

Idiotope-Driven T-Cell/B-Cell Collaboration-Based T-Cell Epitope Prediction Using B-Cell Receptor Repertoire Sequences in Infectious Diseases



Abstract
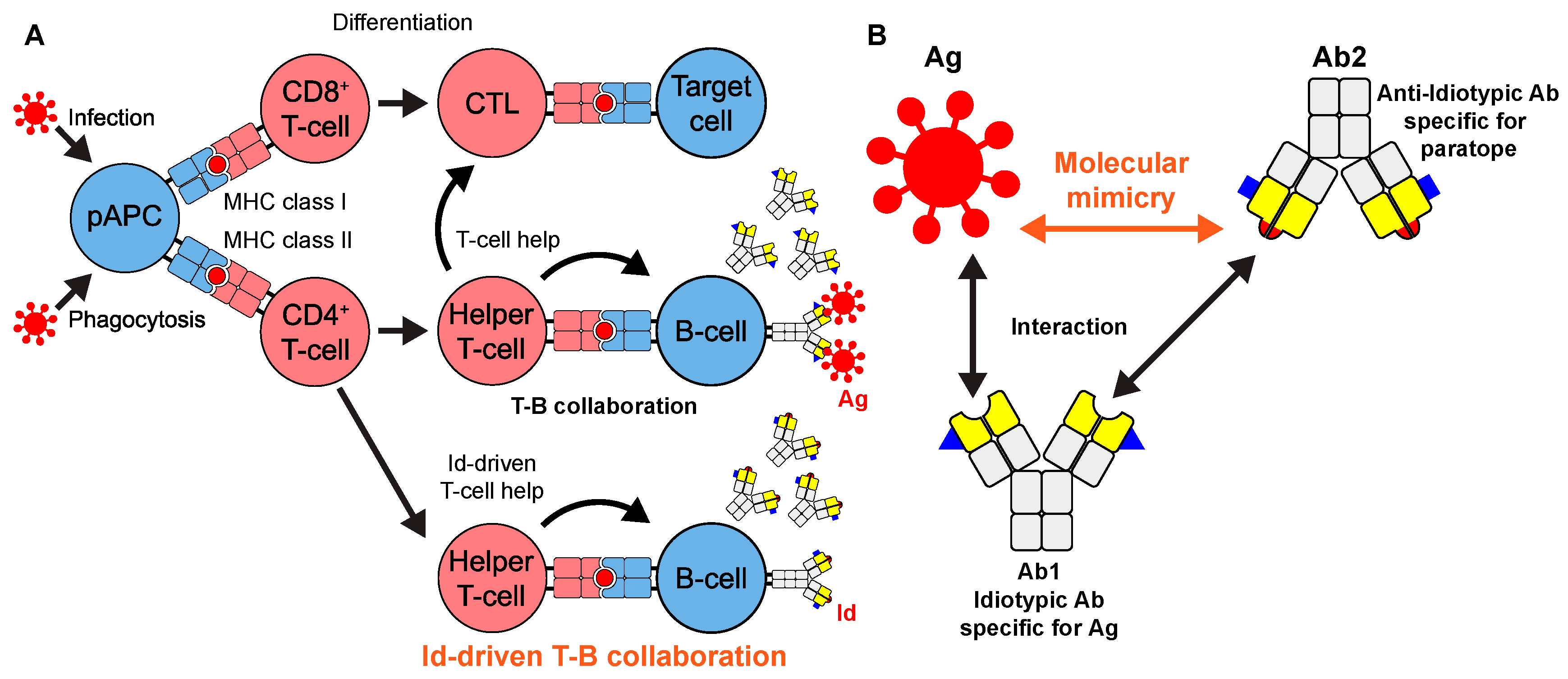
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. HLA Typing
2.3. Peptide Synthesis
2.4. PBMCs
2.5. ELISPOT Assays
2.6. Sequence and Epitope Data Collection
2.7. TCR–Peptide–MHC Structure Analysis
2.8. Repertoire Analysis
2.9. MHC Binding Prediction
2.10. B-Cell Epitope Prediction
3. Results
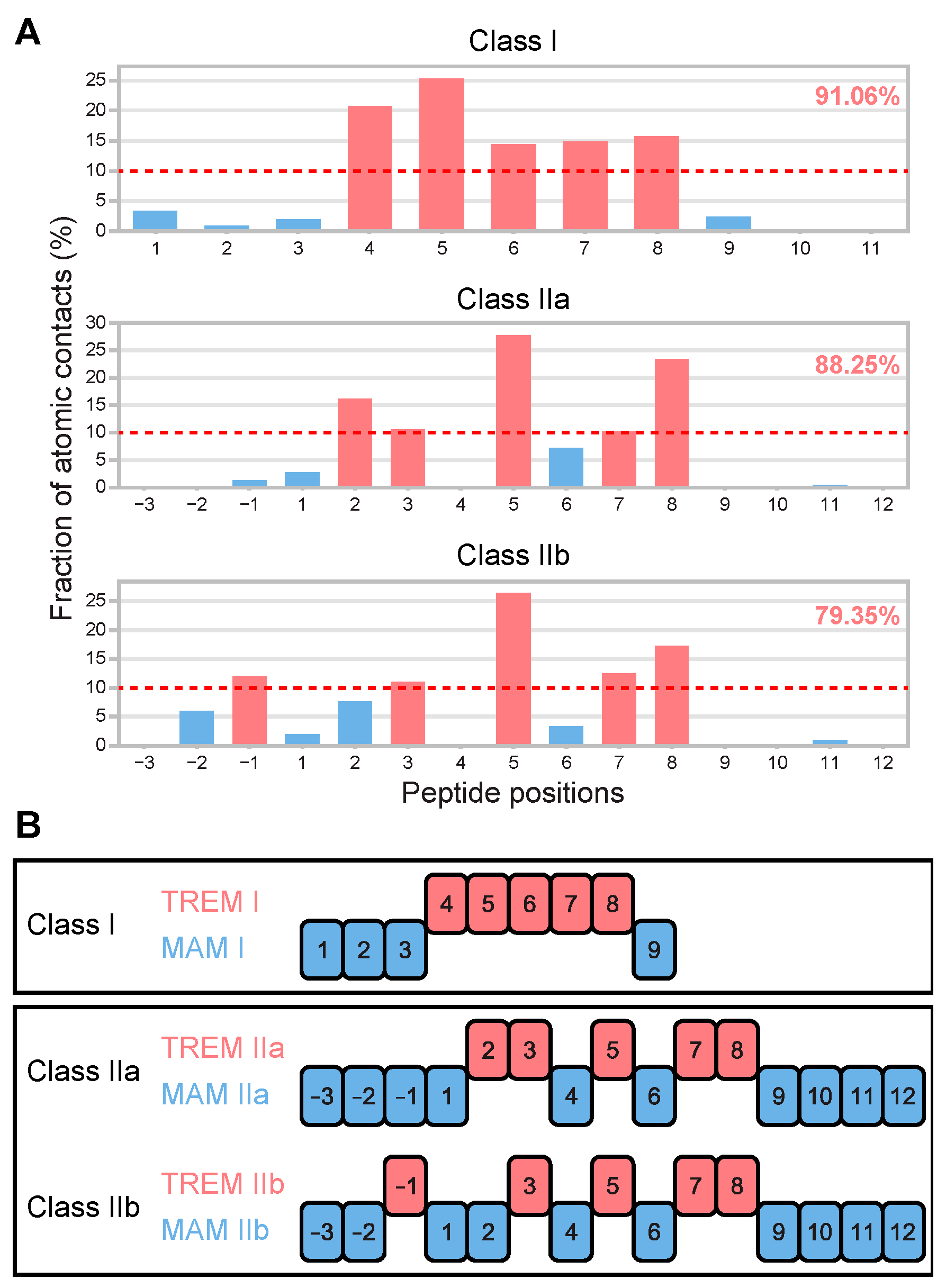
3.1. Determination of T-Cell Epitope Motif Patterns from Peptide–TCR Structures
3.2. TREM Diversity in Germline Ig Variable Regions and the Human Proteome
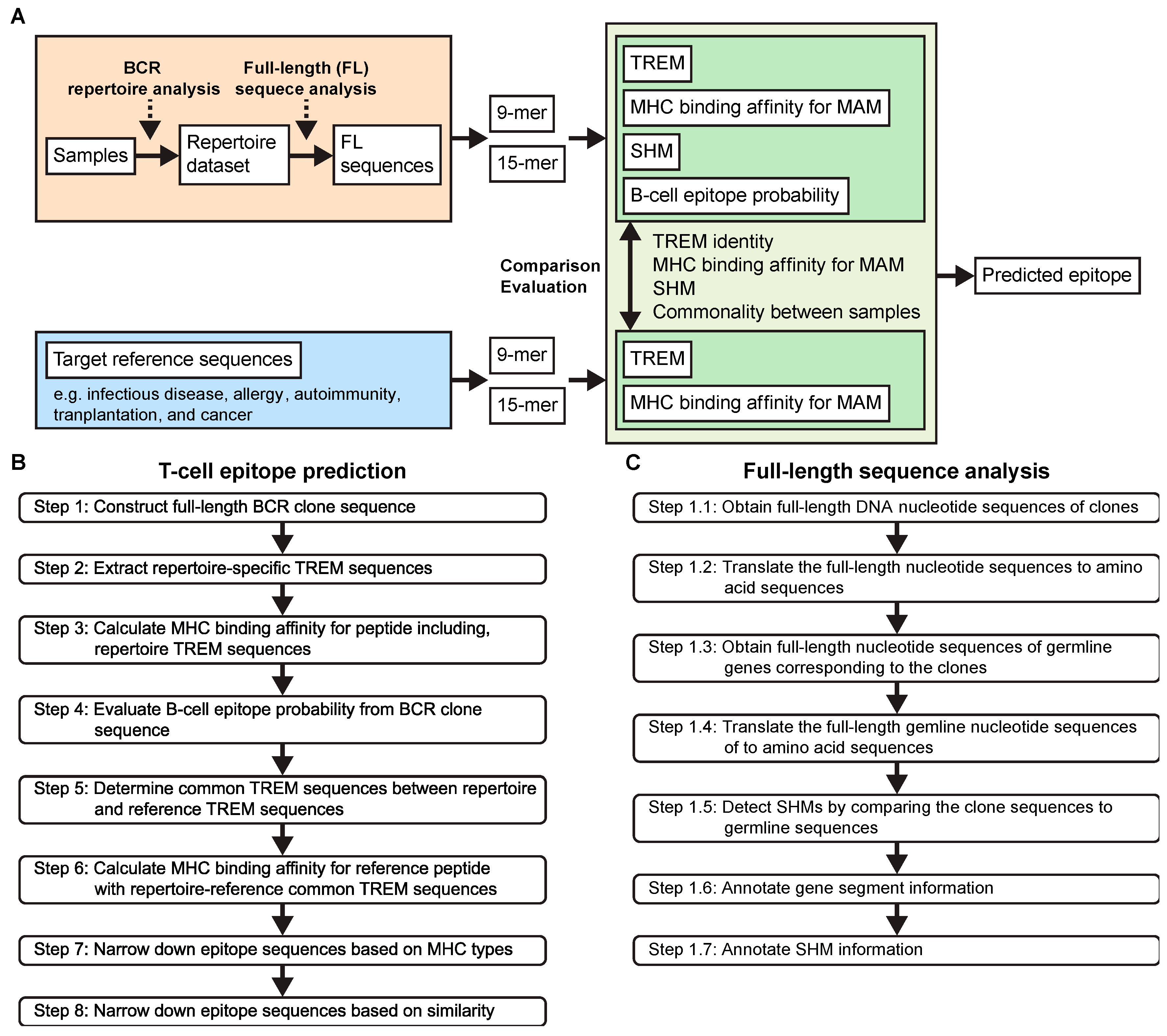
3.3. Developing a New Pipelined T-Cell Epitope Prediction Algorithm
3.3.1. Pipelined Algorithm Overview
3.3.2. Full-Length Sequence Analysis (Step 1)
3.3.3. Extraction of the Repertoire-Specific TREM Sequences (Step 2)
3.3.4. MHC Binding Affinity Determination for TREM-Containing BCR Peptides (Step 3)
3.3.5. Linear B-Cell Epitope Probability Assessment (Step 4)
3.3.6. Molecular Mimicry in BCR and Antigen Reference Sequences (Step 5)
3.3.7. MHC Binding Affinity Determination of the Reference Peptide Sequences (Step 6)
3.3.8. Epitope Sequence Refinement Based on MHC Types (Step 7)
3.3.9. Narrowing down Results Based on Similarity (Step 8)
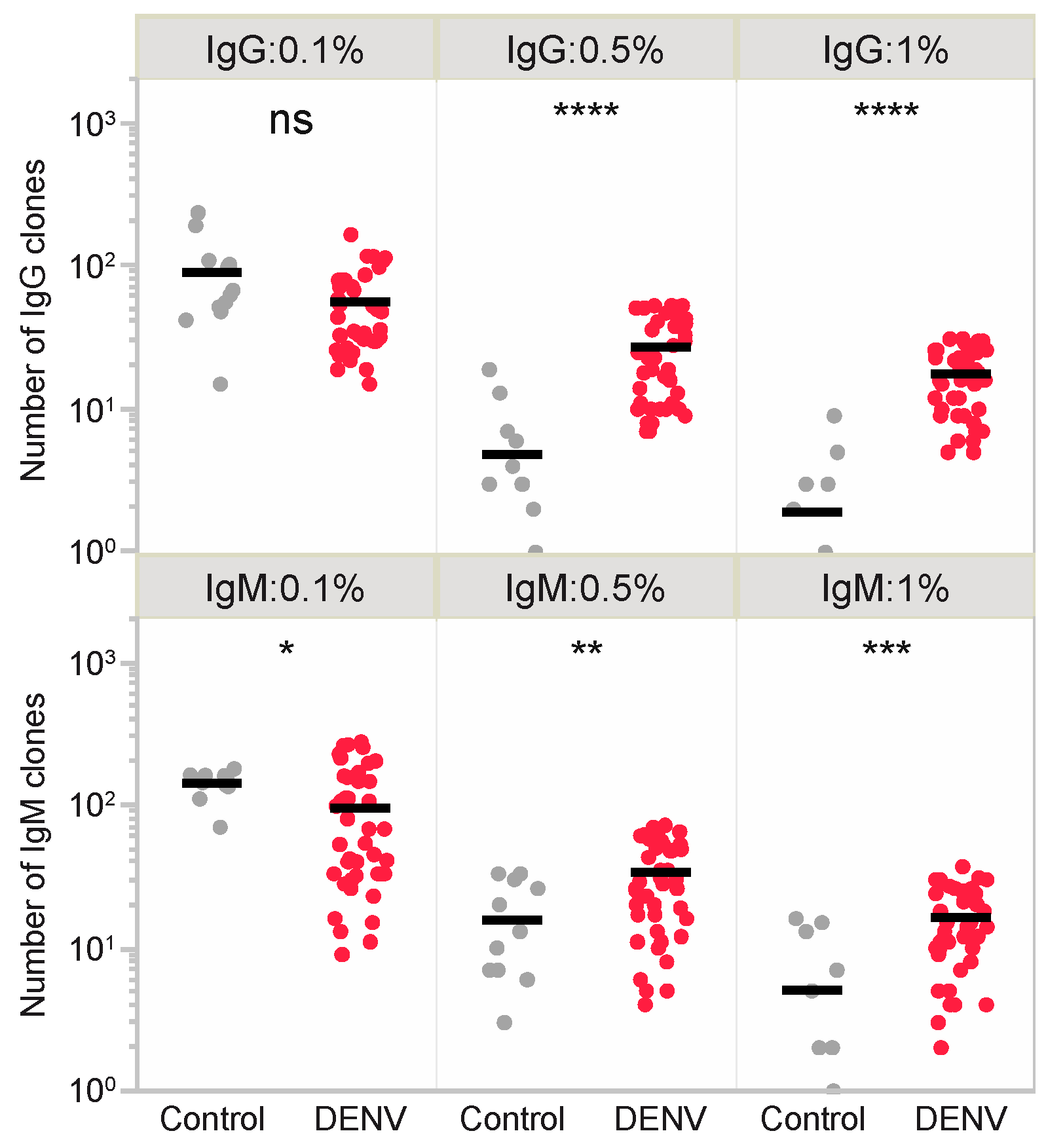
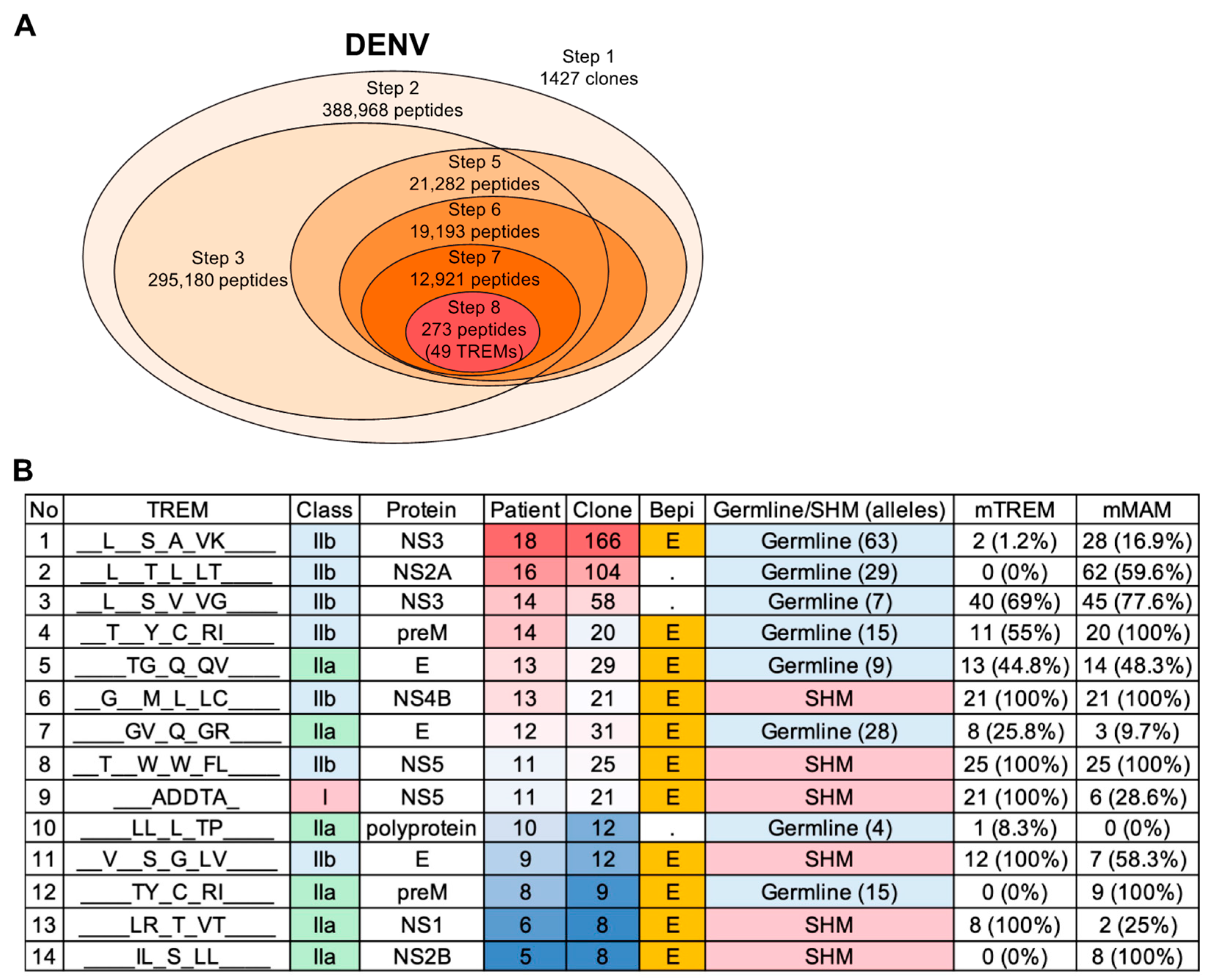
3.4. T-Cell Epitope Prediction in DENV Infection
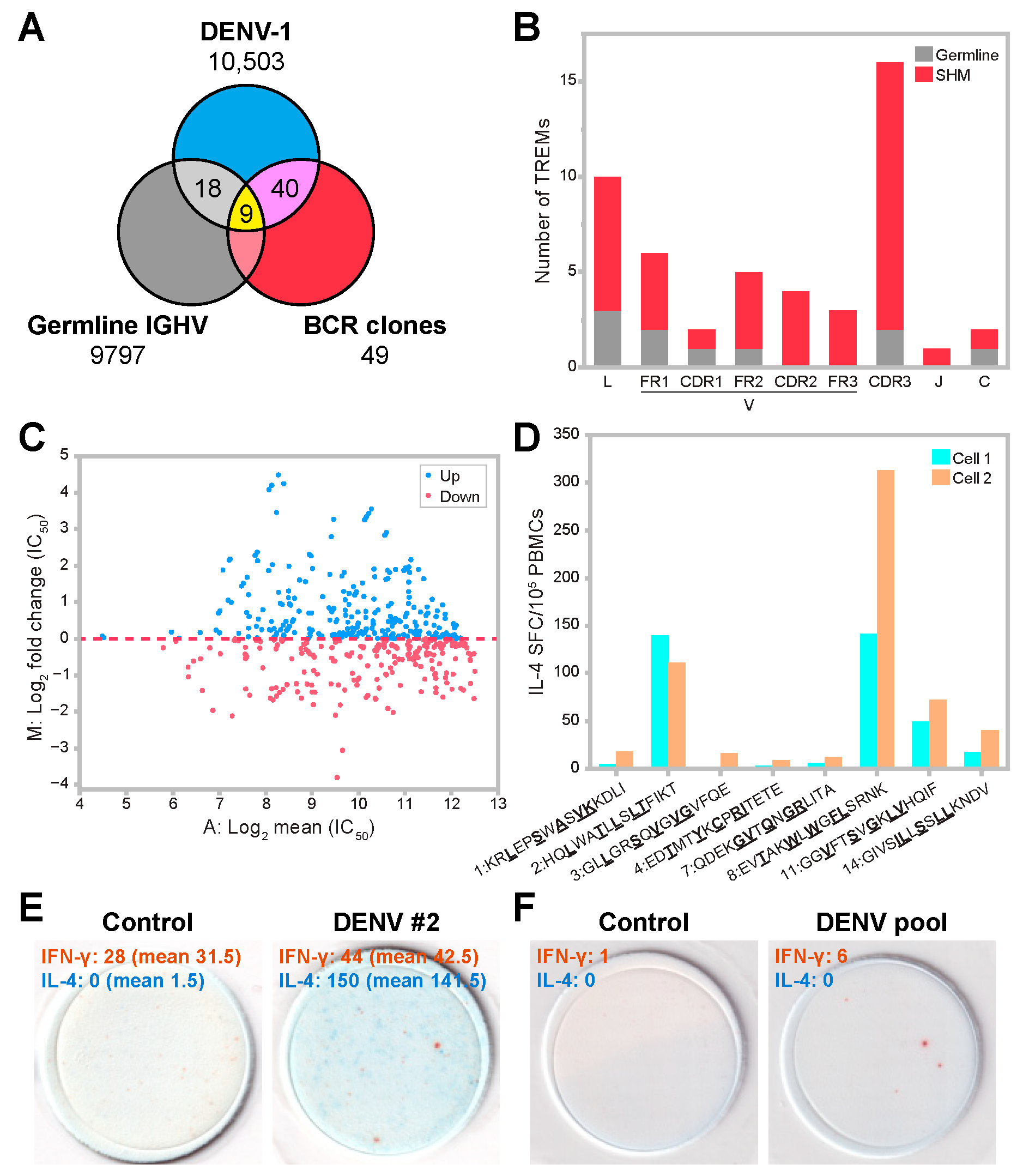
3.5. SHM Contributes to Diversity in Epitope Patterns and Influences HLA Binding
3.6. Predicted TREM Epitopes Exhibit T-Cell Immunogenicity
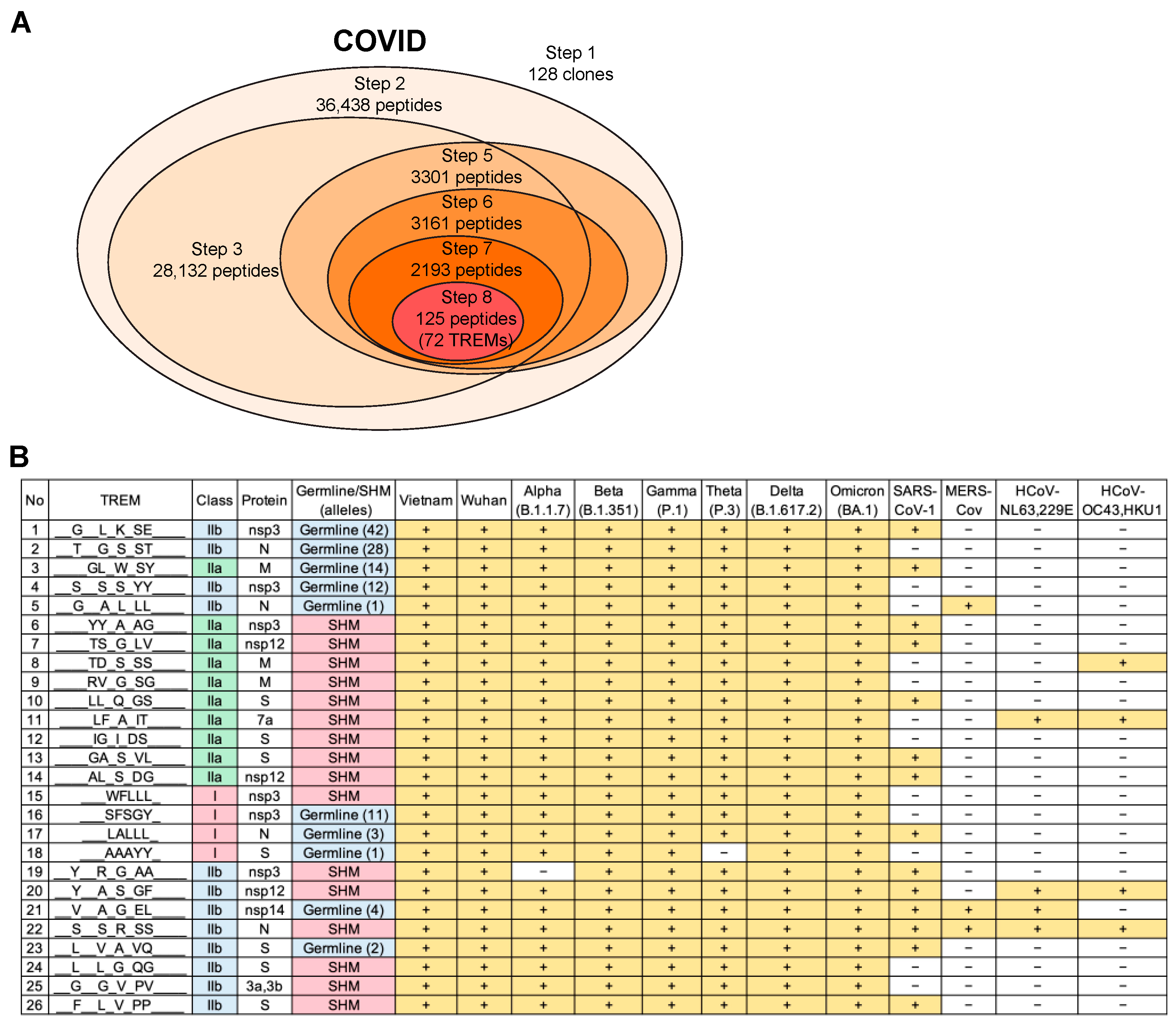
3.7. Validation of the T-Cell Epitope Prediction Method for COVID-19
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Peters, B.; Nielsen, M.; Sette, A. T Cell Epitope Predictions. Annu. Rev. Immunol. 2020, 38, 123–145. [Google Scholar] [CrossRef]
- Rudolph, M.G.; Stanfield, R.L.; Wilson, I.A. How Tcrs Bind Mhcs, Peptides, and Coreceptors. Annu. Rev. Immunol. 2006, 24, 419–466. [Google Scholar] [CrossRef] [PubMed]
- van der Sandt, C.E.; Kreijtz, J.H.C.M.; Rimmelzwaan, G.F. Evasion of Influenza A Viruses from Innate and Adaptive Immune Responses. Viruses 2012, 4, 1438–1476. [Google Scholar] [CrossRef] [PubMed]
