
Discovery of Avian Paramyxoviruses APMV-1 and APMV-6 in Shorebirds and Waterfowl in Southern Ukraine
 , , , , , , , ,
, , , , , , , , 
 and add
Show full author list
and add
Show full author list
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Virus Isolation
2.3. Virus Identification
2.4. RNA Extraction and Diagnostic RT-PCR for APMV
2.5. Dataset Development for Genome Assembly
2.6. Development of Tiling Primers for Full Genome Amplification
2.7. Amplicon Synthesis, cDNA Library Preparation, and Bioinformatics
2.8. Phylogenetic Analysis
3. Results
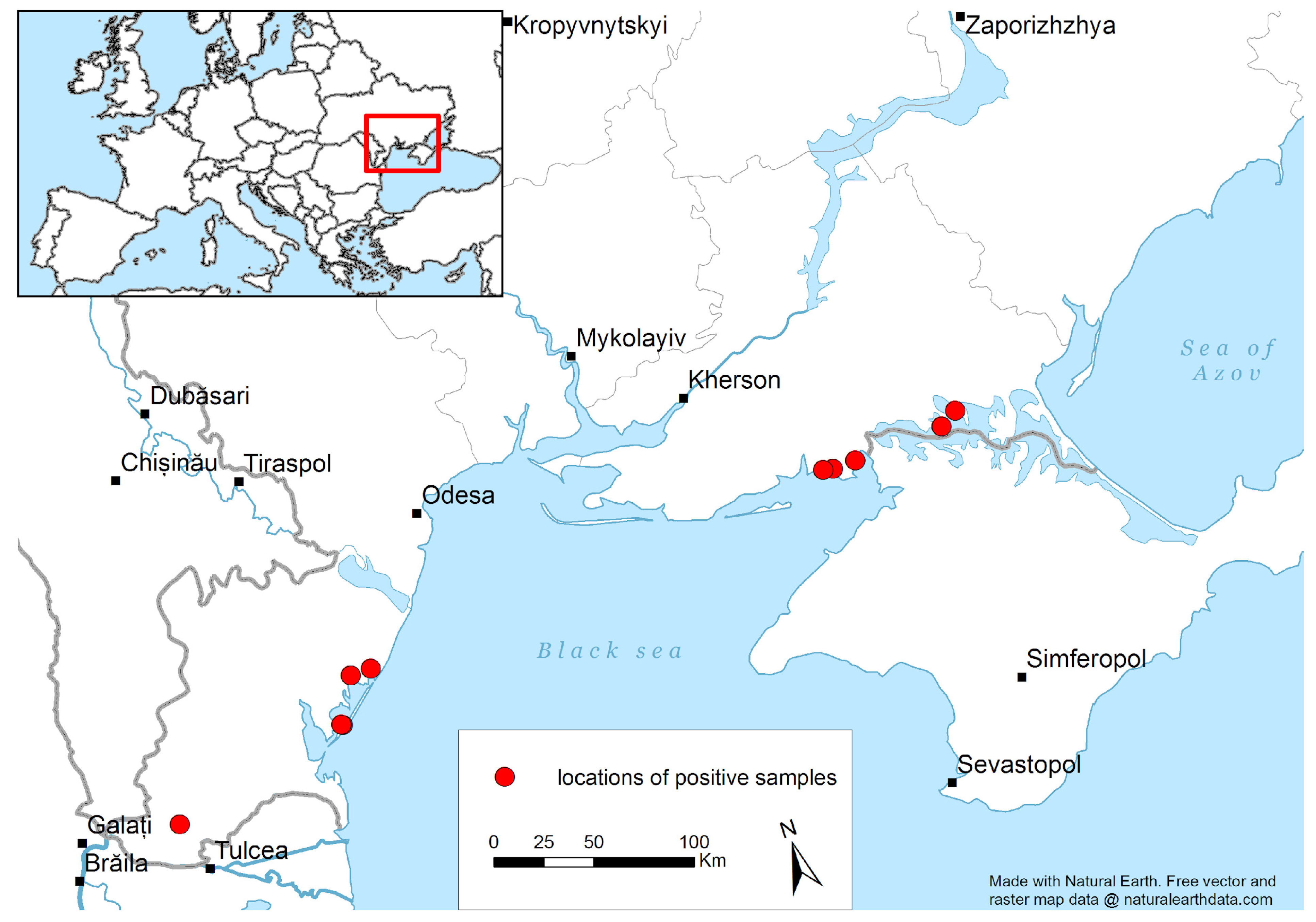
3.1. Avian Avulavirus Detection in Wild Birds in Ukraine
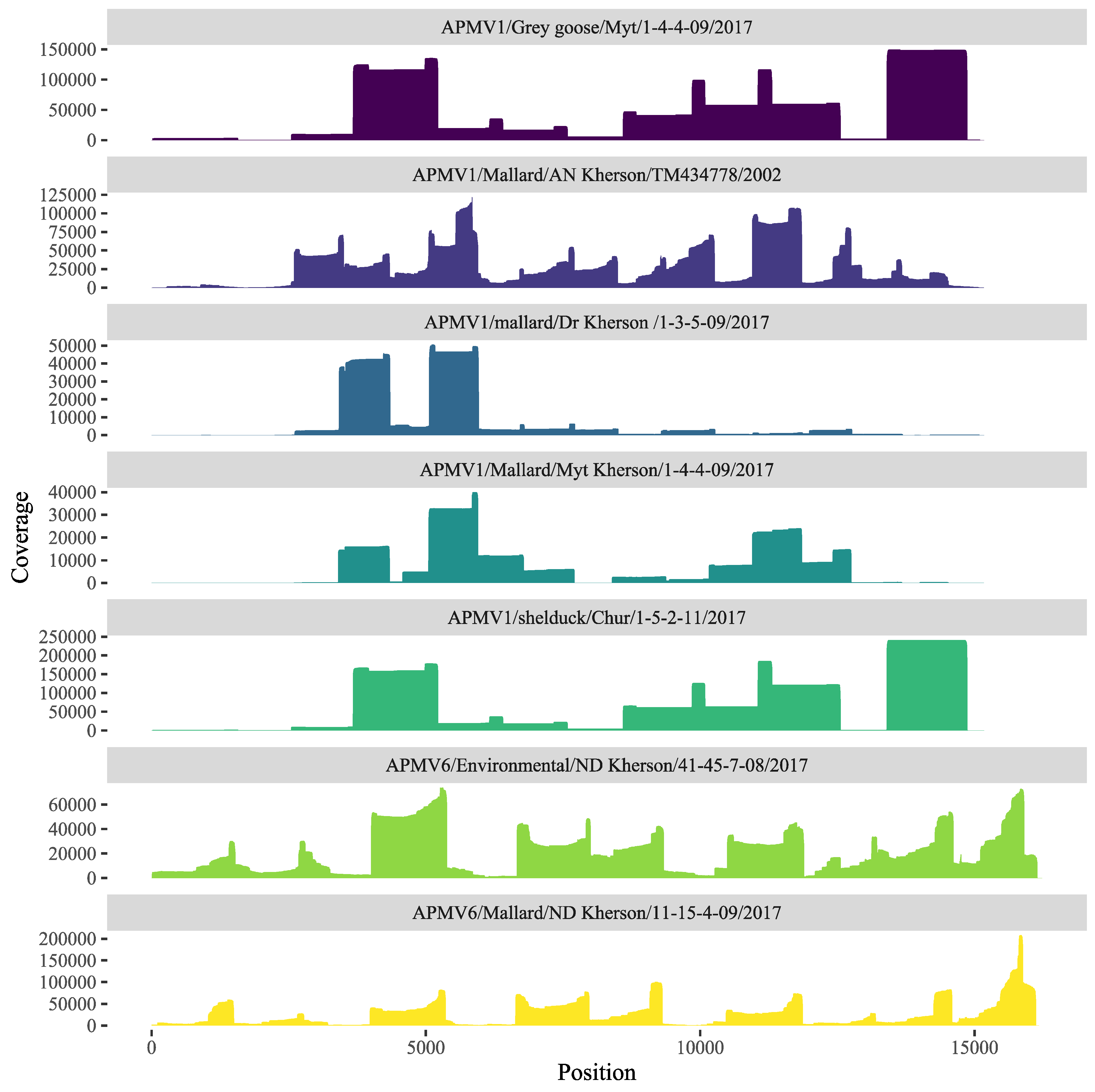
3.2. Sequencing and Phylogenetic Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Maes, P.; Amarasinghe, G.K.; Ayllón, M.A.; Basler, C.F.; Bavari, S. Taxonomy of the order Mononegavirales: Second update 2018. Arch. Virol. 2019, 164, 1233–1244. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Servan, R.; Almeida, D.; Gil, P.; Albina, E. Cleavage site of Newcastle disease virus determines viral fitness in persistent infection cells. Vet. Microbiol. 2018, 216, 123–131. [Google Scholar] [CrossRef]
- Swayne, D.E.; Glisson, J.R.; McDougald, L.R.; Nolan, L.K.; Suarez, D.L.; Nair, V. (Eds.) Diseases of Poultry; John Wiley & Sons: Hoboken, NJ, USA, 2013. [Google Scholar]
- Rehman, Z.U.; Meng, C.; Sun, Y.; Mahrose, K.M.; Umar, S.; Ding, C.; Munir, M. Pathobiology of Avian avulavirus 1: Special focus on waterfowl. Vet. Res. 2018, 49, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Miller, P.J.; Afonso, C.L.; Spackman, E.; Scott, M.A.; Pedersen, J.C.; Senne, D.A.; Brown, J.D.; Fuller, C.M.; Uhart, M.M.; Karesh, W.B.; et al. Evidence for a New Avian Paramyxovirus Serotype 10 Detected in Rockhopper Penguins from the Falkland Islands. J. Virol. 2010, 84, 11496–11504. [Google Scholar] [CrossRef] [PubMed]
- Rahman, A.; Habib, M.; Shabbir, M.Z. Adaptation of Newcastle Disease Virus (NDV) in Feral Birds and their Potential Role in Interspecies Transmission Abstract. Open Virol. J. 2018, 12, 52–68. [Google Scholar] [CrossRef] [PubMed]
- Rahman, A.; Munir, M.; Shabbir, M.Z. Comparative evolutionary and phylogenomic analysis of Avian avulaviruses 1-20. Mol. Phylogenet. Evol. 2018, 127, 931–951. [Google Scholar] [CrossRef] [PubMed]
