

Dynamic Development of Viral and Bacterial Diversity during Grass Silage Preservation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Silage Preparation and Experimental Design
2.2. DNA Extraction, Sequencing, and qPCR
2.3. Prokaryotic Community, Binning, Taxonomic, and Functional Annotation
2.4. Comparison of Lactococcus sp. (C4C_bin8) Defense Mechanism
2.5. Viral Prediction, Taxonomic, and Functional Annotation
2.6. Clustering, Taxonomic Context and Phylogenetic Analysis of vOTUs
2.7. Data Wrangling and Data Availability
3. Results
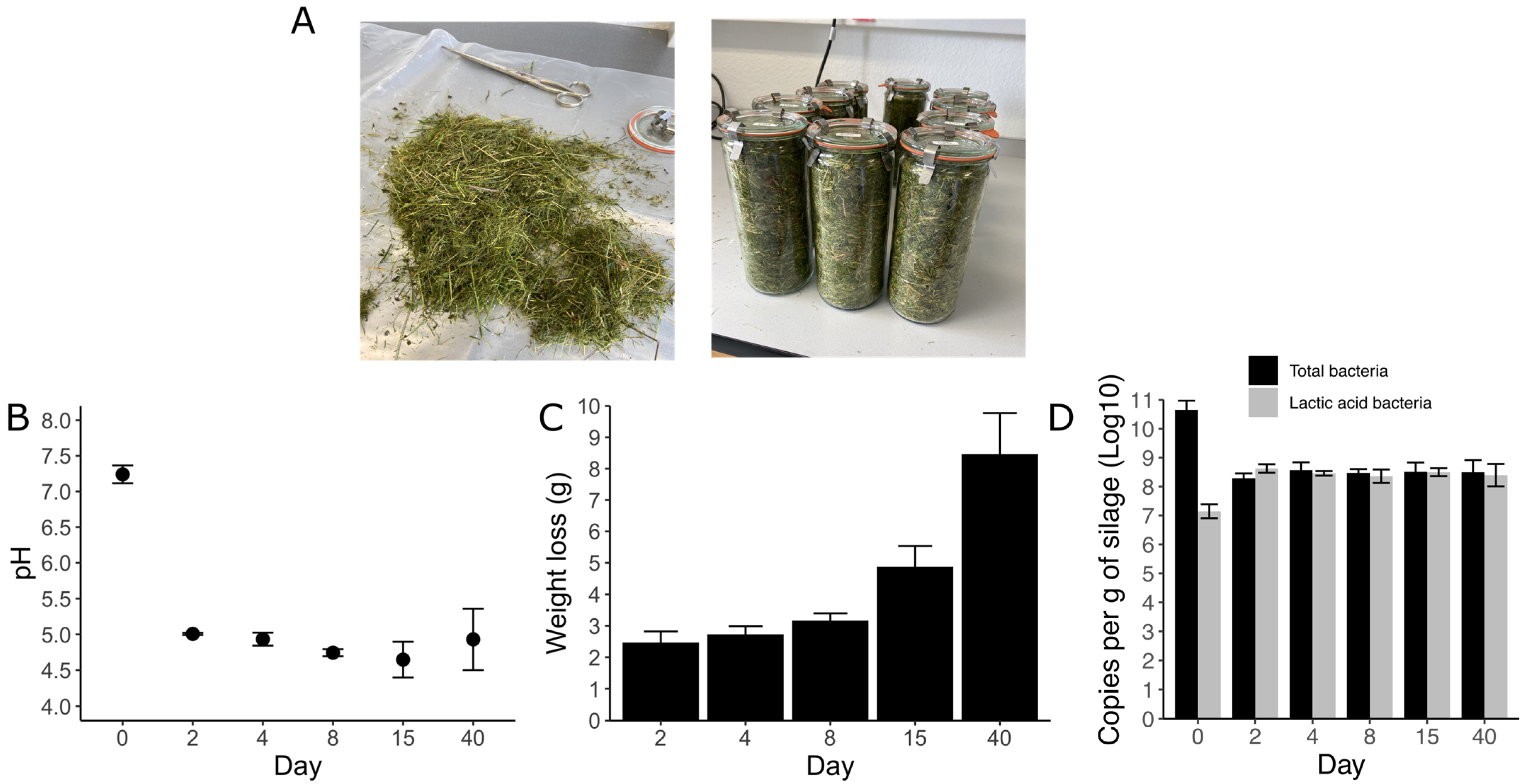
3.1. Silage Characterization
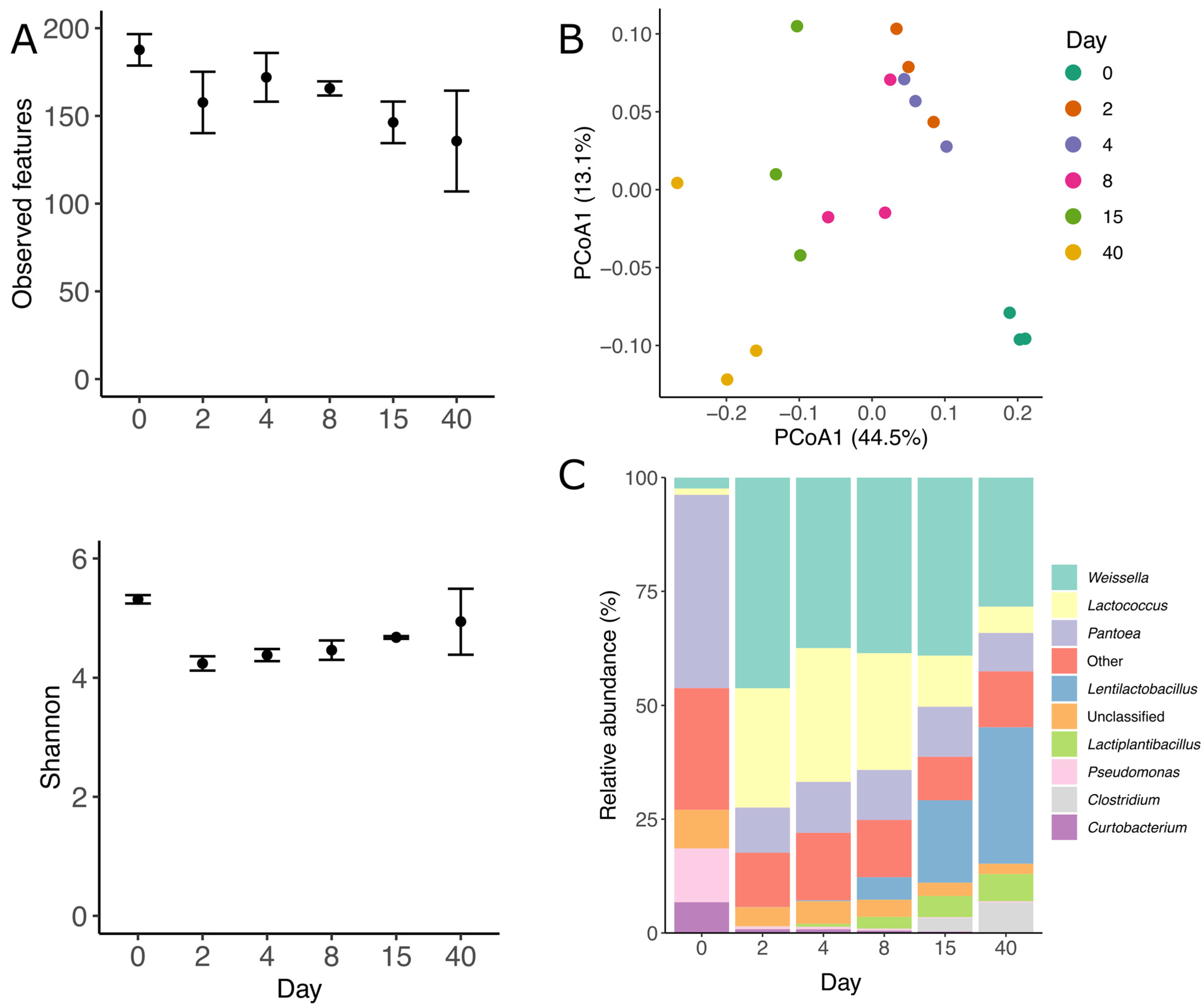
3.2. Prokaryotic Community
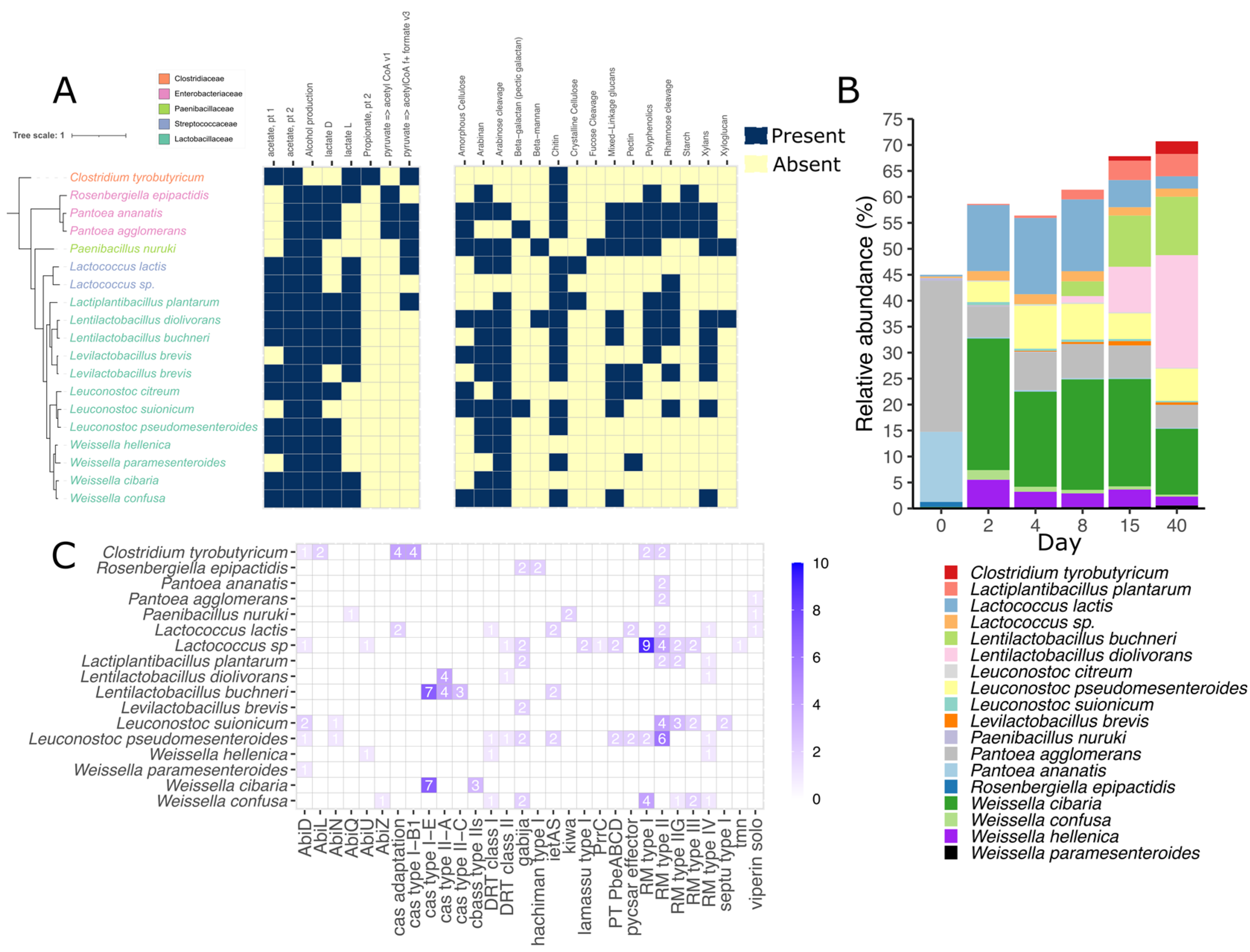
3.3. Metagenomic Binning Reveals the Potential for Antiviral Defense in Silage
3.4. Lactococcus Species Encodes a Variety of Defense Mechanisms in Silage
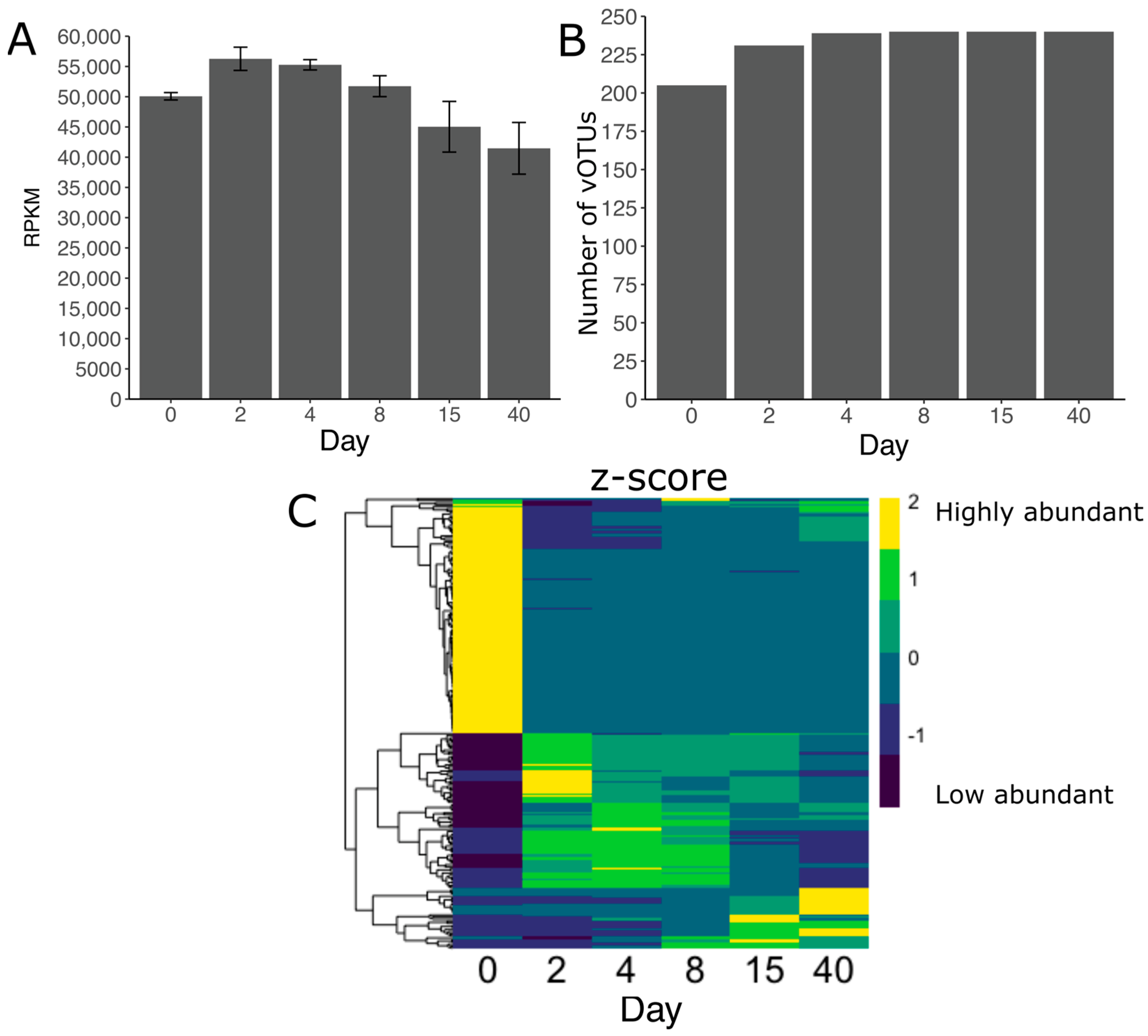
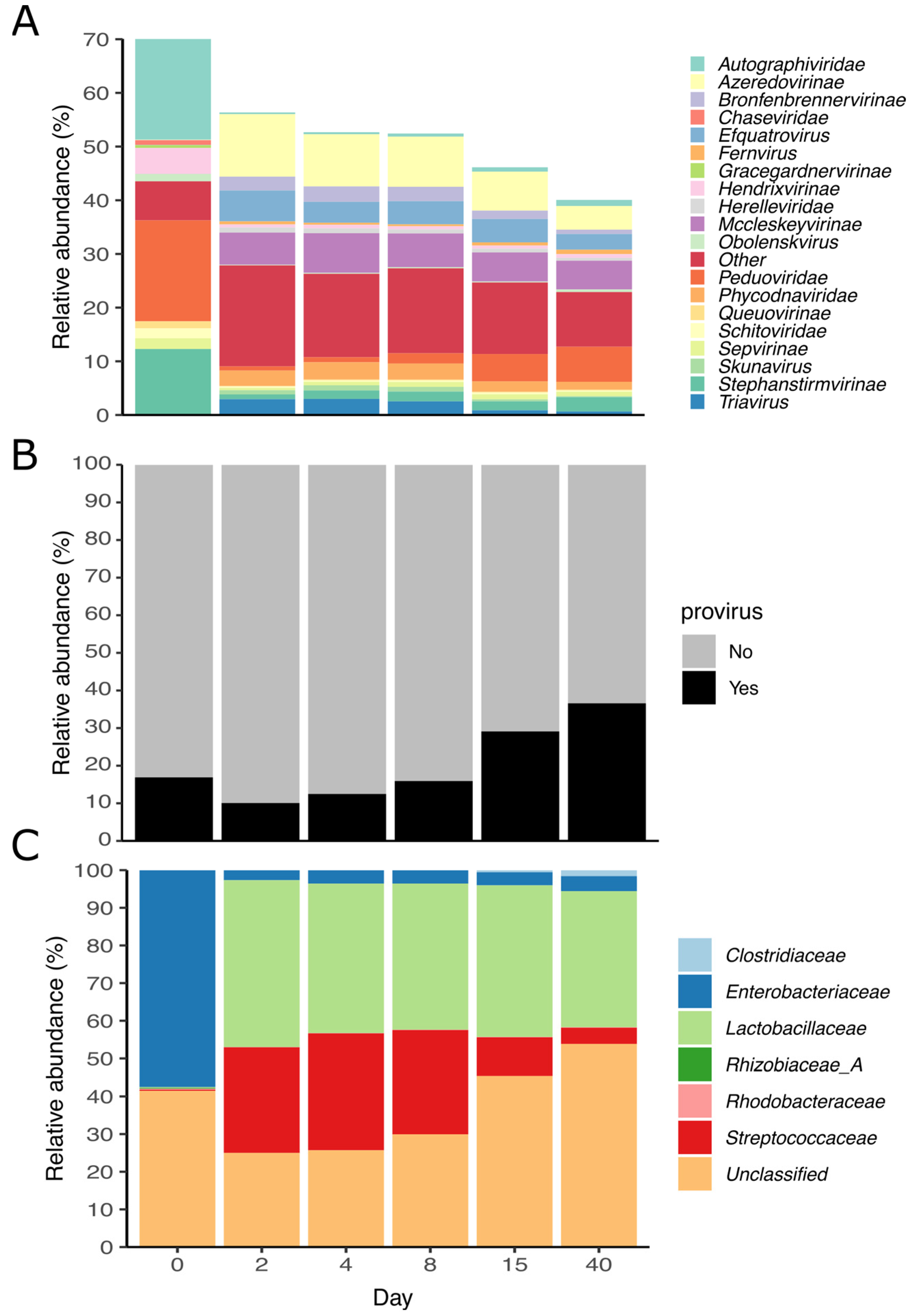
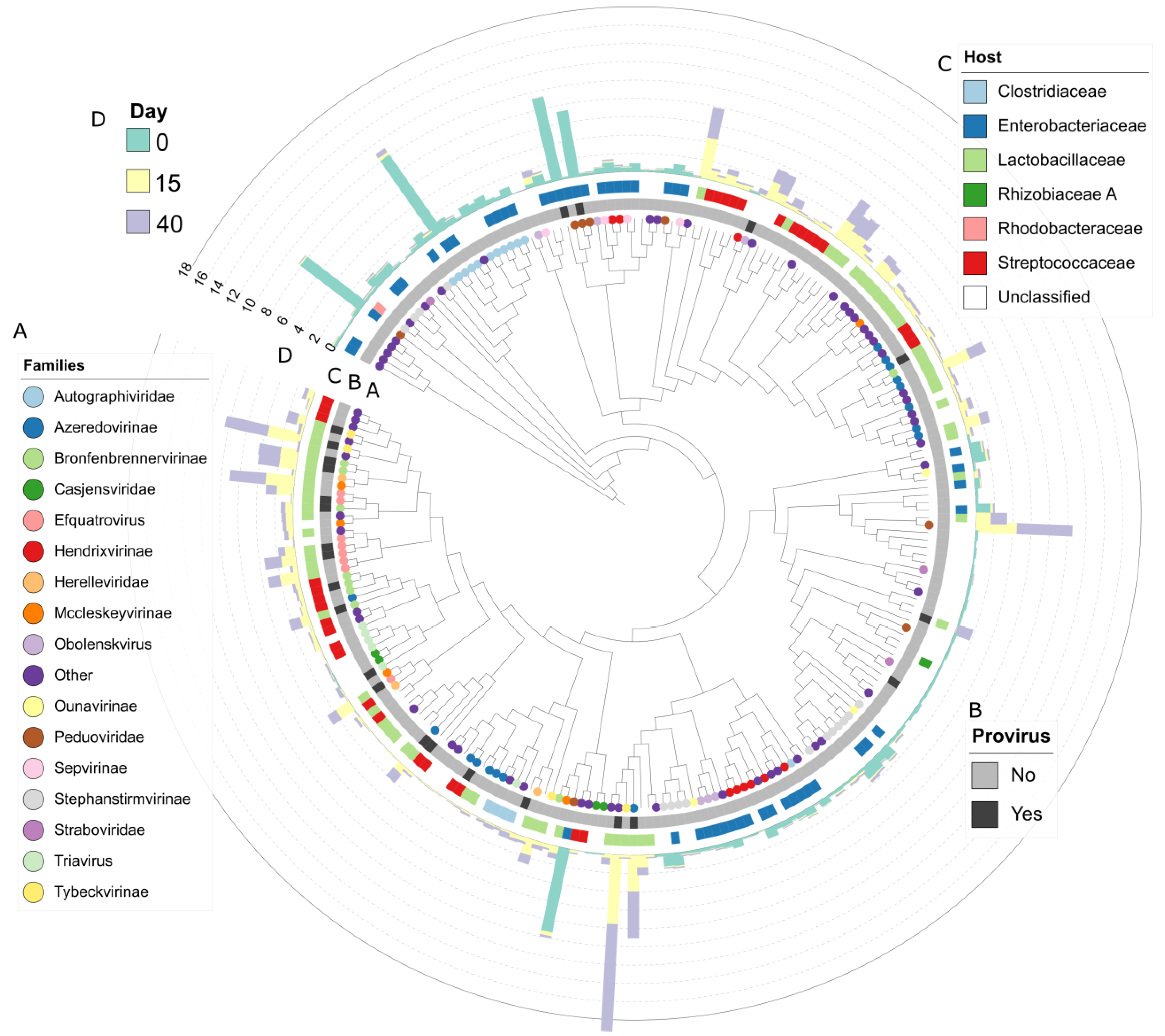
3.5. Viral Diversity during Grass Ensilaging
3.6. Acquired Immunity and Host-Phage Interaction
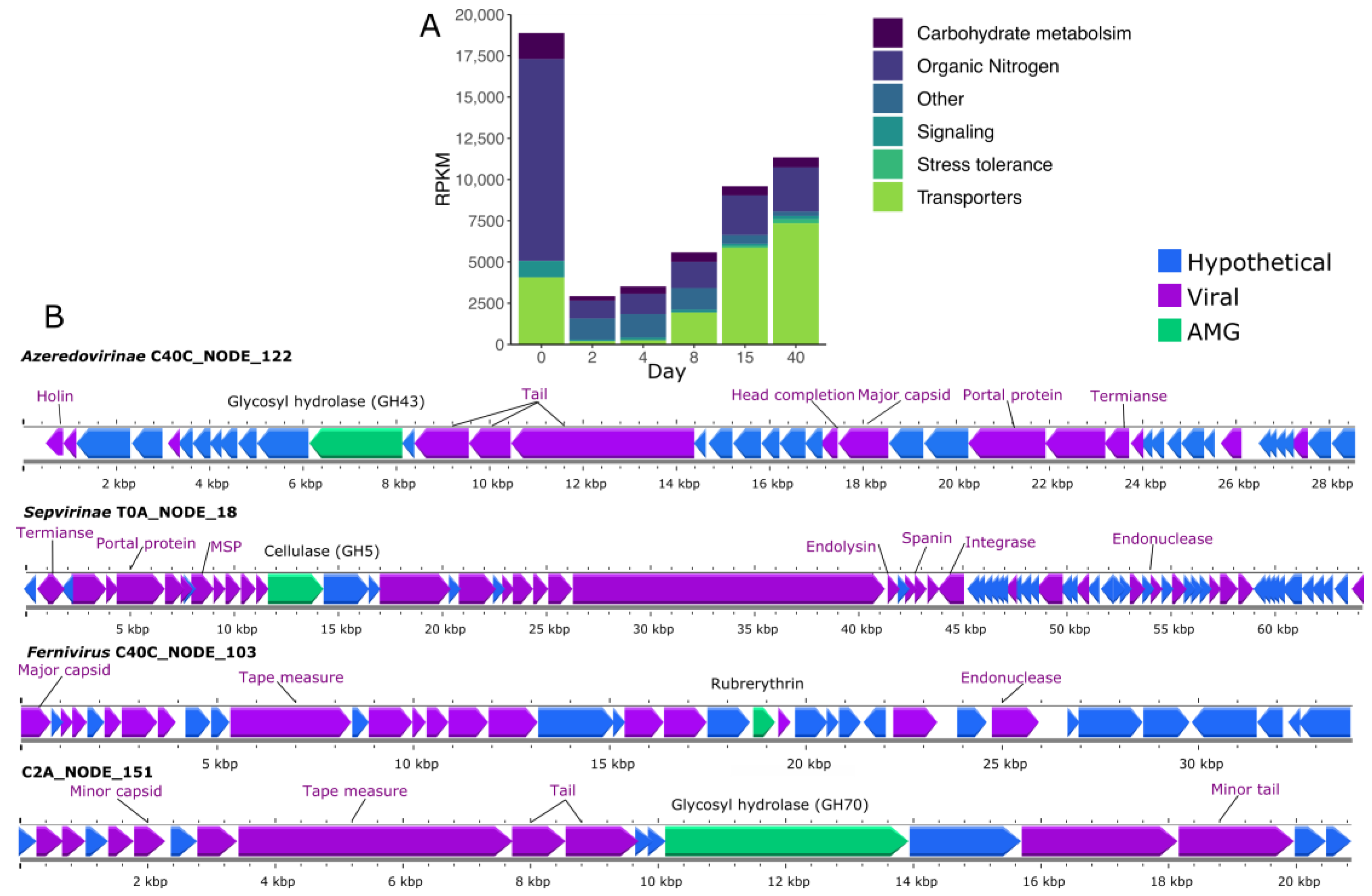
3.7. Auxiliary Metabolic Genes (AMG) Found in vOTUs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Horrocks, R.D.; Dwain Horrocks, R.; Vallentine, J.F. FIELD-HARVESTING SILAGE. In Harvested Forages; Academic Press: Cambridge, MA, USA, 1999; pp. 279–292. [Google Scholar]
- Li, M.; Shan, G.; Zhou, H.; Buescher, W.; Maack, C.; Jungbluth, K.H.; Lipski, A.; Grantz, D.A.; Fan, Y.; Ma, D.; et al. CO2 Production, Dissolution and Pressure Dynamics during Silage Production: Multi-Sensor-Based Insight into Parameter Interactions. Sci. Rep. 2017, 7, 14721. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos Leandro, E.; Ginani, V.C.; de Alencar, E.R.; Pereira, O.G.; Rose, E.C.P.; do Vale, H.M.M.; Pratesi, R.; Hecht, M.M.; Cavalcanti, M.H.; Tavares, C.S.O. Isolation, Identification, and Screening of Lactic Acid Bacteria with Probiotic Potential in Silage of Different Species of Forage Plants, Cocoa Beans, and Artisanal Salami. Probiotics Antimicrob. Proteins 2021, 13, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Grant, R.J.; Ferraretto, L.F. Silage Review: Silage Feeding Management: Silage Characteristics and Dairy Cow Feeding Behavior. J. Dairy Sci. 2018, 101, 4111–4121. [Google Scholar] [CrossRef] [PubMed]
