
The Evolution of Highly Pathogenic Avian Influenza A (H5) in Poultry in Nigeria, 2021–2022

 ,
,  , , , , , , , , and
, , , , , , , , and
Abstract
1. Introduction
2. Materials and Methods
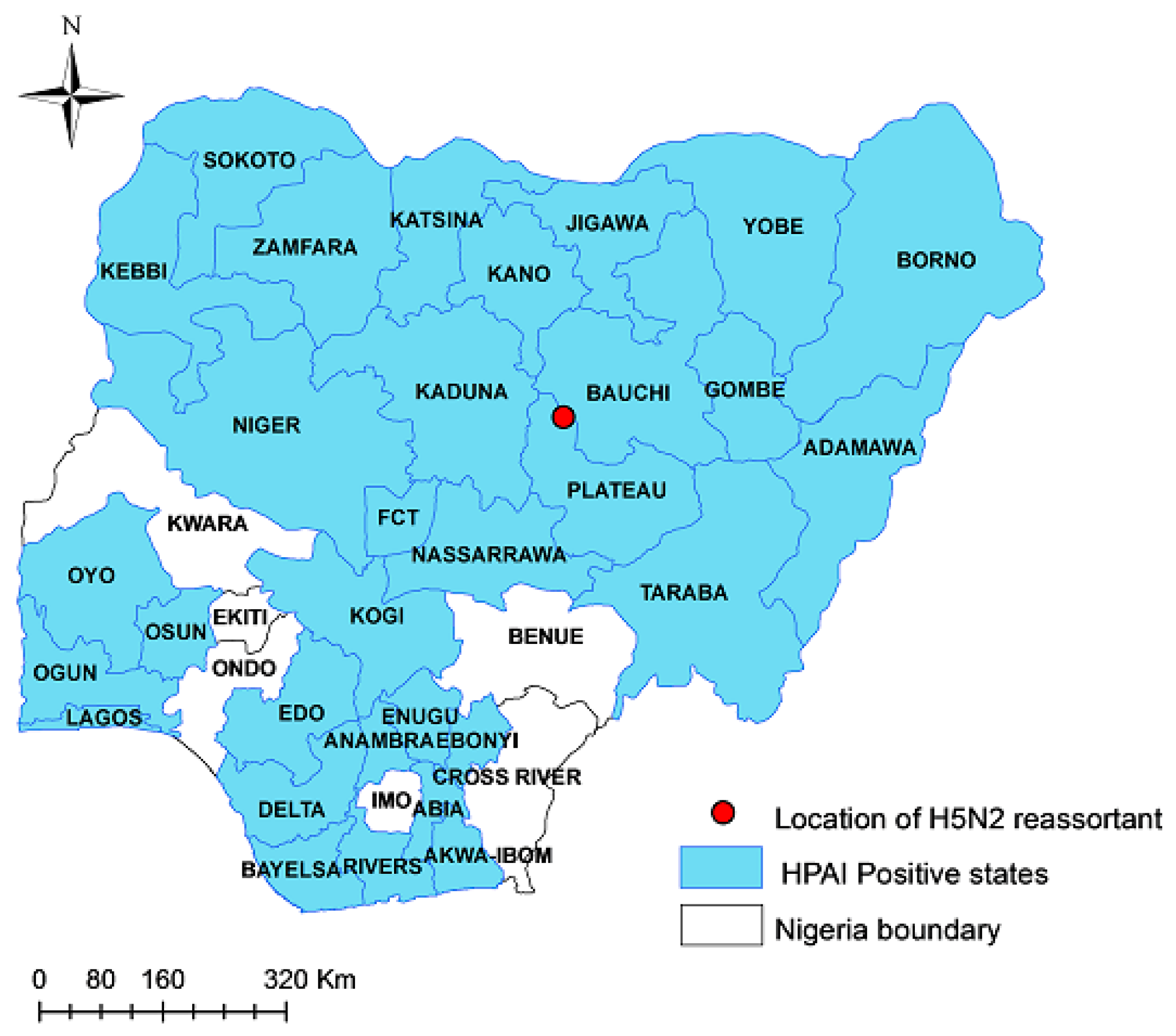
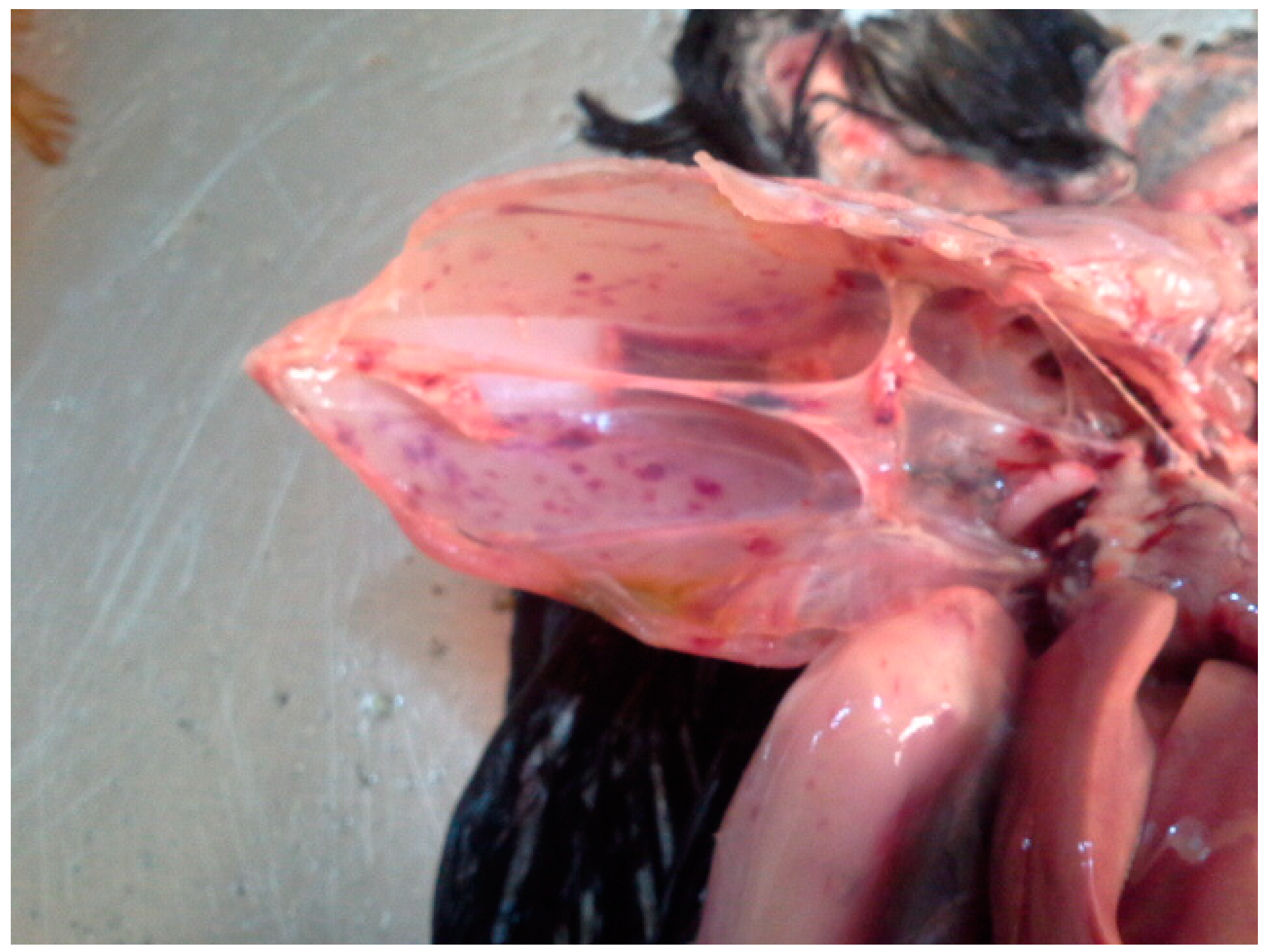
2.1. Sample Collection and Necropsy
2.2. Virus Identification and Isolation
2.3. Genomic Sequencing and Phylogenetic Analysis
3. Results
3.1. Virus Identification and Pathological Findings
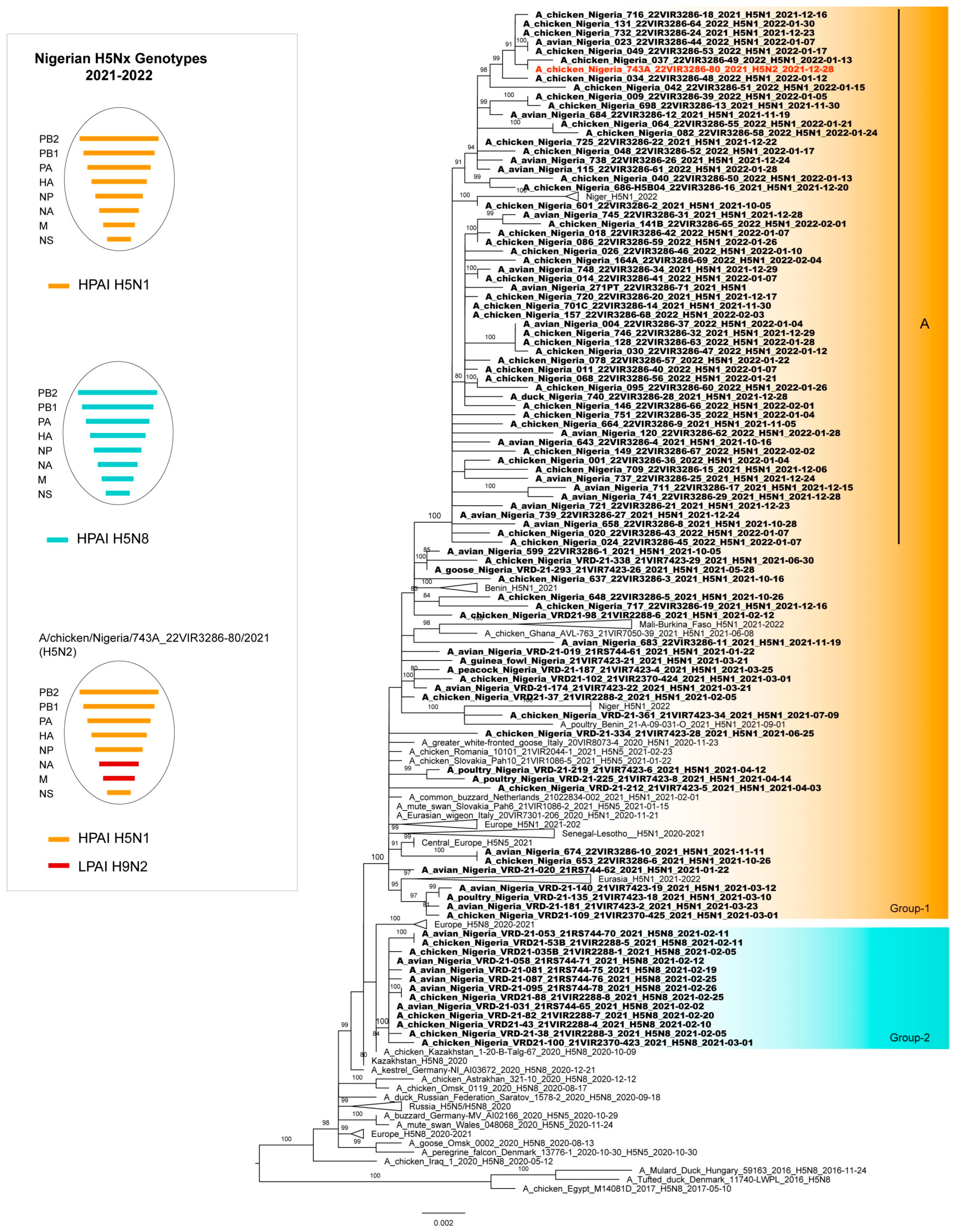
3.2. Phylogenetic Analyses
3.3. Genetic Characterization
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Omotayo, A.O.; Omotoso, A.B.; Daud, S.A.; Omotayo, O.P.; Adeniyi, B.A. Rising Food Prices and Farming Households Food Insecurity during the COVID-19 Pandemic: Policy Implications from SouthWest Nigeria. Agriculture 2022, 12, 363. [Google Scholar] [CrossRef]
- Morse, S.S.; Mazet, J.A.K.; Woolhouse, M.; Parrish, C.R.; Carroll, D.; Karesh, W.B.; Zambrana-Torrelio, C.; Lipkin, W.I.; Daszak, P. Prediction and Prevention of the next Pandemic Zoonosis. Lancet 2012, 380, 1956–1965. [Google Scholar] [CrossRef]
- Lutz, M.M.; Dunagan, M.M.; Kurebayashi, Y.; Takimoto, T. Key Role of the Influenza A Virus PA Gene Segment in the Emergence of Pandemic Viruses. Viruses 2020, 12, 365. [Google Scholar] [CrossRef]
- de Bruin, A.C.M.; Funk, M.; Spronken, M.I.; Gultyaev, A.P.; Fouchier, R.A.M.; Richard, M. Hemagglutinin Subtype Specificity and Mechanisms of Highly Pathogenic Avian Influenza Virus Genesis. Viruses 2022, 14, 1566. [Google Scholar] [CrossRef]
- Webster, R.G.; Bean, W.J.; Gorman, O.T.; Chambers, T.M.; Kawaoka, Y. Evolution and Ecology of Influenza A Viruses. Microbiol. Rev. 1992, 56, 152–179. [Google Scholar] [CrossRef] [PubMed]
- Fusaro, A.; Zecchin, B.; Vrancken, B.; Abolnik, C.; Ademun, R.; Alassane, A.; Arafa, A.; Awuni, J.A.; Couacy-Hymann, E.; Coulibaly, M.B.; et al. Disentangling the Role of Africa in the Global Spread of H5 Highly Pathogenic Avian Influenza. Nat. Commun. 2019, 10, 5310. [Google Scholar] [CrossRef]
- Adlhoch, C.; Fusaro, A.; Gonzales, J.L.; Kuiken, T.; Marangon, S.; Niqueux, É.; Staubach, C.; Terregino, C.; Guajardo, I.M.; Chuzhakina, K.; et al. Avian Influenza Overview June–September 2022. EFSA J. Eur. Food Saf. Auth. 2022, 20, e07597. [Google Scholar] [CrossRef]
- Meseko, C.; Globig, A.; Ijomanta, J.; Joannis, T.; Nwosuh, C.; Shamaki, D.; Harder, T.; Hoffman, D.; Pohlmann, A.; Beer, M.; et al. Evidence of Exposure of Domestic Pigs to Highly Pathogenic Avian Influenza H5N1 in Nigeria. Sci. Rep. 2018, 8, 5900. [Google Scholar] [CrossRef]