- Dimitrov, K.M.; Abolnik, C.; Afonso, C.L.; Albina, E.; Bahl, J.; Berg, M.; Briand, F.X.; Brown, I.H.; Choi, K.S.; Chvala, I.; et al. Updated unified phylogenetic classification system and revised nomenclature for Newcastle disease virus. Infect. Genet. Evol. 2019, 74, 103917. Available online: https://www.sciencedirect.com/science/article/pii/S1567134819301388?via%3Dihub (accessed on 10 September 2022). [CrossRef]
- Diel, D.G.; Luciana, H.A.; Liu, H.; Wang, Z.; Miller, P.J.; Afonso, C.L. Genetic diversity of avian paramyxovirus type 1: Proposal for a unified nomenclature and classification system of Newcastle disease virus genotypes. Infect. Genet. Evol. 2012, 12, 1770–1779. [Google Scholar] [CrossRef]
- Kinde, H.; Hullinger, P.J.; Charlton, B.; Mcfarland, M.; Hietala, S.K.; Velez, V.; Case, J.T.; Garber, L.; Wainwright, S.H.; Mikolon, A.B.; et al. The Isolation of Exotic Newcastle Disease (END) Virus from Nonpoultry Avian Species Associated with the Epidemic of END in Chickens in Southern California: 2002–2003 The Isolation of Exotic Newcastle Disease (END) Virus from Nonpoultry Avian Specie. Avian Dis. 2005, 49, 195–198. [Google Scholar] [CrossRef]
- Reeves, A.B.; Poulson, R.L.; Muzyka, D.; Ogawa, H.; Bui, V.N.; Hall, J.S.; Pantin-jackwood, M.; Stallknecht, D.E.; Ramey, A.M. Limited evidence of intercontinental dispersal of avian paramyxoviruses serotype 4 by migratory birds. Infect. Genet. Evol. 2016, 40, 104–108. [Google Scholar] [CrossRef]
- Muzyka, D.; Pantin-Jackwood, M.; Stegniy, B.; Rula, O.; Bolotin, V.; Stegniy, A.; Gerilovych, A.; Shutchenko, P.; Stegniy, M.; Koshelev, V.; et al. Wild Bird Surveillance for Avian Paramyxoviruses in the Azov-Black Sea Region of Ukraine (2006 to 2011) Reveals Epidemiological. Appl. Environ. Microbiol. 2014, 80, 5427–5438. [Google Scholar] [CrossRef] [PubMed]
- Hicks, J.T.; Dimitrov, K.M.; Afonso, C.L.; Ramey, A.M.; Bahl, J. Global phylodynamic analysis of avian paramyxovirus-1 provides evidence of inter-host transmission and intercontinental spatial diffusion. BMC Evol. Biol. 2019, 19, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.; Subbiah, M.; Kumar, S.; De Nardi, R.; Terregina, C.; Collins, P.L.; Samal, S.K. Complete genome sequences of avain paramyxovirus serotype 6 prototype strain Hong Kong and a recent novel strain from Italy: Evidence for existence of sybgroups with the serotype. Virus Res. 2010, 150, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.; Kim, Y.; An, I.; Jun, S.; Yongkwan, W.; Jeong, H.; Kang, L.; Choi, S.; Pyeong, S.; Wongi, I.; et al. Complete genome sequence of a novel avian paramyxovirus isolated from wild birds in South Korea. Arch. Virol. 