- Schaap-Johansen, A.L.; Vujović, M.; Borch, A.; Hadrup, S.R.; Marcatili, P. T Cell Epitope Prediction and Its Application to Immunotherapy. Front. Immunol. 2021, 12, 712488. [Google Scholar] [CrossRef]
- Jerne, N.K. Towards a Network Theory of the Immune System. Ann. Immunol. 1974, 125C, 373–389. [Google Scholar]
- Mitchison, N.A. T-Cell–B-Cell Cooperation. Nat. Rev. Immunol. 2004, 4, 1599–1601. [Google Scholar] [CrossRef]
- Holmøy, T.; Vartdal, F.; Hestvik, A.L.; Munthe, L.; Bogen, B. The Idiotype Connection: Linking Infection and Multiple Sclerosis. Trends Immunol. 2010, 31, 56–62. [Google Scholar] [CrossRef]
- Tonegawa, S. Somatic Generation of Antibody Diversity. Nature 1983, 302, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Behn, U. Idiotype Network. In eLS; John Wiley & Sons, Ltd.: Chichester, UK, 2011. [Google Scholar]
- López-Requena, A.; Burrone, O.R.; Cesco-Gaspere, M. Idiotypes as Immunogens: Facing the Challenge of Inducing Strong Therapeutic Immune Responses against the Variable Region of Immunoglobulins. Front. Oncol. 2012, 2, 159. [Google Scholar] [CrossRef]
- Naveed, A.; Naz, D.; Rahman, S.u. Idiotype/Anti-Idiotype Antibodies: As a Glorious Savior in COVID-19 Pandemics. Transl. Med. Commun. 2021, 6, 4–8. [Google Scholar] [CrossRef]
- Greenspan, N.S.; Bona, C.A. Idiotypes: Structure and Immunogenicity 1. FASEB J. 1993, 7, 437–444. [Google Scholar] [CrossRef]
- Kieber-Emmons, T.; Monzavi-Karbassi, B.; Pashov, A.; Saha, S.; Murali, R.; Kohler, H. The Promise of the Anti-Idiotype Concept. Front. Oncol. 2012, 2, 196. [Google Scholar] [CrossRef] [PubMed]
- Jørgensen, T.; Bogen, B.; Hannestad, K. T Helper Cells Recognize an Idiotope Located on Peptide 88-114/117 of the Light Chain Variable Domain of an Isologous Myeloma Protein (315). J. Exp. Med. 1983, 158, 2183–2188. [Google Scholar] [CrossRef] [PubMed]
- Weiss, S.; Bogen, B. B-Lymphoma Cells Process and Present Their Endogenous Immunoglobulin to Major Histocompatibility Complex-Restricted T Cells. Proc. Natl. Acad. Sci. USA 1989, 86, 282–286. [Google Scholar] [CrossRef] [PubMed]
- Saeki, Y.; Chen, J.J.; Shi, L.F.; Okuda, Y.; Köhler, H. Idiotype-Specific T Helper Clones Recognize a Variable H Chain Determinant. J. Immunol. 1990, 144, 1625–1628. [Google Scholar] [CrossRef]
