
Mycoviruses in the Rust Fungus Uromyces fabae


Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Material, Fungal Isolate and Spore Propagation
2.2. Double-Stranded RNA Isolation from Urediospores
2.3. DNA Isolation from Urediospores
2.4. Purification of Virus-like Particles and Electron Microscopy
2.5. Sequencing and Assembly
2.6. Sequence Analysis
3. Results
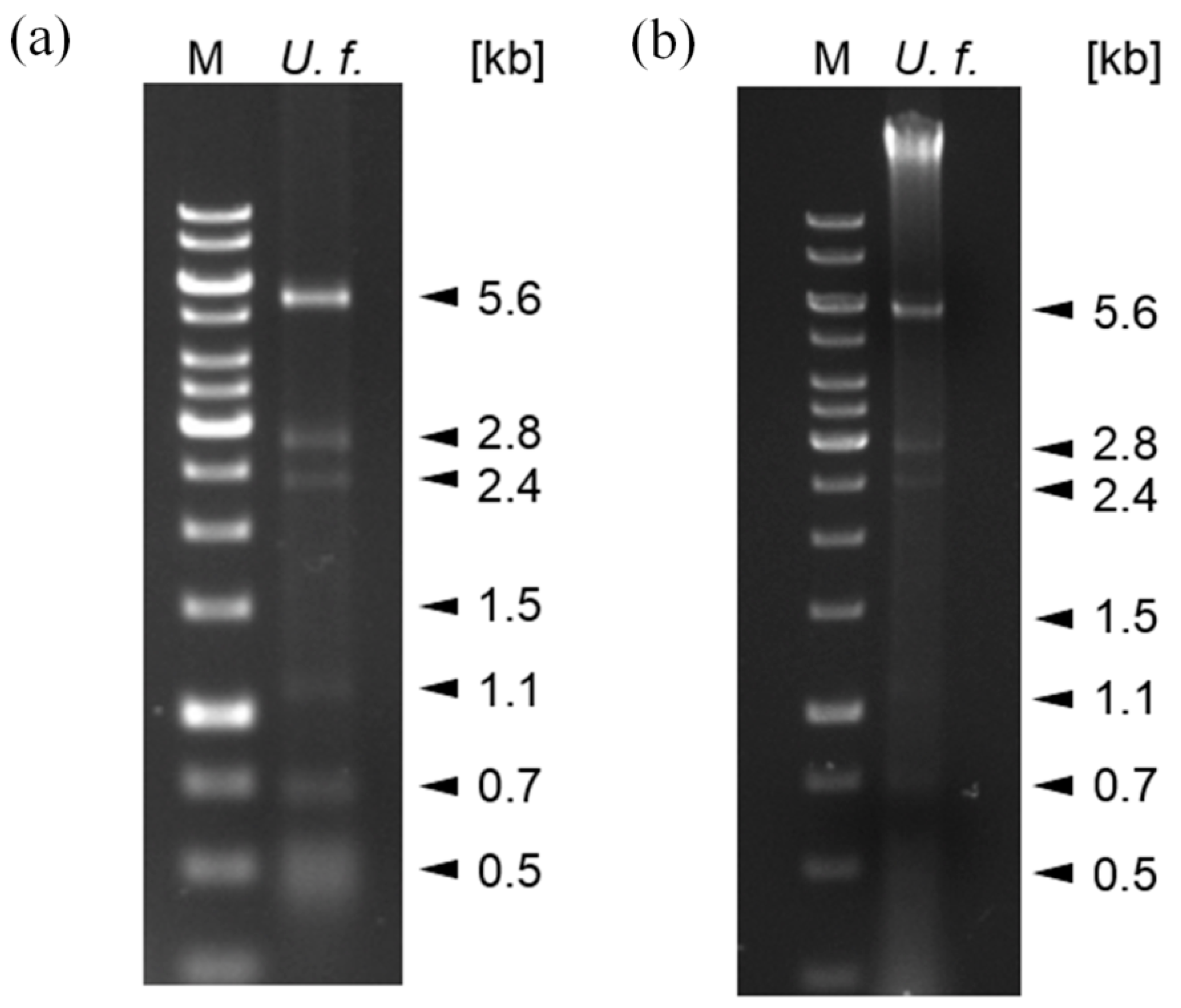
3.1. Seven Fractions of Double-Stranded RNA Can Be Distinguished in U. fabae
3.2. Viruses of U. fabae Have Isometric Particles
3.3. Sequencing of dsRNA Yields Evidence for Several Viral Genomes
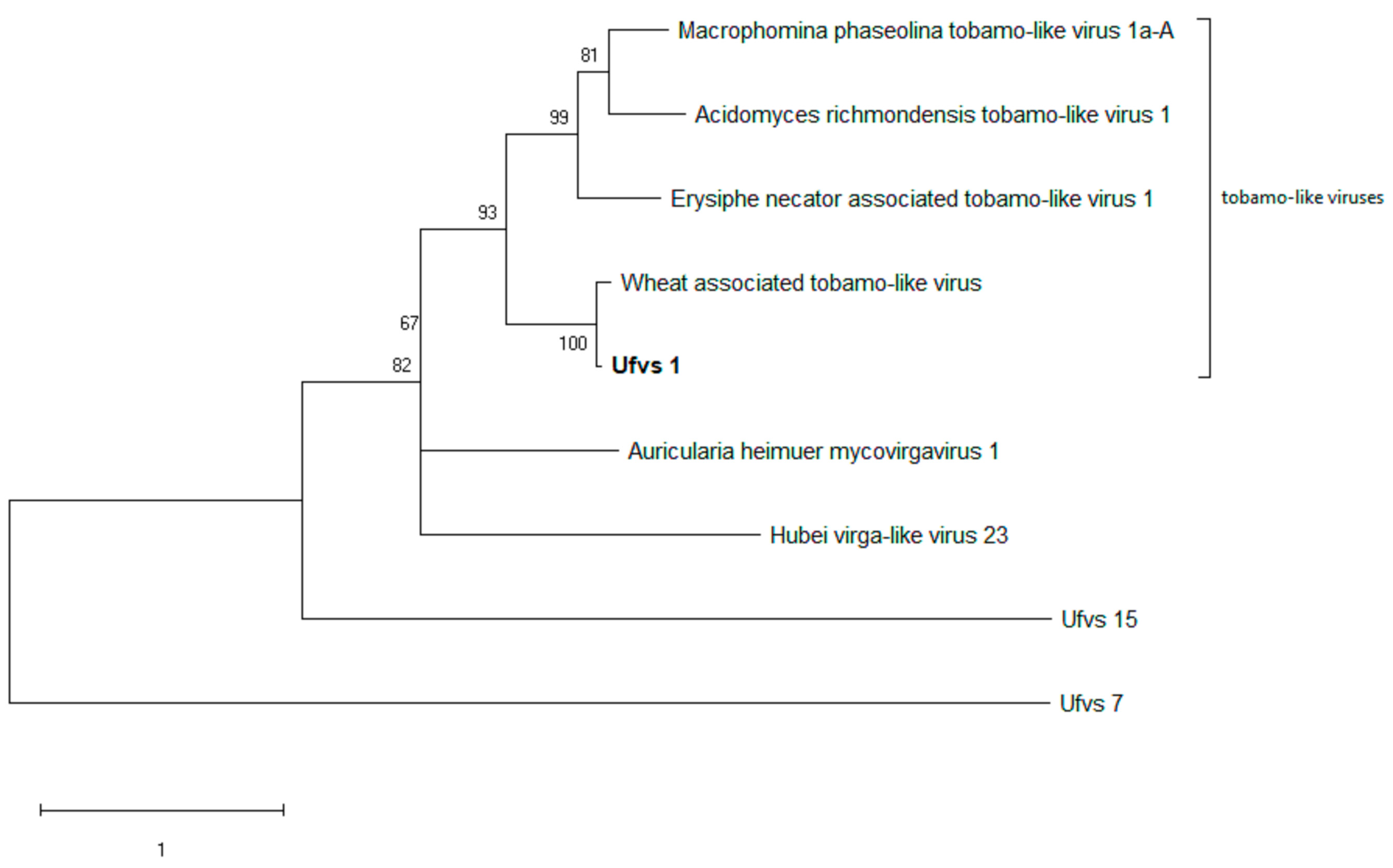
3.4. Phylogenetic Analysis of the Viral Sequences
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Voegele, R.T. Uromyces fabae: Development, metabolism, and interactions with its host Vicia faba. FEMS Microbiol. Lett. 2006, 259, 165–173. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rashid, K.Y.; Bernier, C.C. The effect of rust on yield of faba bean cultivars and slow-rusting populations. Can. J. Plant Sci. 1991, 71, 967–972. [Google Scholar] [CrossRef]
- Conner, R.L.; Bernier, C.C. Host range of Uromyces viciae-fabae. Phytopathology 1982, 72, 687–689. [Google Scholar] [CrossRef]
- Ghabrial, S.A.; Suzuki, N. Viruses of plant pathogenic fungi. Annu. Rev. Phytopathol. 2009, 47, 353–384. [Google Scholar] [CrossRef]
- Hollings, M. Viruses associated with a die-back disease of cultivated mushroom. Nature 1962, 196, 962–965. [Google Scholar] [CrossRef]
- Newton, A.C.; Caten, C.E.; Johnson, R. Variation for isozymes and double-stranded RNA among isolates of Puccinia striiformis and two other cereal rusts. Plant Pathol. 1985, 34, 235–247. [Google Scholar] [CrossRef]
- Zhang, R.; Dickinson, M.J.; Pryor, A. Double-stranded RNAs in the rust fungi. Annu. Rev. Phytopathol. 1994, 32, 115–133. [Google Scholar] [CrossRef]
- Ghabrial, S.A.; Caston, J.R.; Jiang, D.; Nibert, M.L.; Suzuki, N. 50-plus years of fungal viruses. Virology 2015, 479–480, 356–368. [Google Scholar] [CrossRef]
- Pearson, M.N.; Beever, R.E.; Boine, B.; Arthur, K. Mycoviruses of filamentous fungi and their relevance to plant pathology. Mol. Plant Pathol. 2009, 10, 115–128. [Google Scholar] [CrossRef]