- Kung, L., Jr.; Shaver, R.D.; Grant, R.J.; Schmidt, R.J. Silage Review: Interpretation of Chemical, Microbial, and Organoleptic Components of Silages. J. Dairy Sci. 2018, 101, 4020–4033. [Google Scholar] [CrossRef]
- da Silva, T.C.; da Silva, L.D.; Santos, E.M.; Oliveira, J.S.; Perazzo, A.F. Importance of the Fermentation to Produce High-Quality Silage. In Fermentation Process; InTech: Rijeka, Croatia, 2017; pp. 3–21. [Google Scholar]
- Elferink, S.O.; Driehuis, F.; Gottschal, J.; Spoelstra, S. Manipulating Silage Fermentation. Feed Mix 2002, 10, 20–23. [Google Scholar]
- Zheng, J.; Wittouck, S.; Salvetti, E.; Franz, C.M.A.P.; Harris, H.M.B.; Mattarelli, P.; O’Toole, P.W.; Pot, B.; Vandamme, P.; Walter, J.; et al. A Taxonomic Note on the Genus Lactobacillus: Description of 23 Novel Genera, Emended Description of the Genus Lactobacillus Beijerinck 1901, and Union of Lactobacillaceae and Leuconostocaceae. Int. J. Syst. Evol. Microbiol. 2020, 70, 2782–2858. [Google Scholar] [CrossRef] [PubMed]
- Ávila, C.L.S.; Carvalho, B.F. Silage Fermentation-Updates Focusing on the Performance of Micro-Organisms. J. Appl. Microbiol. 2020, 128, 966–984. [Google Scholar] [CrossRef]
- Gharechahi, J.; Kharazian, Z.A.; Sarikhan, S.; Jouzani, G.S.; Aghdasi, M.; Hosseini Salekdeh, G. The Dynamics of the Bacterial Communities Developed in Maize Silage. Microb. Biotechnol. 2017, 10, 1663–1676. [Google Scholar] [CrossRef]
- Shkoporov, A.N.; Hill, C. Bacteriophages of the Human Gut: The “Known Unknown” of the Microbiome. Cell Host Microbe 2019, 25, 195–209. [Google Scholar] [CrossRef]
- Hsu, B.B.; Gibson, T.E.; Yeliseyev, V.; Liu, Q.; Lyon, L.; Bry, L.; Silver, P.A.; Gerber, G.K. Dynamic Modulation of the Gut Microbiota and Metabolome by Bacteriophages in a Mouse Model. Cell Host Microbe 2019, 25, 803–814.e5. [Google Scholar] [CrossRef]
- Clokie, M.R.; Millard, A.D.; Letarov, A.V.; Heaphy, S. Phages in Nature. Bacteriophage 2011, 1, 31–45. [Google Scholar] [CrossRef]
- Doi, K.; Zhang, Y.; Nishizaki, Y.; Umeda, A.; Ohmomo, S.; Ogata, S. A Comparative Study and Phage Typing of Silage-Making Lactobacillus Bacteriophages. J. Biosci. Bioeng. 2003, 95, 518–525. [Google Scholar] [CrossRef]