- Joannis, T.M.; Meseko, C.A.; Oladokun, A.T.; Ularamu, H.G.; Egbuji, A.N.; Solomon, P.; Nyam, D.C.; Gado, D.A.; Luka, P.; Ogedengbe, M.E.; et al. Serologic and Virologic Surveillance of Avian Influenza in Nigeria, 2006–2007. Eurosurveillance 2008, 13, 19007. [Google Scholar] [CrossRef]
- Fusaro, A.; Joannis, T.; Monne, I.; Salviato, A.; Yakubu, B.; Meseko, C.; Oladokun, T.; Fassina, S.; Capua, I.; Cattoli, G. Introduction into Nigeria of a Distinct Genotype of Avian Influenza Virus (H5N1). Emerg. Infect. Dis. 2009, 15, 445–447. [Google Scholar] [CrossRef] [PubMed]
- Oladokun, A.T.; Meseko, C.A.; Ighodalo, E.; John, B.; Ekong, P.S. Effect of Intervention on the Control of Highly Pathogenic Avian Influenza in Nigeria. Pan. Afr. Med. J. 2012, 13, 14. [Google Scholar]
- Coker, T.; Meseko, C.; Odaibo, G.; Olaleye, D. Circulation of the Low Pathogenic Avian Influenza Subtype H5N2 Virus in Ducks at a Live Bird Market in Ibadan, Nigeria. Infect. Dis. Poverty 2014, 3, 38. [Google Scholar] [CrossRef]
- Monne, I.; Meseko, C.; Joannis, T.; Shittu, I.; Ahmed, M.; Tassoni, L.; Fusaro, A.; Cattoli, G. Highly Pathogenic Avian Influenza A(H5N1) Virus in Poultry, Nigeria, 2015. Emerg. Infect. Dis. 2015, 21, 1275–1277. [Google Scholar] [CrossRef]
- Tassoni, L.; Fusaro, A.; Milani, A.; Lemey, P.; Awuni, J.A.; Sedor, V.B.; Dogbey, O.; Commey, A.N.O.; Meseko, C.; Joannis, T.; et al. Genetically Different Highly Pathogenic Avian Influenza A(H5N1) Viruses in West Africa, 2015. Emerg. Infect. Dis. 2016, 22, 2132–2136. [Google Scholar] [CrossRef]
- Laleye, A.T.; Bianco, A.; Shittu, I.; Sulaiman, L.; Fusaro, A.; Inuwa, B.; Oyetunde, J.; Zecchin, B.; Bakam, J.; Pastori, A.; et al. Genetic Characterization of Highly Pathogenic Avian Influenza H5Nx Clade 2.3.4.4b Reveals Independent Introductions in Nigeria. Transbound. Emerg. Dis. 2022, 69, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Shittu, I.; Bianco, A.; Gado, D.; Mkpuma, N.; Sulaiman, L.; Laleye, A.; Gobbo, F.; Bortolami, A.; Bonfante, F.; Vakuru, C.; et al. First Detection of Highly Pathogenic H5N6 Avian Influenza Virus on the African Continent. Emerg. Microbes Infect. 2020, 9, 886–888. [Google Scholar] [CrossRef] [PubMed]
- Sulaiman, L.; Shittu, I.; Fusaro, A.; Inuwa, B.; Zecchin, B.; Gado, D.; Schivo, A.; Bianco, A.; Laleye, A.; Gobbo, F.; et al. Live Bird Markets in Nigeria: A Potential Reservoir for H9N2 Avian Influenza Viruses. Viruses 2021, 13, 1445. [Google Scholar] [CrossRef] [PubMed]