- Hough, B.; Steenkamp, E.; Wingfield, B.; Read, D. Fungal viruses unveiled: A comprehensive review of mycoviruses. Viruses 2023, 15, 1202. [Google Scholar] [CrossRef]
- King, A.M.Q.; Lefkowitz, E.; Adams, M.J.; Carstens, E.B. Virus taxonomy: Classification and nomenclature of viruses. In Ninth Report of the International Committee on Taxonomy of Viruses; Elsevier Academic Press: Amsterdam, The Netherlands; Boston, MA, USA; Heidelberg, Germany; London, UK; New York, NY, USA; Oxford, UK; Paris, France; San Diego, CA, USA; San Francisco, CA, USA; Singapore; Sydney, Australia; Tokyo, Japan, 2011. [Google Scholar]
- Jakupovic, M.; Heintz, M.; Reichmann, P.; Mendgen, K.; Hahn, M. Microarray analysis of expressed sequence tags from haustoria of the rust fungus Uromyces fabae. Fungal Genet. Biol. 2006, 43, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Morris, T.J.; Dodds, J.A. Isolation and analysis of double-stranded RNA from virus-infected plant and fungal tissue. Phytopathology 1979, 69, 854–858. [Google Scholar] [CrossRef]
- Kolmer, J.A.; Liu, J.Q.; Sies, M. Virulence and molecular polymorphism in Puccinia recondita f. sp. tritici in Canada. Phytopathology 1995, 85, 276–285. [Google Scholar] [CrossRef]
- Sanderlin, R.S.; Ghabrial, S.A. Physicochemical properties of two distinct types of virus-like particles from Helminthosporium victoriae. Virology 1978, 87, 142–151. [Google Scholar] [CrossRef]
- Mulisch, M. Präparation für die TEM. In Romeis Mikroskopische Technik; Mulisch, M., Welsch, U., Eds.; Springer: Berlin/Heidelberg, Germany, 2015; pp. 121–152. [Google Scholar]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetic Analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Le, S.Q.; Gascuel, O. An improved general amino acid replacement matrix. Mol. Biol. Evol. 2008, 25, 1307–1320. [Google Scholar] [CrossRef]
- Sato, Y.; Jamal, A.; Kondo, H.; Suzuki, N. Molecular characterization of a novel polymycovirus from Penicillium janthinellum with a focus on its genome-associated PASrp. Front. Microbiol. 2020, 11, 592789. [Google Scholar] [CrossRef]
