauck, R.; Chowdhury, E.U.; Conrad, S.J. Optimizing the Conditions for Whole-Genome Sequencing of Avian Reoviruses. Viruses 2023, 15, 1938. https://doi.org/10.3390/v15091938
Narvaez SA, Harrell TL, Oluwayinka O, Sellers HS, Khalid Z, Hauck R, Chowdhury EU, Conrad SJ. Optimizing the Conditions for Whole-Genome Sequencing of Avian Reoviruses. Viruses. 2023; 15(9):1938. https://doi.org/10.3390/v15091938
Chicago/Turabian StyleNarvaez, Sonsiray Alvarez, Telvin L. Harrell, Olatunde Oluwayinka, Holly S. Sellers, Zubair Khalid, Ruediger Hauck, Erfan U. Chowdhury, and Steven J. Conrad. 2023. "Optimizing the Conditions for Whole-Genome Sequencing of Avian Reoviruses" Viruses 15, no. 9: 1938. https://doi.org/10.3390/v15091938
APA StyleNarvaez, S. A., Harrell, T. L., Oluwayinka, O., Sellers, H. S., Khalid, Z., Hauck, R., Chowdhury, E. U., & Conrad, S. J. (2023). Optimizing the Conditions for Whole-Genome Sequencing of Avian Reoviruses. Viruses, 15(9), 1938. https://doi.org/10.3390/v15091938