2018, 163, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Young, K.T.; Stephens, J.Q.; Poulson, R.L.; Stallknecht, D.E.; Dimitrov, K.M.; Butt, S.L.; Stanton, J.B. Putative Novel Avian Paramyxovirus (AMPV) and Reidentification of APMV-2 and APMV-6 to the Species Level Based on Wild Bird Surveillance (United States, 2016-2018). Appl. Environ. Microbiol. 2022, 88, e0046622. Available online: https://pubmed.ncbi.nlm.nih.gov/35612300/ (accessed on 10 September 2022). [CrossRef]
- Causey, D.; Edwards, S.V. Ecology of avian influenza in birds. J. Infect. Dis. 2008, 197, 29–35. [Google Scholar] [CrossRef]
- Muzyka, D.; Pantin-Jackwood, M.; Spackman, E.; Smith, D.; Rula, O.; Muzyka, N.; Stegniy, B. Isolation and Genetic Characterization of Avian Influenza Viruses Isolated from Wild Birds in the Azov-Black Sea Region of Ukraine (2001–2012). Avian Dis. 2016, 60, 365–377. [Google Scholar] [CrossRef]
- Muzyka, D.; Rula, O.; Tkachenko, S.; Muzyka, N.; Susanne, K.; Pishchanskyi, O.; Stegniy, B.; Pantin-Jackwood, M.; Beer, M. Highly Pathogenic and Low Pathogenic Avian Influenza H5 Subtype Viruses in Wild Birds in Ukraine. Avian Dis. 2019, 63, 235–245. [Google Scholar] [CrossRef]
- Sapachova, M.; Kovalenko, G.; Sushko, M.; Bezymennyi, M.; Muzyka, D.; Usachenko, N.; Abromov, A.; Essen, S.; Lewis, N.S.; Mezhenskyi, A.; et al. Phylogenetic Analysis of H5N8 Highly Pathogenic Avian Influenza Viruses in Ukraine, 2016–2017. Vector-Borne Zoonotic Dis. 2021, 21, 979–988. [Google Scholar] [CrossRef]
- Crossley, B.M.; Rejmanek, D.; Baroch, J.; Stanton, J.B.; Young, K.T.; Killian, M.L.; Torchetti, M.K.; Hietala, S.K. Nanopore sequencing as a rapid tool for identification and pathotyping of avian influenza A viruses. J. Vet. Diagn. Investig. 2021, 33, 253–260. [Google Scholar] [CrossRef]
- Leslie, J. Newcastle disease: Outbreak losses and control policy costs. Vet. Rec. 2000, 146, 603–606. [Google Scholar] [CrossRef] [PubMed]
- Spackman, E.; Pedersen, J.C.; Mckinley, E.T.; Gelb, J.J. Optimal specimen collection and transport methods for the detection of avian influenza virus and Newcastle disease virus. BMC Vet. Res. 2013, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.M.; Dufour-Zavala, L.; Jackwood, M.W.; Lee, M.D.; Lupiani, B.; Reed, W.M.; Spackman, E.; Woolcock, P.R. A Laboratory Manual for the Isolation, Identification, and Characterization of Avian Pathogens, 5th ed.; American Association of Avian Pathologists: Athens, GA, USA, 2008. [Google Scholar]