- Williams, W.M.; Staines, N.A.; Muller, S.; Isenberg, D.A. Human T Cell Responses to Autoantibody Variable Region Peptides. Lupus 1995, 4, 464–471. [Google Scholar] [CrossRef]
- Wysocki, L.J.; Zhanfl, X.; Smith, D.S.; Snyder, C.M.; Bonorino, C. Somatic Origin of T-Cell Epitopes within Antibody Variable Regions: Significance to Monoclonal Therapy and Genesis of Systemic Autoimmune Disease. Immunol. Rev. 1998, 162, 233–246. [Google Scholar] [CrossRef]
- Munthe, L.A.; Kyte, J.A.; Bogen, B. Resting Small B Cells Present Endogenous Immunoglobulin Variable-Region Determinants to Idiotope-Specific CD4+ T Cells in Vivo. Eur. J. Immunol. 1999, 29, 4043–4052. [Google Scholar] [CrossRef]
- Trojan, A.; Schultze, J.L.; Witzens, M.; Vonderheide, R.H.; Ladetto, M.; Donovan, J.W.; Gribben, J.G. Immunoglobulin Framework-Derived Peptides Function as Cytotoxic T-Cell Epitopes Commonly Expressed in B-Cell Malignancies. Nat. Med. 2000, 6, 667–672. [Google Scholar] [CrossRef]
- Hansson, L.; Rabbani, H.; Fagerberg, J.; Österborg, A.; Mellstedt, H. T-Cell Epitopes within the Complementarity-Determining and Framework Regions of the Tumor-Derived Immunoglobulin Heavy Chain in Multiple Myeloma. Blood 2003, 101, 4930–4936. [Google Scholar] [CrossRef]
- Snyder, C.M.; Aviszus, K.; Heiser, R.A.; Tonkin, D.R.; Guth, A.M.; Wysocki, L.J. Activation and Tolerance in CD4+ T Cells Reactive to an Immunoglobulin-Variable Region. J. Exp. Med. 2004, 200, 1–11. [Google Scholar] [CrossRef]
- Khodadoust, M.S.; Olsson, N.; Wagar, L.E.; Haabeth, O.A.W.; Chen, B.; Swaminathan, K.; Rawson, K.; Liu, C.L.; Steiner, D.; Lund, P.; et al. Antigen Presentation Profiling Reveals Recognition of Lymphoma Immunoglobulin Neoantigens. Nature 2017, 543, 723–727. [Google Scholar] [CrossRef] [PubMed]
- Huszthy, P.C.; Gopalakrishnan, R.P.; Jacobsen, J.T.; Haabeth, O.A.W.; Løset, G.Å.; Braathen, R.; Schenck, K.; Tveita, A.A.; Munthe, L.A.; Bogen, B. B Cell Receptor Ligation Induces Display of V-Region Peptides on MHC Class II Molecules to T Cells. Proc. Natl. Acad. Sci. USA 2019, 116, 25850–25859. [Google Scholar] [CrossRef] [PubMed]
- Munthe, L.A.; Os, A.; Zangani, M.; Alerts, E. MHC-Restricted Ig V Region-Driven T-B Lymphocyte Collaboration: B Cell Receptor Ligation Facilitates Switch to IgG Production. J. Immunol. 2022, 172, 7476–7484. [Google Scholar] [CrossRef]