- Vongkamjan, K.; Switt, A.M.; den Bakker, H.C.; Fortes, E.D.; Wiedmann, M. Silage Collected from Dairy Farms Harbors an Abundance of Listeriaphages with Considerable Host Range and Genome Size Diversity. Appl. Environ. Microbiol. 2012, 78, 8666–8675. [Google Scholar] [CrossRef] [PubMed]
- Bernardes, T.F.; Gervásio, J.R.S.; De Morais, G.; Casagrande, D.R. A Comparison of Methods to Determine pH in Silages. J. Dairy Sci. 2019, 102, 9039–9042. [Google Scholar] [CrossRef]
- Roth, C.; Sims, T.; Rodehutscord, M.; Seifert, J.; Camarinha-Silva, A. The Active Core Microbiota of Two High-Yielding Laying Hen Breeds Fed with Different Levels of Calcium and Phosphorus. Front. Physiol. 2022, 13, 951350. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, D.M.; Weimer, P.J. Dominance of Prevotella and Low Abundance of Classical Ruminal Bacterial Species in the Bovine Rumen Revealed by Relative Quantification Real-Time PCR. Appl. Microbiol. Biotechnol. 2007, 75, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Gong, J.; Yu, H.; Jin, Y.; Zhu, J.; Han, Y. Identification of Changes in the Composition of Ileal Bacterial Microbiota of Broiler Chickens Infected with Clostridium Perfringens. Vet. Microbiol. 2010, 140, 116–121. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, Interactive, Scalable and Extensible Microbiome Data Science Using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-Resolution Sample Inference from Illumina Amplicon Data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A Versatile Open Source Tool for Metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef] [PubMed]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-Learn: Machine Learning in PYthon. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA Ribosomal RNA Gene Database Project: Improved Data Processing and Web-Based Tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Robeson, M.S., 2nd; O’Rourke, D.R.; Kaehler, B.D.; Ziemski, M.; Dillon, M.R.; Foster, J.T.; Bokulich, N.A. RESCRIPt: Reproducible Sequence Taxonomy Reference Database Management. PLoS Comput. Biol. 2021, 17, e1009581. [Google Scholar] [CrossRef] [PubMed]
- Menzel, P.; Ng, K.L.; Krogh, A. Fast and Sensitive Taxonomic Classification for Metagenomics with Kaiju. Nat. Commun. 2016, 7, 11257. [Google Scholar] [CrossRef]
- Nurk, S.; Meleshko, D.; Korobeynikov, A.; Pevzner, P.A. metaSPAdes: A New Versatile Metagenomic Assembler. Genome Res. 2017, 27, 824–834. [Google Scholar] [CrossRef]