- Swayne, D.; Brown, E. (Eds.) The Manual of Diagnostic Tests and Vaccines for Terrestrial Animals (Mammals, Birds, Bees), 6th ed.; World Organization for Animal Health (WOAH): Paris, France, 2008. [Google Scholar]
- Spackman, E. A Brief Introduction to Avian Influenza Virus. In Avian Influenza Virus Methods; Humana Press: Totowa, NJ, USA, 2008; pp. 1–6. [Google Scholar]
- Czegledi, A.; Ujvari, D.; Somogyi, E.; Wehmann, E.; Werner, O.; Lomniczi, B. Third genome size category of avian paramyxovirus serotype 1 (Newcastle disease virus) and evolutionary implications. Virus Res. 2006, 120, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Wise, M.G.; Suarez, D.L.; Seal, B.S.; Pedersen, J.C.; Senne, D.A.; King, D.J.; Kapczynski, D.R.; Spackman, E. Development of a Real-Time Reverse-Transcription PCR for Detection of Newcastle Disease Virus RNA in Clinical Samples. J. Clin. Microbiol. 2004, 42, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Quick, J.; Grubaugh, N.D.; Pullan, S.T.; Claro, I.M.; Smith, A.D.; Gangavarapu, K.; Oliveira, G.; Robles-Sikisaka, R.; Rogers, T.F.; Beutler, N.A.; et al. Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat. Protoc. 2017, 12, 1261–1276. [Google Scholar] [CrossRef]
- Quick, J.; Loman, N.J.; Duraffour, S.; Simpson, J.T.; Severi, E.; Cowley, L.; Bore, J.A.; Koundouno, R.; Dudas, G.; Mikhail, A.; et al. Real-time, portable genome sequencing for Ebola surveillance. Nature 2016, 530, 228–232. [Google Scholar] [CrossRef]
- Li, H. Sequence analysis Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Katoh, K.; Kuma, K.; Toh, H.; Miyata, T. MAFFT version 5: Improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 2005, 33, 511–518. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Toh, H. Recent developments in the MAFFT multiple sequence alignment program. Brief. Bioinform. 2008, 9, 286–298. [Google Scholar] [CrossRef]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Ho, S.Y.W.; Phillips, M.J.; Rambaut, A. Relaxed Phylogenetics and Dating with Confidence. PLoS Biol. 2006, 4, 699–710. [Google Scholar] [CrossRef]
- Hasegawa, M.; Kishin, H.; Yano, T. Dating of the Human-Ape Splitting by a Molecular Clock of Mitochondrial DNA. J. Mol. Evol. 1985, 22, 160–174. [Google Scholar] [CrossRef]
- Martin, D.P.; Varsani, A.; Roumagnac, P.; Botha, G.; Maslamoney, S.; Schwab, T.; Kelz, Z.; Kumar, V.; Murrell, B. RDP5: A computer program for analyzing recombination in, and removing signals of recombination from, nucleotide sequence datasets. Virus Evol. 2020, 7, veaa087. Available online: https://pubmed.ncbi.nlm.nih.gov/33936774/ (accessed on 10 September 2022). [CrossRef]