- Gaidet, N.; Cattoli, G.; Hammoumi, S.; Newman, S.H.; Hagemeijer, W.; Takekawa, J.Y.; Cappelle, J.; Dodman, T.; Joannis, T.; Gil, P.; et al. Evidence of Infection by H5N2 Highly Pathogenic Avian Influenza Viruses in Healthy Wild Waterfowl. PLoS Pathog. 2008, 4, e1000127. [Google Scholar] [CrossRef]
- Snoeck, C.J.; Adeyanju, A.T.; de Landtsheer, S.; Ottosson, U.; Manu, S.; Hagemeijer, W.; Mundkur, T.; Muller, C.P. Reassortant Low-Pathogenic Avian Influenza H5N2 Viruses in African Wild Birds. J. Gen. Virol. 2011, 92, 1172–1183. [Google Scholar] [CrossRef]
- Meseko, C.A.; Ehizibolo, D.O.; Vakuru, C. Migratory Waterfowls from Europe as Potential Source of Highly Pathogenic Avian Influenza Infection to Nigeria Poultry. Niger. Vet. J. 2018, 39, 1. [Google Scholar] [CrossRef]
- Spackman, E.; Senne, D.A.; Myers, T.J.; Bulaga, L.L.; Garber, L.P.; Perdue, M.L.; Lohman, K.; Daum, L.T.; Suarez, D.L. Development of a Real-Time Reverse Transcriptase PCR Assay for Type A Influenza Virus and the Avian H5 and H7 Hemagglutinin Subtypes. J. Clin. Microbiol. 2002, 40, 3256–3260. [Google Scholar] [CrossRef]
- Slomka, M.J.; Pavlidis, T.; Banks, J.; Shell, W.; McNally, A.; Essen, S.; Brown, I.H. Validated H5 Eurasian Real-Time Reverse Transcriptase-Polymerase Chain Reaction and Its Application in H5N1 Outbreaks in 2005–2006. Avian Dis. 2007, 51, 373–377. [Google Scholar] [CrossRef]
- Monne, I.; Ormelli, S.; Salviato, A.; De Battisti, C.; Bettini, F.; Salomoni, A.; Drago, A.; Zecchin, B.; Capua, I.; Cattoli, G. Development and Validation of a One-Step Real-Time PCR Assay for Simultaneous Detection of Subtype H5, H7, and H9 Avian Influenza Viruses. J. Clin. Microbiol. 2008, 46, 1769–1773. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, B.; Hoffmann, D.; Henritzi, D.; Beer, M.; Harder, T.C. Riems Influenza a Typing Array (RITA): An RT-QPCR-Based Low Density Array for Subtyping Avian and Mammalian Influenza a Viruses. Sci. Rep. 2016, 6, 27211. [Google Scholar] [CrossRef]
- Panzarin, V.; Marciano, S.; Fortin, A.; Brian, I.; D’amico, V.; Gobbo, F.; Bonfante, F.; Palumbo, E.; Sakoda, Y.; Le, K.T.; et al. Redesign and Validation of a Real-Time RT-PCR to Improve Surveillance for Avian Influenza Viruses of the H9 Subtype. Viruses 2022, 14, 1263. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ Data to High Confidence Variant Calls: The Genome Analysis Toolkit Best Practices Pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef]