- Kang, D.D.; Li, F.; Kirton, E.; Thomas, A.; Egan, R.; An, H.; Wang, Z. MetaBAT 2: An Adaptive Binning Algorithm for Robust and Efficient Genome Reconstruction from Metagenome Assemblies. PeerJ 2019, 7, e7359. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-W.; Simmons, B.A.; Singer, S.W. MaxBin 2.0: An Automated Binning Algorithm to Recover Genomes from Multiple Metagenomic Datasets. Bioinformatics 2016, 32, 605–607. [Google Scholar] [CrossRef] [PubMed]
- Alneberg, J.; Bjarnason, B.S.; de Bruijn, I.; Schirmer, M.; Quick, J.; Ijaz, U.Z.; Lahti, L.; Loman, N.J.; Andersson, A.F.; Quince, C. Binning Metagenomic Contigs by Coverage and Composition. Nat. Methods 2014, 11, 1144–1146. [Google Scholar] [CrossRef]
- Uritskiy, G.V.; DiRuggiero, J.; Taylor, J. MetaWRAP-a Flexible Pipeline for Genome-Resolved Metagenomic Data Analysis. Microbiome 2018, 6, 158. [Google Scholar] [CrossRef]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the Quality of Microbial Genomes Recovered from Isolates, Single Cells, and Metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Chaumeil, P.-A.; Mussig, A.J.; Hugenholtz, P.; Parks, D.H. GTDB-Tk: A Toolkit to Classify Genomes with the Genome Taxonomy Database. Bioinformatics 2019, 36, 1925–1927. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Chuvochina, M.; Chaumeil, P.-A.; Rinke, C.; Mussig, A.J.; Hugenholtz, P. A Complete Domain-to-Species Taxonomy for Bacteria and Archaea. Nat. Biotechnol. 2020, 38, 1079–1086. [Google Scholar] [CrossRef] [PubMed]
- Payne, L.J.; Todeschini, T.C.; Wu, Y.; Perry, B.J.; Ronson, C.W.; Fineran, P.C.; Nobrega, F.L.; Jackson, S.A. Identification and Classification of Antiviral Defence Systems in Bacteria and Archaea with PADLOC Reveals New System Types. Nucleic Acids Res. 2021, 49, 10868–10878. [Google Scholar] [CrossRef]
- Russel, J.; Pinilla-Redondo, R.; Mayo-Muñoz, D.; Shah, S.A.; Sørensen, S.J. CRISPRCasTyper: Automated Identification, Annotation, and Classification of CRISPR-Cas Loci. CRISPR J. 2020, 3, 462–469. [Google Scholar] [CrossRef]
- Olm, M.R.; Brown, C.T.; Brooks, B.; Banfield, J.F. dRep: A Tool for Fast and Accurate Genomic Comparisons That Enables Improved Genome Recovery from Metagenomes through de-Replication. ISME J. 2017, 11, 2864–2868. [Google Scholar] [CrossRef]