- Fukuta, M.; Nguyen, C.T.; Nguyen, T.T.T.; Nguyen, T.T.N.; Vu, T.B.H.; Takemura, T.; Nguyen, L.K.H.; Inoue, S.; Morita, K.; Le, T.Q.M.; et al. Discrepancies in Infectivity of Flavivirus and SARS-CoV-2 Clinical Samples: An Improved Assay for Infectious Virus Shedding and Viremia Assessment. Int. J. Environ. Res. Public Health 2021, 18, 9845. [Google Scholar] [CrossRef]
- Takemura, T.; Nguyen, C.T.; Pham, H.C.; Nguyen, T.T.; Hoang, V.M.P.; Nguyen, L.K.H.; Nabeshima, T.; Nguyen, T.T.T.; Le, T.Q.M.; Moi, M.L.; et al. The 2017 Dengue Virus 1 Outbreak in Northern Vietnam Was Caused by a Locally Circulating Virus Group. Trop. Med. Health 2022, 50, 3–7. [Google Scholar] [CrossRef]
- Le, T.Q.M.; Takemura, T.; Moi, M.L.; Nabeshima, T.; Nguyen, L.K.H.; Hoang, V.M.P.; Ung, T.H.T.; Le, T.T.; Nguyen, V.S.; Pham, H.Q.A.; et al. Severe Acute Respiratory Syndrome Coronavirus 2 Shedding by Travelers, Vietnam, 2020. Emerg. Infect. Dis. 2020, 26, 1624–1626. [Google Scholar] [CrossRef] [PubMed]
- Shiina, T.; Suzuki, S.; Ozaki, Y.; Taira, H.; Kikkawa, E.; Shigenari, A.; Oka, A.; Umemura, T.; Joshita, S.; Takahashi, O.; et al. Super High Resolution for Single Molecule-Sequence-Based Typing of Classical HLA Loci at the 8-Digit Level Using next Generation Sequencers. Tissue Antigens 2012, 80, 305–316. [Google Scholar] [CrossRef]
- Ozaki, Y.; Suzuki, S.; Kashiwase, K.; Shigenari, A.; Okudaira, Y.; Ito, S.; Masuya, A.; Azuma, F.; Yabe, T.; Morishima, S.; et al. Cost-Efficient Multiplex PCR for Routine Genotyping of up to Nine Classical HLA Loci in a Single Analytical Run of Multiple Samples by next Generation Sequencing. BMC Genom. 2015, 16, 318. [Google Scholar] [CrossRef]
- Ramachandran, H.; Laux, J.; Moldovan, I.; Caspell, R.; Lehmann, P.V.; Subbramanian, R.A. Optimal Thawing of Cryopreserved Peripheral Blood Mononuclear Cells for Use in High-Throughput Human Immune Monitoring Studies. Cells 2012, 1, 313–324. [Google Scholar] [CrossRef]
- Hayashi, S.; Imanishi, R.; Adachi, M.; Ikejima, S.; Nakata, J.; Morimoto, S.; Fujiki, F.; Nishida, S.; Tsuboi, A.; Hosen, N.; et al. Reader-Free Elispot Assay for Immuno-Monitoring in Peptide-Based Cancer Vaccine Immunotherapy. Biomed. Rep. 2020, 12, 244–250. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Giudicelli, V.; Chaume, D.; Lefranc, M.P. IMGT/GENE-DB: A Comprehensive Database for Human and Mouse Immunoglobulin and T Cell Receptor Genes. Nucleic Acids Res. 2005, 33, 256–261. [Google Scholar] [CrossRef] [PubMed]