- Wilm, A.; Aw, P.P.K.; Bertrand, D.; Yeo, G.H.T.; Ong, S.H.; Wong, C.H.; Khor, C.C.; Petric, R.; Hibberd, M.L.; Nagarajan, N. LoFreq: A Sequence-Quality Aware, Ultra-Sensitive Variant Caller for Uncovering Cell-Population Heterogeneity from High-Throughput Sequencing Datasets. Nucleic Acids Res. 2012, 40, 11189–11201. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; Von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- European Food Safety Authority; European Centre for Disease Prevention and Control; European Union Reference Laboratory for Avian Influenza; Adlhoch, C.; Fusaro, A.; Gonzales, J.L.; Kuiken, T.; Marangon, S.; Mirinaviciute, G.; Niqueux, É.; et al. Avian Influenza Overview December 2022–March 2023. EFSA J. 2023, 21, e07917. [Google Scholar] [CrossRef]
- Yang, Z.-Y.; Wei, C.-J.; Kong, W.-P.; Wu, L.; Xu, L.; Smith, D.F.; Nabel, G.J. Immunization by Avian H5 Influenza Hemagglutinin Mutants with Altered Receptor Binding Specificity. Science 2007, 317, 825–828. [Google Scholar] [CrossRef] [PubMed]
- Suttie, A.; Deng, Y.M.; Greenhill, A.R.; Dussart, P.; Horwood, P.F.; Karlsson, E.A. Inventory of Molecular Markers Affecting Biological Characteristics of Avian Influenza A Viruses. Virus Genes 2019, 55, 739–768. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Gu, M.; Liu, K.; Li, Q.; Li, J.; Shi, L.; Li, X.; Wang, X.; Hu, J.; Liu, X.; et al. T160A Mutation-Induced Deglycosylation at Site 158 in Hemagglutinin Is a Critical Determinant of the Dual Receptor Binding Properties of Clade 2.3.4.4 H5NX Subtype Avian Influenza Viruses. Vet. Microbiol. 2018, 217, 158–166. [Google Scholar] [CrossRef]
- Sun, H.; Deng, G.; Sun, H.; Song, J.; Zhang, W.; Li, H.; Wei, X.; Li, F.; Zhang, X.; Liu, J.; et al. N-Linked Glycosylation Enhances Hemagglutinin Stability in Avian H5N6 Influenza Virus to Promote Adaptation in Mammals. PNAS Nexus 2022, 1, pgac085. [Google Scholar] [CrossRef] [PubMed]
- Munier, S.; Larcher, T.; Cormier-Aline, F.; Soubieux, D.; Su, B.; Guigand, L.; Labrosse, B.; Cherel, Y.; Quéré, P.; Marc, D.; et al. A Genetically Engineered Waterfowl Influenza Virus with a Deletion in the Stalk of the Neuraminidase Has Increased Virulence for Chickens. J. Virol. 2010, 84, 940–952. [Google Scholar] [CrossRef]
- Banks, J.; Speidel, E.S.; Moore, E.; Plowright, L.; Piccirillo, A.; Capua, I.; Cordioli, P.; Fioretti, A.; Alexander, D.J. Changes in the Haemagglutinin and the Neuraminidase Genes Prior to the Emergence of Highly Pathogenic H7N1 Avian Influenza Viruses in Italy. Arch. Virol. 2001, 146, 963–973. [Google Scholar] [CrossRef]
- Croville, G.; Soubies, S.M.; Barbieri, J.; Klopp, C.; Mariette, J.; Bouchez, O.; Camus-Bouclainville, C.; Guérin, J.L. Field Monitoring of Avian Influenza Viruses: Whole-Genome Sequencing and Tracking of Neuraminidase Evolution Using 454 Pyrosequencing. J. Clin. Microbiol. 2012, 50, 2881–2887. [Google Scholar] [CrossRef]