- Kuiken, T.; Leighton, F.A.; Wobeser, G.; Danesik, K.L.; Riva, J.; Heckert, R.A. An Epidemic of Newcastle Disease in Double-Crested Cormorants from Saskatchewan. J. Wildl. Dis. 1998, 34, 457–471. [Google Scholar] [CrossRef]
- Diel, D.G.; Miller, P.J.; Wolf, P.C.; Mickley, R.M.; Anthony, R.; Emanueli, D.C.; Shively, K.J.; Pedersen, K.; Afonso, C.L.; Diel, D.G.; et al. Characterization of Newcastle Disease Viruses Isolated from Cormorant and Gull Species in the United States in 2010. Avian Dis. 2012, 56, 128–133. [Google Scholar] [CrossRef]
- Yakovleva, A.; Kovalenko, G.; Redlinger, M.; Liulchuk, M.G.; Bortz, E.; Zadorozhna, V.I.; Scherbinska, A.M.; Wertheim, J.O.; Goodfellow, I.; Meredith, L.; et al. Tracking SARS-COV-2 variants using Nanopore sequencing in Ukraine in 2021. Sci. Rep. 2022, 12, 15749. Available online: https://pubmed.ncbi.nlm.nih.gov/36131001/ (accessed on 22 September 2022). [CrossRef] [PubMed]
- Glickman, R.L.; Syddall, R.J.; Iorio, R.M.; Sheehan, J.P.; Bratt, M.A. Quantitative Basic Residue Requirements in the Cleavage-Activation Site of the Fusion Glycoprotein as a Determinant of Virulence for Newcastle Disease Virus. J. Virol. 1988, 62, 354–356. [Google Scholar] [CrossRef]
- Miller, P.J.; Lucio, E.; Afonso, C.L. Newcastle disease: Evolution of genotypes and the related diagnostic challenges. Infect. Genet. Evol. 2010, 10, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Nayak, B.; Militino, F.; Kumar, S.; Paldurai, A.; Collins, P.L.; Samal, S.K. Avian paramyxovirus serotypes 2-9 (APMV-2-9) vary in the ability to induce protective immunity in chickens against challenge with virulent Newcastle disease virus (APMV-1). Vaccine 2012, 30, 2220–2227. [Google Scholar] [CrossRef] [PubMed]
- Boere, G.C.; Stroud, D.A. The Flyway Concept: What It Is and What It Isn’t; Boere, G.C., Galbraith, C.A., Stroud, D.A., Eds.; The Stationery Office: Edinburgh, UK, 2016. [Google Scholar]
- Young, K.T.; Lahmers, K.K.; Sellers, H.S.; Stallknecht, D.E.; Poulson, R.L.; Saliki, J.T.; Tompkins, S.M.; Padykula, I.; Siepker, C.; Howerth, E.W.; et al. Randomly primed, strand-switching, MinION-based sequencing for the detection and characterization of cultured RNA viruses. J. Vet. Diagn. Investig. 2021, 33, 202–215. Available online: https://pubmed.ncbi.nlm.nih.gov/33357075/ (accessed on 22 September 2022). [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name | HI Test Results | Sample Description |
|---|---|---|