- Frankish, A.; Diekhans, M.; Jungreis, I.; Lagarde, J.; Loveland, J.E.; Mudge, J.M.; Sisu, C.; Wright, J.C.; Armstrong, J.; Barnes, I.; et al. Gencode 2021. Nucleic Acids Res. 2021, 49, D916–D923. [Google Scholar] [CrossRef] [PubMed]
- Brister, J.R.; Bao, Y.; Zhdanov, S.A.; Ostapchuck, Y.; Chetvernin, V.; Kiryutin, B.; Zaslavsky, L.; Kimelman, M.; Tatusova, T.A. Virus Variation Resource—Recent Updates and Future Directions. Nucleic Acids Res. 2014, 42, 660–665. [Google Scholar] [CrossRef]
- Khare, S.; Gurry, C.; Freitas, L.; Schultz, M.B.; Bach, G.; Diallo, A.; Akite, N.; Ho, J.; Lee, R.T.C.; Yeo, W.; et al. GISAID’s Role in Pandemic Response. China CDC Wkly. 2021, 3, 1049–1051. [Google Scholar] [CrossRef]
- Vita, R.; Mahajan, S.; Overton, J.A.; Dhanda, S.K.; Martini, S.; Cantrell, J.R.; Wheeler, D.K.; Sette, A.; Peters, B. The Immune Epitope Database (IEDB): 2018 Update. Nucleic Acids Res. 2019, 47, D339–D343. [Google Scholar] [CrossRef]
- Bekker, G.J.; Yokochi, M.; Suzuki, H.; Ikegawa, Y.; Iwata, T.; Kudou, T.; Yura, K.; Fujiwara, T.; Kawabata, T.; Kurisu, G. Protein Data Bank Japan: Celebrating Our 20th Anniversary during a Global Pandemic as the Asian Hub of Three Dimensional Macromolecular Structural Data. Protein Sci. 2022, 31, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Kitaura, K.; Yamashita, H.; Ayabe, H.; Shini, T.; Matsutani, T.; Suzuki, R. Different Somatic Hypermutation Levels among Antibody Subclasses Disclosed by a New Next-Generation Sequencing-Based Antibody Repertoire Analysis. Front. Immunol. 2017, 8, 389. [Google Scholar] [CrossRef]
- Jurtz, V.; Paul, S.; Andreatta, M.; Marcatili, P.; Peters, B.; Nielsen, M. NetMHCpan-4.0: Improved Peptide–MHC Class I Interaction Predictions Integrating Eluted Ligand and Peptide Binding Affinity Data. J. Immunol. 2017, 199, 3360–3368. [Google Scholar] [CrossRef]
- Jensen, K.K.; Andreatta, M.; Marcatili, P.; Buus, S.; Greenbaum, J.A.; Yan, Z.; Sette, A.; Peters, B.; Nielsen, M. Improved Methods for Predicting Peptide Binding Affinity to MHC Class II Molecules. Immunology 2018, 154, 394–406. [Google Scholar] [CrossRef] [PubMed]
- Jespersen, M.C.; Peters, B.; Nielsen, M.; Marcatili, P. BepiPred-2.0: Improving Sequence-Based B-Cell Epitope Prediction Using Conformational Epitopes. Nucleic Acids Res. 2017, 45, W24–W29. [Google Scholar] [CrossRef]