- Li, J.; Dohna, H.Z.; Cardona, C.J.; Miller, J.; Carpenter, T.E. Emergence and Genetic Variation of Neuraminidase Stalk Deletions in Avian Influenza Viruses. PLoS ONE 2011, 6, e14722. [Google Scholar] [CrossRef]
- Gao, Y.; Zhang, Y.; Shinya, K.; Deng, G.; Jiang, Y.; Li, Z.; Guan, Y.; Tian, G.; Li, Y.; Shi, J.; et al. Identification of Amino Acids in HA and PB2 Critical for the Transmission of H5N1 Avian Influenza Viruses in a Mammalian Host. PLoS Pathog. 2009, 5, e1000709. [Google Scholar] [CrossRef] [PubMed]
- Le, Q.M.; Sakai-Tagawa, Y.; Ozawa, M.; Ito, M.; Kawaoka, Y. Selection of H5N1 Influenza Virus PB2 during Replication in Humans. J. Virol. 2009, 83, 5278–5281. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Chen, H.; Jiao, P.; Deng, G.; Tian, G.; Li, Y.; Hoffmann, E.; Webster, R.G.; Matsuoka, Y.; Yu, K. Molecular Basis of Replication of Duck H5N1 Influenza Viruses in a Mammalian Mouse Model. J. Virol. 2005, 79, 12058–12064. [Google Scholar] [CrossRef]
- Steel, J.; Lowen, A.C.; Mubareka, S.; Palese, P. Transmission of Influenza Virus in a Mammalian Host Is Increased by PB2 Amino Acids 627K or 627E/701N. PLoS Pathog. 2009, 5, e1000252. [Google Scholar] [CrossRef] [PubMed]
- Taft, A.S.; Ozawa, M.; Fitch, A.; Depasse, J.V.; Halfmann, P.J.; Hill-Batorski, L.; Hatta, M.; Friedrich, T.C.; Lopes, T.J.S.; Maher, E.A.; et al. Identification of Mammalian-Adapting Mutations in the Polymerase Complex of an Avian H5N1 Influenza Virus. Nat. Commun. 2015, 6, 7491. [Google Scholar] [CrossRef] [PubMed]
- Yamayoshi, S.; Kiso, M.; Yasuhara, A.; Ito, M.; Shu, Y.; Kawaoka, Y. Enhanced Replication of Highly Pathogenic Influenza A(H7N9) Virus in Humans. Emerg. Infect. Dis. 2018, 24, 746–750. [Google Scholar] [CrossRef]
- Pinto, R.M.; Bakshi, S.; Lytras, S.; Zakaria, M.K.; Swingler, S.; Worrell, J.C.; Herder, V.; Varjak, M.; Cameron-Ruiz, N.; Rodriguez, M.C.; et al. Zoonotic Avian Influenza Viruses Evade Human BTN3A3 Restriction. bioRxiv 2022. [Google Scholar] [CrossRef]
- Fan, S.; Deng, G.; Song, J.; Tian, G.; Suo, Y.; Jiang, Y.; Guan, Y.; Bu, Z.; Kawaoka, Y.; Chen, H. Two Amino Acid Residues in the Matrix Protein M1 Contribute to the Virulence Difference of H5N1 Avian Influenza Viruses in Mice. Virology 2009, 384, 28–32. [Google Scholar] [CrossRef]
- Guo, J.; Chen, J.; Li, Y.; Li, Y.; Deng, G.; Shi, J.; Liu, L.; Chen, H.; Li, X. SUMOylation of Matrix Protein M1 and Filamentous Morphology Collectively Contribute to the Replication and Virulence of Highly Pathogenic H5N1 Avian Influenza Viruses in Mammals. J. Virol. 2022, 96, e0163021. [Google Scholar] [CrossRef]