| APMV1|Mallard|Myt_Kherson|1-4-4-09|2017 | APMV1 | Genetic material (RNA) collected in September 2017 in Kherson Oblast from a clinically healthy wild mallard. |
| APMV1|Mallard|AN_Kherson|TM434778|2002 | APMV1 | Genetic material (RNA) collected in December 2002 in Kherson Oblast from a clinically healthy wild mallard. |
| APMV-6|Environmental|ND Kherson|41-45|7-08|2017 | APMV6 | Genetic material (RNA) collected in August 2017 in Kherson Oblast from the environment. |
| APMV-6|Mallard|ND Kherson|11-15|4-09|2017 | APMV6 | Genetic material (RNA) collected in September 2017 in Kherson Oblast from a clinically healthy wild mallard. |
| APMV-1|Mallard|Dr_Kherson|1-3|5-09|2017 | APMV1 | Genetic material (RNA) collected in September 2017 in Kherson Oblast from a clinically healthy wild mallard. |
| APMV-1|Grey_Goose|Myt_Kherson|1-4|4-09|2017 | APMV1 | Genetic material (RNA) collected in September 2017 in Kherson Oblast from a clinically healthy wild grey goose. |
| APMV-1|Shelduck|Chur_Kherson|1-5|2-11|2017 | APMV1 | Genetic material (RNA) collected in November 2017 in Kherson Oblast from a clinically healthy wild shelduck. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klink, A.C.; Rula, O.; Sushko, M.; Bezymennyi, M.; Mezinov, O.; Gaidash, O.; Bai, X.; Stegniy, A.; Sapachova, M.; Datsenko, R.; et al. Discovery of Avian Paramyxoviruses APMV-1 and APMV-6 in Shorebirds and Waterfowl in Southern Ukraine. Viruses 2023, 15, 699. https://doi.org/10.3390/v15030699
Klink AC, Rula O, Sushko M, Bezymennyi M, Mezinov O, Gaidash O, Bai X, Stegniy A, Sapachova M, Datsenko R, et al. Discovery of Avian Paramyxoviruses APMV-1 and APMV-6 in Shorebirds and Waterfowl in Southern Ukraine. Viruses. 2023; 15(3):699. https://doi.org/10.3390/v15030699
Chicago/Turabian StyleKlink, Amy C., Oleksandr Rula, Mykola Sushko, Maksym Bezymennyi, Oleksandr Mezinov, Oleksandr Gaidash, Xiao Bai, Anton Stegniy, Maryna Sapachova, Roman Datsenko, and et al. 2023. "Discovery of Avian Paramyxoviruses APMV-1 and APMV-6 in Shorebirds and Waterfowl in Southern Ukraine" Viruses 15, no. 3: 699. https://doi.org/10.3390/v15030699
APA StyleKlink, A. C., Rula, O., Sushko, M., Bezymennyi, M., Mezinov, O., Gaidash, O., Bai, X., Stegniy, A., Sapachova, M., Datsenko, R., Skorokhod, S., Nedosekov, V., Hill, N. J., Ninua, L., Kovalenko, G., Ducluzeau, A. L., Mezhenskyi, A., Buttler, J., Drown, D. M., ... Muzyka, D. (2023). Discovery of Avian Paramyxoviruses APMV-1 and APMV-6 in Shorebirds and Waterfowl in Southern Ukraine. Viruses, 15(3), 699. https://doi.org/10.3390/v15030699