- Bogen, B.; Ruffini, P. Review: To What Extent Are T Cells Tolerant to Immunoglobulin Variable Regions? Scand. J. Immunol. 2009, 70, 526–530. [Google Scholar] [CrossRef] [PubMed]
- Klein, L.; Kyewski, B.; Allen, P.M.; Hogquist, K.A. Positive and Negative Selection of the T Cell Repertoire: What Thymocytes See (and Don’t See). Nat. Rev. Immunol. 2014, 14, 377–391. [Google Scholar] [CrossRef]
- Galanis, K.A.; Nastou, K.C.; Papandreou, N.C.; Petichakis, G.N.; Pigis, D.G.; Iconomidou, V.A. Linear B-Cell Epitope Prediction for in Silico Vaccine Design: A Performance Review of Methods Available via Command-Line Interface. Int. J. Mol. Sci. 2021, 22, 3210. [Google Scholar] [CrossRef]
- Bhatt, P.; Sabeena, S.P.; Varma, M.; Arunkumar, G. Current Understanding of the Pathogenesis of Dengue Virus Infection. Curr. Microbiol. 2021, 78, 17–32. [Google Scholar] [CrossRef]
- Vaughan, K.; Greenbaum, J.; Blythe, M.; Peters, B.; Sette, A. Meta-Analysis of All Immune Epitope Data in the Flavivirus Genus: Inventory of Current Immune Epitope Data Status in the Context of Virus Immunity and Immunopathology. Viral Immunol. 2010, 23, 259–284. [Google Scholar] [CrossRef]
- Azkur, A.K.; Akdis, M.; Azkur, D.; Sokolowska, M.; van de Veen, W.; Brüggen, M.C.; O’Mahony, L.; Gao, Y.; Nadeau, K.; Akdis, C.A. Immune Response to SARS-CoV-2 and Mechanisms of Immunopathological Changes in COVID-19. Allergy Eur. J. Allergy Clin. Immunol. 2020, 75, 1564–1581. [Google Scholar] [CrossRef] [PubMed]
- Sohail, M.S.; Ahmed, S.F.; Quadeer, A.A.; McKay, M.R. In Silico T Cell Epitope Identification for SARS-CoV-2: Progress and Perspectives. Adv. Drug Deliv. Rev. 2021, 171, 29–47. [Google Scholar] [CrossRef]
- Mateus, J.; Grifoni, A.; Tarke, A.; Sidney, J.; Ramirez, S.I.; Dan, J.M.; Burger, Z.C.; Rawlings, S.A.; Smith, D.M.; Phillips, E.; et al. Selective and Cross-Reactive SARS-CoV-2 T Cell Epitopes in Unexposed Humans. Science 2020, 370, 89–94. [Google Scholar] [CrossRef]
- Moise, L.; Gutierrez, A.H.; Bailey-Kellogg, C.; Terry, F.; Leng, Q.; Abdel Hady, K.M.; VerBerkmoes, N.C.; Sztein, M.B.; Losikoff, P.T.; Martin, W.D.; et al. The Two-Faced T Cell Epitope: Examining the Host-Microbe Interface with JanusMatrix. Hum. Vaccines Immunother. 2013, 9, 1577–1586. [Google Scholar] [CrossRef] [PubMed]