- Cheung, C.L.; Rayner, J.M.; Smith, G.J.D.; Wang, P.; Naipospos, T.S.P.; Zhang, J.; Yuen, K.Y.; Webster, R.G.; Peiris, J.S.M.; Guan, Y.; et al. Distribution of Amantadine-Resistant H5N1 Avian Influenza Variants in Asia. J. Infect. Dis. 2006, 193, 1626–1629. [Google Scholar] [CrossRef]
- Lan, Y.; Zhang, Y.; Dong, L.; Wang, D.; Huang, W.; Xin, L.; Yang, L.; Zhao, X.; Li, Z.; Wang, W.; et al. A Comprehensive Surveillance of Adamantane Resistance among Human Influenza A Virus Isolated from Mainland China between 1956 and 2009. Antivir. Ther. 2010, 15, 853–859. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Young, J.S.; Jeung, H.P.; Lee, J.H.; Yun, H.B.; Song, M.S.; Oh, T.K.; Han, H.S.; Pascua, P.N.Q.; Choi, Y.K. Emergence of Amantadine-Resistant H3N2 Avian Influenza A Virus in South Korea. J. Clin. Microbiol. 2008, 46, 3788–3790. [Google Scholar] [CrossRef]
- Jiao, P.; Tian, G.; Li, Y.; Deng, G.; Jiang, Y.; Liu, C.; Liu, W.; Bu, Z.; Kawaoka, Y.; Chen, H. A Single-Amino-Acid Substitution in the NS1 Protein Changes the Pathogenicity of H5N1 Avian Influenza Viruses in Mice. J. Virol. 2008, 82, 1146–1154. [Google Scholar] [CrossRef] [PubMed]
- Kuo, R.-L.; Krug, R.M. Influenza a Virus Polymerase Is an Integral Component of the CPSF30-NS1A Protein Complex in Infected Cells. J. Virol. 2009, 83, 1611–1616. [Google Scholar] [CrossRef]
- Imai, H.; Shinya, K.; Takano, R.; Kiso, M.; Muramoto, Y.; Sakabe, S.; Murakami, S.; Ito, M.; Yamada, S.; thi Quynh Le, M.; et al. The HA and NS Genes of Human H5N1 Influenza A Virus Contribute to High Virulence in Ferrets. PLoS Pathog. 2010, 6, e1001106. [Google Scholar] [CrossRef]
- Miotto, O.; Heiny, A.T.; Tan, T.W.; Thomas, J.T.; Brusic, V. Identification of Human-to-Human Transmissibility Factors in PB2 Proteins of Influenza A by Large-Scale Mutual Information Analysis. BMC Bioinform. 2008, 9, S18. [Google Scholar] [CrossRef]
- Finkelstein, D.B.; Mukatira, S.; Mehta, P.K.; Obenauer, J.C.; Su, X.; Webster, R.G.; Naeve, C.W. Persistent Host Markers in Pandemic and H5N1 Influenza Viruses. J. Virol. 2007, 81, 10292–10299. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.E.; Gardner, S.N.; Vitalis, E.A.; Slezak, T.R. Conserved Amino Acid Markers from Past Influenza Pandemic Strains. BMC Microbiol. 2009, 9, 77. [Google Scholar] [CrossRef] [PubMed]
- Heikkinen, L.S.; Kazlauskas, A.; Melén, K.; Wagner, R.; Ziegler, T.; Julkunen, I.; Saksela, K. Avian and 1918 Spanish Influenza a Virus NS1 Proteins Bind to Crk/CrkL Src Homology 3 Domains to Activate Host Cell Signaling. J. Biol. Chem. 2008, 283, 5719–5727. [Google Scholar] [CrossRef] [PubMed]