- Redila, C.D.; Prakash, V.; Nouri, S. Metagenomics analysis of the wheat virome identifies novel plant and fungal-associated viral sequences. Viruses 2021, 13, 2457. [Google Scholar] [CrossRef]
- Marzano, S.L.; Nelson, B.D.; Ajayi-Oyetunde, O.; Bradley, C.A.; Hughes, T.J.; Hartman, G.L.; Eastburn, D.M.; Domier, L.L. Identification of diverse mycoviruses through metatranscriptomics characterization of the viromes of five major fungal plant pathogens. J. Virol. 2016, 90, 6846–6863. [Google Scholar] [CrossRef]
- Gilbert, K.B.; Holcomb, E.E.; Allscheid, R.L.; Carrington, J.C. Hiding in plain sight: New virus genomes discovered via a systematic analysis of fungal public transcriptomes. PLoS ONE 2019, 14, e0219207. [Google Scholar] [CrossRef] [PubMed]
- Link, T.I.; Lang, P.; Scheffler, B.E.; Duke, M.V.; Graham, M.A.; Cooper, B.; Tucker, M.L.; Van de Mortel, M.; Voegele, R.T.; Mendgen, K.; et al. The haustorial transcriptomes of Uromyces appendiculatus and Phakopsora pachyrhizi and their candidate effector families. Mol. Plant Pathol. 2014, 15, 379–393. [Google Scholar] [CrossRef] [PubMed]
- Jo, Y.; Choi, H.; Chu, H.; Cho, W.K. Identification of viruses from fungal transcriptomes. bioRxiv 2020. [Google Scholar] [CrossRef]
- Zheng, L.; Lu, X.; Liang, X.; Jiang, S.; Zhao, J.; Zhan, G.; Liu, P.; Wu, J.; Kang, Z. Molecular characterization of novel totivirus-like double-stranded RNAs from Puccinia striiformis f. sp. tritici, the causal agent of wheat stripe rust. Front. Microbiol. 2017, 8, 1960. [Google Scholar] [CrossRef]
- Kondo, H.; Hisano, S.; Chiba, S.; Maruyama, K.; Andika, I.B.; Toyoda, K.; Fujimori, F.; Suzuki, N. Sequence and phylogenetic analyses of novel totivirus-like double-stranded RNAs from field-collected powdery mildew fungi. Virus Res. 2016, 213, 353–364. [Google Scholar] [CrossRef]
- Son, M.; Yu, J.; Kim, K.-H. Five questions about mycoviruses. PLoS Path. 2015, 11, e1005172. [Google Scholar] [CrossRef]
- Shi, M.; Lin, X.D.; Tian, J.H.; Chen, L.J.; Chen, X.; Li, C.X.; Qin, X.C.; Li, J.; Cao, J.P.; Eden, J.S.; et al. Redefining the invertebrate RNA virosphere. Nature 2016, 540, 539–543. [Google Scholar] [CrossRef]