- Shaffer, M.; Borton, M.A.; McGivern, B.B.; Zayed, A.A.; La Rosa, S.L.; Solden, L.M.; Liu, P.; Narrowe, A.B.; Rodríguez-Ramos, J.; Bolduc, B.; et al. DRAM for Distilling Microbial Metabolism to Automate the Curation of Microbiome Function. Nucleic Acids Res. 2020, 48, 8883–8900. [Google Scholar] [CrossRef]
- Asnicar, F.; Thomas, A.M.; Beghini, F.; Mengoni, C.; Manara, S.; Manghi, P.; Zhu, Q.; Bolzan, M.; Cumbo, F.; May, U.; et al. Precise Phylogenetic Analysis of Microbial Isolates and Genomes from Metagenomes Using PhyloPhlAn 3.0. Nat. Commun. 2020, 11, 2500. [Google Scholar] [CrossRef]
- Kim, M.; Oh, H.-S.; Park, S.-C.; Chun, J. Towards a Taxonomic Coherence between Average Nucleotide Identity and 16S rRNA Gene Sequence Similarity for Species Demarcation of Prokaryotes. Int. J. Syst. Evol. Microbiol. 2014, 64, 346–351. [Google Scholar] [CrossRef]
- Sangal, V.; Goodfellow, M.; Jones, A.L.; Schwalbe, E.C.; Blom, J.; Hoskisson, P.A.; Sutcliffe, I.C. Next-Generation Systematics: An Innovative Approach to Resolve the Structure of Complex Prokaryotic Taxa. Sci. Rep. 2016, 6, 38392. [Google Scholar] [CrossRef]
- Qin, Q.-L.; Xie, B.-B.; Zhang, X.-Y.; Chen, X.-L.; Zhou, B.-C.; Zhou, J.; Oren, A.; Zhang, Y.-Z. A Proposed Genus Boundary for the Prokaryotes Based on Genomic Insights. J. Bacteriol. 2014, 196, 2210–2215. [Google Scholar] [CrossRef]
- Pritchard, L.; Glover, R.H.; Humphris, S.; Elphinstone, J.G.; Toth, I.K. Genomics and Taxonomy in Diagnostics for Food Security: Soft-Rotting Enterobacterial Plant Pathogens. Anal. Methods 2015, 8, 12–24. [Google Scholar] [CrossRef]
- Kim, D.; Park, S.; Chun, J. Introducing EzAAI: A Pipeline for High Throughput Calculations of Prokaryotic Average Amino Acid Identity. J. Microbiol. 2021, 59, 476–480. [Google Scholar] [CrossRef]
- Guo, J.; Bolduc, B.; Zayed, A.A.; Varsani, A.; Dominguez-Huerta, G.; Delmont, T.O.; Pratama, A.A.; Gazitúa, M.C.; Vik, D.; Sullivan, M.B.; et al. VirSorter2: A Multi-Classifier, Expert-Guided Approach to Detect Diverse DNA and RNA Viruses. Microbiome 2021, 9, 37. [Google Scholar] [CrossRef]
- Kieft, K.; Zhou, Z.; Anantharaman, K. VIBRANT: Automated Recovery, Annotation and Curation of Microbial Viruses, and Evaluation of Viral Community Function from Genomic Sequences. Microbiome 2020, 8, 90. [Google Scholar] [CrossRef]