- Hassan, K.E.; Saad, N.; Abozeid, H.H.; Shany, S.; El-Kady, M.F.; Arafa, A.; EL-Sawah, A.A.A.; Pfaff, F.; Hafez, H.M.; Beer, M.; et al. Genotyping and Reassortment Analysis of Highly Pathogenic Avian Influenza Viruses H5N8 and H5N2 from Egypt Reveals Successive Annual Replacement of Genotypes. Infect. Genet. Evol. 2020, 84, 104375. [Google Scholar] [CrossRef]
- Chen, J.; Xu, L.; Liu, T.; Xie, S.; Li, K.; Li, X.; Zhang, M.; Wu, Y.; Wang, X.; Wang, J.; et al. Novel Reassortant Avian Influenza A(H5N6) Virus, China, 2021. Emerg. Infect. Dis. 2022, 28, 1701–1707. [Google Scholar] [CrossRef] [PubMed]
- Ouoba, L.B.; Habibata-Zerbo, L.; Zecchin, B.; Barbierato, G.; Hamidou-Ouandaogo, S.; Palumbo, E.; Giussani, E.; Bortolami, A.; Niang, M.; Traore-Kam, A.; et al. Emergence of a Reassortant 2.3.4.4b Highly Pathogenic H5N1 Avian Influenza Virus Containing H9N2 PA Gene in Burkina Faso, West Africa, in 2021. Viruses 2022, 14, 1901. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Zhang, X.; Liu, F.; Yao, H.; Wu, N.; Wu, H. Rapid Emergence of a PB2 D701N Substitution during Adaptation of an H9N2 Avian Influenza Virus in Mice. Arch. Virol. 2022, 167, 2299–2303. [Google Scholar] [CrossRef] [PubMed]
- Peacock, T.P.; James, J.; Sealy, J.E.; Iqbal, M. A Global Perspective on H9N2 Avian Influenza Virus. Viruses 2019, 11, 620. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Protein | Genetic Marker | Phenotypic Effect | Virus Name | References |
|---|---|---|---|---|
| PB2 | T105V | May be associated with human adaptation (statistical analysis) | A/chicken/Nigeria/648_22VIR3286-5/2021 | [56] |
| D701N | Mammalian adaptive marker | A/chicken/Nigeria/743A_22VIR3286-80/2021 (H5N2) | [41,42,43,44,45] | |
| K482R | Increase polymerase activity in mammalian cell line | A/avian/Nigeria/271PT_22VIR3286-71/2021 | [46] | |
| PA | D55N | Host specificity marker through statistical methods (D in avian, N in human) | A/guinea_fowl/Nigeria/VRD-21-169_21VIR7423-21/2021 | [57] |
| S409N | Host specificity marker through statistical methods (S in avian, N in human) | A/chicken/Nigeria/040_22VIR3286-50/2022 | [57] | |
| HA | PLREKRRKR/ GLF | HPAI cleavage site | All viruses | Not available |
| S137A | Increased α2-6 sialic acid virus binding | All viruses except for A/avian/Nigeria/VRD-21-019_21RS744-61/2021 | [33] | |
| S158N | Increased α2-6 sialic acid virus binding | All viruses | [34] | |
| T160A | Increased α2-6 sialic acid virus binding | In all the H5N8 strains, and in eighteen H5N1 viruses | [34,35] | |
| NA | 44-65 del in the stalk region | Enhanced virulence, marker of viral adaptation from wild bird to poultry | In 73 out of 82 H5N1 viruses | [37,38,39,40] |