- Nuss, D.L. Biological control of chestnut blight: An example of virus-mediated attenuation of fungal pathogenesis. Microbiol. Rev. 1992, 56, 561–576. [Google Scholar] [CrossRef]
- Nuss, D.L. Hypovirulence: Mycoviruses at the fungal-plant interface. Nat. Rev. Microbiol. 2005, 3, 632–642. [Google Scholar] [CrossRef]
- Zhang, Y.; Guo, H.; Zhou, S.; Chen, D.; Xu, G.; Kang, Z.; Zheng, L. A novel mitovirus PsMV2 facilitates the virulence of wheat stripe rust fungus. Viruses 2023, 15, 1265. [Google Scholar] [CrossRef]
- Zhang, Y.; Liang, X.; Zhao, M.; Qi, T.; Guo, H.; Zhao, J.; Zhan, G.; Kang, Z.; Zheng, L. A novel ambigrammatic mycovirus, PsV5, works hand in glove with wheat stripe rust fungus to facilitate infection. Plant Commun. 2023, 4, 100505. [Google Scholar] [CrossRef] [PubMed]
- Barilli, E.; Rubiales, D. Identification and characterization of resistance to rust in lentil and its wild relatives. Plants 2023, 12, 626. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus | Accession | Length (nt) | Open Reading Frames | |||||
|---|---|---|---|---|---|---|---|---|
| Position | Frame | AA | Top BLAST Hit | E Value | %ID | |||
| Ufvs_1 | OQ995224 | 10755 | 33–5366 | +3 | 1777 | methyltransferase, partial (Wheat-associated tobamo-like virus) UIN24825.1 | 0.0 | 64.36 |
| 6959–9529 | +2 | 856 | DEAD-like helicase (Wheat-associated tobamo-like virus) UIN24854.1 | 0.0 | 60.56 | |||
| 5589–6935 | +3 | 448 | RNA-dependent RNA polymerase (Wheat-associated tobamo-like virus) UIN24853.1 | 0.0 | 90.75 | |||
| 9618–10511 | +3 | 297 | hypothetical protein (Wheat-associated tobamo-like virus) UIN24855.1 | 4 × 10−164 | 77.74 | |||
| Ufvs_2 | OQ995225 | 5133 | 136–2607 | +1 | 823 | putative CP (Uromyces totivirus A) QED43025.1 | 0.0 | 70.81 |
| 2691–5108 | +3 | 805 | RdRp, partial (Uromyces totivirus A) QED43026.1 | 0.0 | 71.58 | |||
| Ufvs_3 | OQ995226 | 5001 | 63–2519 | +3 | 818 | capsid protein (Helianthus annuus leaf-associated totivirus 6) UMQ74227.1 | 0.0 | 70.29 |
| 3068–4981 | +2 | 637 | putative RdRp (Uromyces totivirus B) QED42952.1 | 0.0 | 70.65 | |||
| Ufvs_4 | OQ995227 | 4983 | 49–2532 | +1 | 827 | coat protein (Erysiphales-associated totivirus 6) QIP68055.1 | 0.0 | 43.70 |