- Nayfach, S.; Camargo, A.P.; Schulz, F.; Eloe-Fadrosh, E.; Roux, S.; Kyrpides, N.C. CheckV Assesses the Quality and Completeness of Metagenome-Assembled Viral Genomes. Nat. Biotechnol. 2021, 39, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.-Z.; Yuan, W.-G.; Shang, J.; Shi, Y.-H.; Yang, L.-L.; Liu, M.; Zhu, P.; Jin, T.; Sun, Y.; Yuan, L.-H. Virus Classification for Viral Genomic Fragments Using PhaGCN2. Brief. Bioinform. 2022, 24, 1–9. [Google Scholar] [CrossRef]
- Bin Jang, H.; Bolduc, B.; Zablocki, O.; Kuhn, J.H.; Roux, S.; Adriaenssens, E.M.; Brister, J.R.; Kropinski, A.M.; Krupovic, M.; Lavigne, R.; et al. Taxonomic Assignment of Uncultivated Prokaryotic Virus Genomes Is Enabled by Gene-Sharing Networks. Nat. Biotechnol. 2019, 37, 632–639. [Google Scholar] [CrossRef] [PubMed]
- Cook, R.; Brown, N.; Redgwell, T.; Rihtman, B.; Barnes, M.; Clokie, M.; Stekel, D.J.; Hobman, J.; Jones, M.A.; Millard, A. INfrastructure for a PHAge REference Database: Identification of Large-Scale Biases in the Current Collection of Cultured Phage Genomes. PHAGE 2021, 2, 214–223. [Google Scholar] [CrossRef]
- Hyatt, D.; Chen, G.-L.; Locascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic Gene Recognition and Translation Initiation Site Identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Göker, M. VICTOR: Genome-Based Phylogeny and Classification of Prokaryotic Viruses. Bioinformatics 2017, 33, 3396–3404. [Google Scholar] [CrossRef] [PubMed]
- Göker, M.; García-Blázquez, G.; Voglmayr, H.; Tellería, M.T.; Martín, M.P. Molecular Taxonomy of Phytopathogenic Fungi: A Case Study in Peronospora. PLoS ONE 2009, 4, e6319. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, J.M.; Rinne, M. Highlights of Progress in Silage Conservation and Future Perspectives. Grass Forage Sci. 2018, 73, 40–52. [Google Scholar] [CrossRef]
- Hayes, S.; Mahony, J.; Nauta, A.; van Sinderen, D. Metagenomic Approaches to Assess Bacteriophages in Various Environmental Niches. Viruses 2017, 9, 127. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.; Clooney, A.G.; Stockdale, S.R.; Buttimer, C.; Draper, L.A.; Ross, R.P.; Hill, C. Isolation of a Novel Jumbo Bacteriophage Effective Against Klebsiella Aerogenes. Front. Med. 2020, 7, 67. [Google Scholar] [CrossRef]