- Bremel, R.D.; Homan, E.J. Frequency Patterns of T-Cell Exposed Amino Acid Motifs in Immunoglobulin Heavy Chain Peptides Presented by MHCs. Front. Immunol. 2014, 5, 541. [Google Scholar] [CrossRef]
- Calis, J.J.A.; de Boer, R.J.; Keşmir, C. Degenerate T-Cell Recognition of Peptides on MHC Molecules Creates Large Holes in the T-Cell Repertoire. PLoS Comput. Biol. 2012, 8, e1002412. [Google Scholar] [CrossRef] [PubMed]
- Breme, R.D.; Homan, E.J. Extensive T-Cell Epitope Repertoire Sharing among Human Proteome, Gastrointestinal Microbiome, and Pathogenic Bacteria: Implications for the Definition of Self. Front. Immunol. 2015, 6, 538. [Google Scholar] [CrossRef]
- Arthur, J.M.; Forrest, J.C.; Boehme, K.W.; Kennedy, J.L.; Owens, S.; Herzog, C.; Liu, J.; Harville, T.O. Development of ACE2 Autoantibodies after SARS-CoV-2 Infection. PLoS ONE 2021, 16, e0257016. [Google Scholar] [CrossRef]
- Murphy, W.J.; Ph, D.; Longo, D.L. A Possible Role for Anti-Idiotype Antibodies in SARS-CoV-2 Infection and Vaccination. N. Engl. J. Med. 2022, 386, 394–396. [Google Scholar] [CrossRef]
- Muri, J.; Cecchinato, V.; Cavalli, A.; Shanbhag, A.A.; Matkovic, M.; Biggiogero, M.; Maida, P.A.; Moritz, J.; Toscano, C.; Ghovehoud, E.; et al. Autoantibodies against Chemokines Post-SARS-CoV-2 Infection Correlate with Disease Course. Nat. Immunol. 2023, 24, 604–611. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. of Alleles | TREM I (3258) | TREM IIa (3251) | TREM IIb (3288) | Average (3265.7) |
|---|---|---|---|---|
| 1 (0.3%) | 33.6% (1095) | 33.7% (1097) | 33.8% (1111) | 33.7% (1101) |
| 2–5 (0.6–1.49%) | 35% (1140) | 34.5% (1123) | 34.4% (1132) | 34.7% (1131.7) |
| 6–9 (1.79–2.68%) | 9.6% (314) | 10.5% (340) | 10.3% (340) | 10.1% (331.3) |
| 10–20 (2.98–5.95%) | 9.8% (318) | 9.4% (306) | 9.3% (307) | 9.5% (310.3) |
| 21–41 (6.25–12.5%) | 5.2% (171) | 5.5% (179) | 5.8% (191) | 5.5% (180.3) |
| 42–83 (12.5–25%) | 4.2% (138) | 4% (131) | 4.3% (141) | 4.2% (136.7) |
| 84–167 (25–50%) | 2.2% (72) | 2% (66) | 1.9% (61) | 2% (66.3) |
| 168+ (>50%) | 0.3% (10) | 0.3% (9) | 0.2% (5) | 0.2% (8) |
| Types | IgG | IgM | IgG and IgM |
|---|---|---|---|
| ≥1% clones a | 725 (5–31) | 702 (2–37) | 1427 (2–37) |
| TREMs | 29 | 37 | 49 |
| TREM clones a | 129 (0–12) | 292 (0–17) | 421 (0–17) |
| SHM clones a | 77 (0–9) | 111 (0–7) | 188 (0–9) |