| 2721–4979 | +3 | 752 | RNA-dependent RNA polymerase (Erysiphales-associated totivirus 6) QIP68054.1 | 0.0 | 51.07 | |||
| Ufvs_5 | OQ995228 | 4971 | 24–2222 | +3 | 732 | putative capsid protein (Poaceae Liege totivirus 8) UVG55934.1 | 2 × 10−175 | 39.53 |
| 2222–4951 | +2 | 909 | putative RNA-dependent RNA polymerase (Poaceae Liege totivirus 8) UVG55935.1 | 0.0 | 47.87 | |||
| Ufvs_6 | OQ995229 | 4967 | 76–2481 | +1 | 801 | putative capsid protein (Puccinia striiformis totivirus 4) ATO91012.1 | 0.0 | 72.03 |
| 2532–4922 | +3 | 796 | putative RNA-dependent RNA polymerase (Puccinia striiformis totivirus 4) ATO91013.1 | 0.0 | 67.93 | |||
| Ufvs_7 | OQ995230 | 4967 | 47–2521 | +2 | 824 | putative capsid protein (Puccinia striiformis totivirus 1) ATO91006.1 | 0.0 | 39.35 |
| 2560–4941 | +1 | 793 | putative RdRp (Phakopsora totivirus B) QED42972.1 | 0.0 | 68.70 | |||
| Ufvs_8 | OQ995231 | 4778 | 25–2268 | +1 | 747 | capsid protein (Helianthus annus leaf-associated totivirus 7) UMQ74228.1 | 0.0 | 88.95 |
| 2271–4751 | +3 | 826 | hypothetical protein 2 (Wuhan insect virus 26) YP_009342428.1 | 0.0 | 46.20 | |||
| Ufvs_9 | OQ995232 | 4758 | 276–2204 | +3 | 642 | hypothetical protein (Uromyces totivirus D) QED43018.1 | 0.0 | 80.53 |
| 2243–4735 | +2 | 830 | hypothetical protein, partial (Uromyces totivirus D) QED43019.1 | 0.0 | 80.29 | |||
| Ufvs_10 | OQ995233 | 4708 | 84–2144 | +3 | 686 | putative capsid protein (Puccinia striiformis totivirus 5) ATO91014.1 | 0.0 | 80.47 |
| 2327–4693 | +2 | 788 | putative RNA-dependent RNA polymerase (Puccinia striiformis totivirus 5) ATO91015.1 | 0.0 | 72.88 | |||
| Ufvs_11 | OQ995234 | 3054 | 89–2053 | +2 | 654 | putative capsid protein (Puccinia striiformis totivirus 1) ATO91006.1 | 2 × 10−131 | 35.19 |
| 2059–3012 | +1 | 317 | putative RdRp (Phakopsora totivirus E) QED42935.1 | 4 × 10−75 | 41.70 | |||
| Ufvs_12 | OQ995235 | 2885 | 196–2847 | +1 | 883 | RNA-dependent RNA polymerase (Erysiphales associated totivirus 19) QIP68078.1 | 0.0 | 46.36 |
| Ufvs_13 | OQ995236 | 2430 | 34–1881 | +1 | 615 | putative RdRp (Phakopsora totivirus E) QED42935.1 | 0.0 | 55.90 |