- Breitbart, M.; Rohwer, F. Here a Virus, There a Virus, Everywhere the Same Virus? Trends Microbiol. 2005, 13, 278–284. [Google Scholar] [CrossRef]
- Queiroz, L.L.; Lacorte, G.A.; Isidorio, W.R.; Landgraf, M.; de Melo Franco, B.D.G.; Pinto, U.M.; Hoffmann, C. High Level of Interaction between Phages and Bacteria in an Artisanal Raw Milk Cheese Microbial Community. mSystems 2022, 8, e00564-22. [Google Scholar] [CrossRef]
- Yu, Z.; Ma, Y.; Guan, Y.; Zhu, Y.; Wang, K.; Wang, Y.; Liu, P.; Chen, J.; Yu, Y. Metagenomics of Virus Diversities in Solid-State Brewing Process of Traditional Chinese Vinegar. Foods 2022, 11, 3296. [Google Scholar] [CrossRef]
- Walker, P.J.; Siddell, S.G.; Lefkowitz, E.J.; Mushegian, A.R.; Adriaenssens, E.M.; Alfenas-Zerbini, P.; Dempsey, D.M.; Dutilh, B.E.; García, M.L.; Curtis Hendrickson, R.; et al. Recent Changes to Virus Taxonomy Ratified by the International Committee on Taxonomy of Viruses (2022). Arch. Virol. 2022, 167, 2429–2440. [Google Scholar] [CrossRef]
- You, L.; Yang, C.; Jin, H.; Kwok, L.-Y.; Sun, Z.; Zhang, H. Metagenomic Features of Traditional Fermented Milk Products. LWT 2022, 155, 112945. [Google Scholar] [CrossRef]
- Koskella, B.; Brockhurst, M.A. Bacteria-Phage Coevolution as a Driver of Ecological and Evolutionary Processes in Microbial Communities. FEMS Microbiol. Rev. 2014, 38, 916–931. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Breidt, F.; Plengvidhya, V.; Fleming, H.P. Bacteriophage Ecology in Commercial Sauerkraut Fermentations. Appl. Environ. Microbiol. 2003, 69, 3192–3202. [Google Scholar] [CrossRef] [PubMed]
- Jończyk, E.; Kłak, M.; Międzybrodzki, R.; Górski, A. The Influence of External Factors on Bacteriophages—Review. Folia Microbiol. 2011, 56, 191–200. [Google Scholar] [CrossRef]
- Ledormand, P.; Desmasures, N.; Schlusselhuber, M.; Sesboüé, A.; Ledauphin, J.; Dalmasso, M. Phages Shape Microbial Dynamics and Metabolism of a Model Community Mimicking Cider, a Fermented Beverage. Viruses 2022, 14, 2283. [Google Scholar] [CrossRef]
- Somerville, V.; Schowing, T.; Chabas, H.; Schmidt, R.S.; von Ah, U.; Bruggmann, R.; Engel, P. Extensive Diversity and Rapid Turnover of Phage Defense Repertoires in Cheese-Associated Bacterial Communities. Microbiome 2022, 10, 137. [Google Scholar] [CrossRef]