| NP | Y52H | Confer resistance to BTN3A3 (butyrophilin subfamily 3 member A3) inhibitor | In 82 out of 83 H5N1 viruses | [47] |
| M1 | N30D, T215A | Increased virulence in mice | All viruses | [48,49] |
| M2 | V27I | Associated with amantadine resistance | A/avian/Nigeria/741_22VIR3286-29/2021 A/chicken/Nigeria/751_22VIR3286-35/2022 | [52] |
| S31N | Associated with amantadine resistance | A/chicken/Nigeria/743A_22VIR3286-80/2021 (H5N2) | [50,51] | |
| NS1 | P42S | Increase virulence in mice and could be critical to antagonize the interferon induction | All viruses | [53] |
| P87S | Host specificity marker through statistical methods (S in human, P in avian) | All viruses with the exception of A/chicken/Nigeria/725_22VIR3286-22/2021 A/avian/Nigeria/115_22VIR3286-61/2022 | [58] | |
| L103F | Increase virulence in mice | All viruses | [54] | |
| I106M | Increase virulence in mice | All viruses | [54] | |
| K217E | Decrease replicative or pathogenic potential of the virus | A/chicken/Nigeria/157_22VIR3286-68/2022 | [59] | |
| N205S | Decreased antiviral response in host | All H5N1 viruses | [55] | |
| NS2 | T48A | Decreased antiviral response in host | All H5N1 viruses | [55] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meseko, C.; Milani, A.; Inuwa, B.; Chinyere, C.; Shittu, I.; Ahmed, J.; Giussani, E.; Palumbo, E.; Zecchin, B.; Bonfante, F.; et al. The Evolution of Highly Pathogenic Avian Influenza A (H5) in Poultry in Nigeria, 2021–2022. Viruses 2023, 15, 1387. https://doi.org/10.3390/v15061387
Meseko C, Milani A, Inuwa B, Chinyere C, Shittu I, Ahmed J, Giussani E, Palumbo E, Zecchin B, Bonfante F, et al. The Evolution of Highly Pathogenic Avian Influenza A (H5) in Poultry in Nigeria, 2021–2022. Viruses. 2023; 15(6):1387. https://doi.org/10.3390/v15061387
Chicago/Turabian StyleMeseko, Clement, Adelaide Milani, Bitrus Inuwa, Chinonyerem Chinyere, Ismaila Shittu, James Ahmed, Edoardo Giussani, Elisa Palumbo, Bianca Zecchin, Francesco Bonfante, and et al. 2023. "The Evolution of Highly Pathogenic Avian Influenza A (H5) in Poultry in Nigeria, 2021–2022" Viruses 15, no. 6: 1387. https://doi.org/10.3390/v15061387
APA StyleMeseko, C., Milani, A., Inuwa, B., Chinyere, C., Shittu, I., Ahmed, J., Giussani, E., Palumbo, E., Zecchin, B., Bonfante, F., Maniero, S., Angot, A., Niang, M., Fusaro, A., Gobbo, F., Terregino, C., Olasoju, T., Monne, I., & Muhammad, M. (2023). The Evolution of Highly Pathogenic Avian Influenza A (H5) in Poultry in Nigeria, 2021–2022. Viruses, 15(6), 1387. https://doi.org/10.3390/v15061387