| 1826–2392 | +2 | 188 | RNA-dependent RNA polymerase (Red clover powdery mildew-associated totivirus 5) YP_009182188.1 | 7 × 10−13 | 26.37 | |||
| Ufvs_14 | OQ995237 | 2130 | 31–2055 | +1 | 674 | RdRp, partial (Uromyces virus B) QED43024.1 | 2 × 10−171 | 43.35 |
| Ufvs_15 | OQ995238 | 2097 | 211–2049 | +1 | 612 | RdRp, partial (Uromyces virus B) QED43024.1 | 0.0 | 94.92 |
| Ufvs_16 | OQ995239 | 2007 | 141–1925 | +3 | 594 | RNA-dependent RNA polymerase, partial (Helianthus annuus leaf-associated totivirus 3) UMQ74221.1 | 0.0 | 75.38 |
| Ufvs_17 | OQ995240 | 1592 | 27–1577 | +3 | 516 | putative RNA-dependent RNA polymerase (Puccinia striiformis totivirus 5) ATO91015.1 | 0.0 | 81.57 |
| Ufvs_18 | OQ995241 | 1560 | 45–1313 | +3 | 422 | p46 (Citrus concave gum-associated virus) UDN65939.1 | 4 × 10−70 | 36.56 |
| Ufvs_19 | OQ995242 | 1123 | 994–212 | −1 | 260 | nucleocapsid (Laurel Lake virus) YP_009667030.1 | 2 × 10−39 | 35.51 |
| Ufvs_20 | OQ995243 | 1113 | beg–753 | +1 | 250 | Movement protein (blackberry line pattern virus) WDD63193.1 | 5 × 10−31 | 36.41 |
| Virus Name | NCBI Accession No. of RdRp |
|---|---|
| Acidomyces richmondensis tobamo-like virus 1 | AZT88673:1 |
| Aspergillus fumigatus polymycovirus 1 | BCH36613.1 |
| Aspergillus spelaeus tetramycovirus 1 | YP_010839683.1 |
| Auricularia heimuer mycovirgavirus 1 | QIM57886.1 |
| Beauveria bassiana polymycovirus 1 | VCV25414.1 |
| Delisea pulchra totivirus IndA | AMB17468 |
| Delisea pulchra totivirus IndA | AMB17473 |
| Delisea pulchra totivirus IndA | AMB17477 |
| Delisea pulchra totivirus IndA | AMB17478 |
| Delisea pulchra totivirus IndA | AMB17470 |
| Diplodia seriata polymycovirus 1 | UOK20165.1 |
| Erysiphales associated totivirus 3 | QIP68048.1 |
| Erysiphales associated totivirus 4 | QIP68050.1 |
| Erysiphales associated totivirus 6 | QIP68054.1 |
| Erysiphe necator associated tobamo-like virus 1 | QKN22701.1 |
| Hubei toti-like virus 2 | YP_009336496.1 |
| Hubei toti-like virus 3 | APG76078.1 |
| Hubei toti-like virus 4 | APG76044.1 |
| Hubei virga-like virus 23 | YP_009337439.1 |
| Macrophomina phaseolina tobamo-like virus 1a-A | QOE55599.1 |
| Maize associated totivirus 1 | AWD38954.1 |
| Maize-associated totivirus 2 | YP_009259486.1 |