| TREM clones in ≥1% clones (%) b | 18.1 (0–57.1) | 42.8 (0–100) | 30.6 (0–100) |
| SHM clones in ≥1% clones (%) c | 10.6 (0–57.1) | 16.5 (0–66.7) | 13.6 (0–66.7) |
| SHM clones in TREM clones (%) d | 61.9 (0–100) | 40.4 (0–100) | 50.4 (0–100) |
| Maximum TREM e | 1.8 (0–5) | 1.7 (0–4) | 1.8 (0–5) |
| Maximum SHM e | 1 (0–4) | 1 (0–2) | 1 (0–4) |
| No | TREM | Germline/ SHM | Protein | Epitope Sequence | Versatility a | Cell 1 IC50 Min b | Cell 2 IC50 Min b | Cultured ELISPOT Assays |
|---|---|---|---|---|---|---|---|---|
| 1 | __L__S_A_VK____ | Germline | NS3 | KRLEPSWASVKKDLI | ++ | 266.82 | 530.23 | no |
| 2 | __L__T_L_LT____ | Germline | NS2A | HQLWATLLSLTFIKT | +++ | 37.97 | 56.26 | yes |
| 3 | __L__S_V_VG____ | Germline | NS3 | GLLGRSQVGVGVFQE | ++ | 884.5 | 390.99 | no |
| 4 | __T__Y_C_RI____ | Germline | preM | EDTMTYKCPRITETE | ++ | 2496.2 | 1098.35 | no |
| 7 | ____GV_Q_GR____ | Germline | E | QDEKGVTQNGRLITA | ++ | 1571.02 | 376.13 | no |
| 8 | __T__W_W_FL____ | SHM | NS5 | EVTAKWLWGFLSRNK | +++ | 122.72 | 290.34 | yes |
| 11 | __V__S_G_LV____ | SHM | E | GGVFTSVGKLVHQIF | +++ | 50.14 | 77.93 | yes |
| 14 | ____IL_S_LL____ | SHM | NS2B | GIVSILLSSLLKNDV | +++ | 92.23 | 80.6 | yes |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakamura, Y.; Moi, M.L.; Shiina, T.; Shin-I, T.; Suzuki, R. Idiotope-Driven T-Cell/B-Cell Collaboration-Based T-Cell Epitope Prediction Using B-Cell Receptor Repertoire Sequences in Infectious Diseases. Viruses 2023, 15, 1186. https://doi.org/10.3390/v15051186
Nakamura Y, Moi ML, Shiina T, Shin-I T, Suzuki R. Idiotope-Driven T-Cell/B-Cell Collaboration-Based T-Cell Epitope Prediction Using B-Cell Receptor Repertoire Sequences in Infectious Diseases. Viruses. 2023; 15(5):1186. https://doi.org/10.3390/v15051186
Chicago/Turabian StyleNakamura, Yukio, Meng Ling Moi, Takashi Shiina, Tadasu Shin-I, and Ryuji Suzuki. 2023. "Idiotope-Driven T-Cell/B-Cell Collaboration-Based T-Cell Epitope Prediction Using B-Cell Receptor Repertoire Sequences in Infectious Diseases" Viruses 15, no. 5: 1186. https://doi.org/10.3390/v15051186
APA StyleNakamura, Y., Moi, M. L., Shiina, T., Shin-I, T., & Suzuki, R. (2023). Idiotope-Driven T-Cell/B-Cell Collaboration-Based T-Cell Epitope Prediction Using B-Cell Receptor Repertoire Sequences in Infectious Diseases. Viruses, 15(5), 1186. https://doi.org/10.3390/v15051186