- Tesson, F.; Hervé, A.; Mordret, E.; Touchon, M.; d’Humières, C.; Cury, J.; Bernheim, A. Systematic and Quantitative View of the Antiviral Arsenal of Prokaryotes. Nat. Commun. 2022, 13, 2561. [Google Scholar] [CrossRef] [PubMed]
- Crawley, A.B.; Henriksen, E.D.; Stout, E.; Brandt, K.; Barrangou, R. Characterizing the Activity of Abundant, Diverse and Active CRISPR-Cas Systems in Lactobacilli. Sci. Rep. 2018, 8, 11544. [Google Scholar] [CrossRef]
- Ter Horst, A.M.; Santos-Medellín, C.; Sorensen, J.W.; Zinke, L.A.; Wilson, R.M.; Johnston, E.R.; Trubl, G.G.; Pett-Ridge, J.; Blazewicz, S.J.; Hanson, P.J.; et al. Minnesota Peat Viromes Reveal Terrestrial and Aquatic Niche Partitioning for Local and Global Viral Populations. Microbiome 2021, 9, 233. [Google Scholar] [CrossRef]
- Hwang, Y.; Rahlff, J.; Schulze-Makuch, D.; Schloter, M.; Probst, A.J. Diverse Viruses Carrying Genes for Microbial Extremotolerance in the Atacama Desert Hyperarid Soil. mSystems 2021, 6, e00385-21. [Google Scholar] [CrossRef]
- Nelson, A.R.; Narrowe, A.B.; Rhoades, C.C.; Fegel, T.S.; Daly, R.A.; Roth, H.K.; Chu, R.K.; Amundson, K.K.; Young, R.B.; Steindorff, A.S.; et al. Wildfire-Dependent Changes in Soil Microbiome Diversity and Function. Nat Microbiol. 2022, 7, 1419–1430. [Google Scholar] [CrossRef]
- Islam, M.M.; Fernando, S.C.; Saha, R. Metabolic Modeling Elucidates the Transactions in the Rumen Microbiome and the Shifts Upon Virome Interactions. Front. Microbiol. 2019, 10, 2412. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sáenz, J.S.; Rios-Galicia, B.; Rehkugler, B.; Seifert, J. Dynamic Development of Viral and Bacterial Diversity during Grass Silage Preservation. Viruses 2023, 15, 951. https://doi.org/10.3390/v15040951
Sáenz JS, Rios-Galicia B, Rehkugler B, Seifert J. Dynamic Development of Viral and Bacterial Diversity during Grass Silage Preservation. Viruses. 2023; 15(4):951. https://doi.org/10.3390/v15040951
Chicago/Turabian StyleSáenz, Johan S., Bibiana Rios-Galicia, Bianca Rehkugler, and Jana Seifert. 2023. "Dynamic Development of Viral and Bacterial Diversity during Grass Silage Preservation" Viruses 15, no. 4: 951. https://doi.org/10.3390/v15040951
APA StyleSáenz, J. S., Rios-Galicia, B., Rehkugler, B., & Seifert, J. (2023). Dynamic Development of Viral and Bacterial Diversity during Grass Silage Preservation. Viruses, 15(4), 951. https://doi.org/10.3390/v15040951