| Penicillium brevicompactum tetramycovirus 1 | YP_010086053.1 |
| Phakopsora pachyrhizi mycovirus | ALO81041.1 |
| Phakopsora totivirus A | QED42974.1 |
| Phakopsora totivirus C | QED43023.1 |
| Poaceae Liege totivirus 9 | UVG55937.1 |
| Poaceae Liege totivirus 10 | UVG55939.1 |
| Puccinia striiformis totivirus 1 | ATO91007.1 |
| Puccinia striiformis totivirus 2 | ATO91009.1 |
| Puccinia striiformis totivirus 3 | ATO91011.1 |
| Puccinia striiformis totivirus 4 | ATO91013.1 |
| Red clover powdery mildew-associated totivirus 1 | BAT62478.1 |
| Red clover powdery mildew-associated totivirus 2 | YP_009182176.1 |
| Red clover powdery mildew-associated totivirus 3 | YP_009182181.1 |
| Red clover powdery mildew-associated totivirus 4 | BAT62484.1 |
| Red clover powdery mildew-associated totivirus 6 | YP_009182190.1 |
| Red clover powdery mildew-associated totivirus 7 | YP_009182195.1 |
| Red clover powdery mildew-associated totivirus 8 | BAT62492.1 |
| Saccharomyces paradoxus virus L-A-45 | ATL63182.1 |
| Scheffersomyces segobiensis virus L | YP_009507831.1 |
| Sclerotinia sclerotiorum tetramycovirus-1 | AWY10945.1 |
| Trichoderma koningiopsis totivirus 1 | QGA70771.1 |
| Tuber aestivum virus 1 | YP_009507833.1 |
| Wuhan insect virus 26 | YP_009342428.1 |
| Wuhan insect virus 27 | YP_009342434.1 |
| Xanthophyllomyces dendrorhous virus L1A | YP_007697651.1 |
| Xanthophyllomyces dendrorhous virus L1B | YP_009507835.1 |
| XiangYun toti-like virus 8 | UUG74262.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seitz, J.M.; Voegele, R.T.; Link, T.I. Mycoviruses in the Rust Fungus Uromyces fabae. Viruses 2023, 15, 1692. https://doi.org/10.3390/v15081692
Seitz JM, Voegele RT, Link TI. Mycoviruses in the Rust Fungus Uromyces fabae. Viruses. 2023; 15(8):1692. https://doi.org/10.3390/v15081692
Chicago/Turabian StyleSeitz, Janina M., Ralf T. Voegele, and Tobias I. Link. 2023. "Mycoviruses in the Rust Fungus Uromyces fabae" Viruses 15, no. 8: 1692. https://doi.org/10.3390/v15081692
APA StyleSeitz, J. M., Voegele, R. T., & Link, T. I. (2023). Mycoviruses in the Rust Fungus Uromyces fabae. Viruses, 15(8), 1692. https://doi.org/10.3390/v15081692