
The Viromes of Six Ecosystem Service Provider Parasitoid Wasps

 , ,
, ,  , ,
, , 
Abstract
1. Introduction
2. Materials and Methods
- Recovery and processing of RNA-seq libraries
- Manual curation and improvement of putative viral genomes
- Phylogenetic analyses
- Quantification of viral sequences
3. Results
3.1. Metagenomics Analyses
3.2. Characterization of the Wasps’ Virome
- Ampulex compressa
- Cotesia vestalis
- Diachasma alloeum
- Ectemnius lituratus
- Pemphredon lugubris
- Telenomus podisi
3.3. Quantification of Viral Transcripts
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Purkiss, T.; Lach, L. Pathogen spillover from Apis mellifera to a stingless bee. Proc. R. Soc. B Boil. Sci. 2019, 286, 20191071. [Google Scholar] [CrossRef] [PubMed]
- Gorbalenya, A.E.; Lauber, C. Bioinformatics of virus taxonomy: Foundations and tools for developing sequence-based hierarchical classification. Curr. Opin. Virol. 2022, 52, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Li, C.-X.; Shi, M.; Tian, J.-H.; Lin, X.-D.; Kang, Y.-J.; Chen, L.-J.; Qin, X.-C.; Xu, J.; Holmes, E.C.; Zhang, Y.-Z. Unprecedented genomic diversity of RNA viruses in arthropods reveals the ancestry of negative-sense RNA viruses. eLife 2015, 4, e05378. [Google Scholar] [CrossRef] [PubMed]
- Herniou, E.A.; Huguet, E.; Thézé, J.; Bézier, A.; Periquet, G.; Drezen, J.-M. When parasitic wasps hijacked viruses: Genomic and functional evolution of polydnaviruses. Philos. Trans. R. Soc. B Biol. Sci. 2013, 368, 20130051. [Google Scholar] [CrossRef] [PubMed]
- Blomström, A.-L. Viral metagenomics as an emerging and powerful tool in veterinary medicine. Vet. Q. 2011, 31, 107–114. [Google Scholar] [CrossRef]
- Käfer, S.; Paraskevopoulou, S.; Zirkel, F.; Wieseke, N.; Donath, A.; Petersen, M.; Jones, T.C.; Liu, S.; Zhou, X.; Middendorf, M.; et al. Re-assessing the diversity of negative strand RNA viruses in insects. PLoS Pathog. 2019, 15, e1008224. [Google Scholar] [CrossRef]
- Stork, N.E. How Many Species of Insects and Other Terrestrial Arthropods Are There on Earth? Annu. Rev. Entomol. 2018, 63, 31–45. [Google Scholar] [CrossRef]
- Ballal, C.R.; Verghese, A. Role of Parasitoids and Predators in the Management of Insect Pests. In New Horizons in Insect Science: Towards Sustainable Pest Management; Chakravarthy, A.K., Ed.; Springer: New Delhi, India, 2015. [Google Scholar] [CrossRef]
- Hiroyoshi, S.; Harvey, J.A.; Nakamatsu, Y.; Nemoto, H.; Mitsuhashi, J.; Mitsunaga, T.; Tanaka, T. Potential Host Range of the Larval Endoparasitoid Cotesia vestalis (=plutellae) (Hymenoptera: Braconidae). Int. J. Insect Sci. 2017, 9, 1179543317715623. [Google Scholar] [CrossRef]
- Brock, R.E.; Cini, A.; Sumner, S. Ecosystem services provided by aculeate wasps. Biol. Rev. 2021, 96, 1645–1675. [Google Scholar] [CrossRef]
- Raven, P.H.; Wagner, D.L. Agricultural intensification and climate change are rapidly decreasing insect biodiversity. Proc. Natl. Acad. Sci. USA 2021, 118, e2002548117. [Google Scholar] [CrossRef]
- Forister, M.L.; Pelton, E.M.; Black, S.H. Declines in insect abundance and diversity: We know enough to act now. Conserv. Sci. Pract. 2019, 1, e80. [Google Scholar] [CrossRef]
- Paukkunen, J.; Pöyry, J.; Kuussaari, M. Species traits explain long-term population trends of Finnish cuckoo wasps (Hymenoptera: Chrysididae). Insect Conserv. Divers. 2017, 11, 58–71. [Google Scholar] [CrossRef]
- Sánchez-Bayo, F.; Wyckhuys, K.A. Worldwide decline of the entomofauna: A review of its drivers. Biol. Conserv. 2019, 232, 8–27. [Google Scholar] [CrossRef]
- Bohart, R.M.; Menke, A.S. Sphecid Wasps of the World; University of California Press: Oakland, CA, USA, 1976; No. 1. [Google Scholar] [CrossRef]
- Arvidson, R.; Landa, V.; Frankenberg, S.; Adams, M.E. Life History of the Emerald Jewel Wasp Ampulex compressa. J. Hymenopt. Res. 2018, 63, 1–13. [Google Scholar] [CrossRef]
- Furlong, M.J.; Wright, D.J.; Dosdall, L.M. Diamondback Moth Ecology and Management: Problems, Progress, and Prospects. Annu. Rev. Èntomol. 2013, 58, 517–541. [Google Scholar] [CrossRef]
- Forbes, A.A.; Powell, T.H.Q.; Lobo, N.F.; Noor, M.A.F.; Feder, J.L. Permanent genetic resources: Polymorphic microsatellite loci for Diachasma alloeum (Hymenoptera: Braconidae). Mol. Ecol. Resour. 2008, 8, 373–376. [Google Scholar] [CrossRef]
- Bees Wasps & Ants Recording Society, “Ectemnius lituratus”. 2014. Available online: https://www.bwars.com/wasp/crabronidae/crabroninae/ectemnius-lituratus (accessed on 11 February 2023).
- Bees Wasps & Ants Recording Society, “Pemphredon lugubris”. Available online: https://www.bwars.com/wasp/crabronidae/inae/pemphredon-lugubris (accessed on 11 February 2023).
- Nalam, V.; Louis, J.; Shah, J. Plant defense against aphids, the pest extraordinaire. Plant Sci. 2018, 279, 96–107. [Google Scholar] [CrossRef]
- Schmidt, M.H.; Lauer, A.; Purtauf, T.; Thies, C.; Schaefer, M.; Tscharntke, T. Relative importance of predators and parasitoids for cereal aphid control. Proc. R. Soc. B Biol. Sci. 2003, 270, 1905–1909. [Google Scholar] [CrossRef]
- Borges, M.; Colazza, S.; Ramirez-Lucas, P.; Chauhan, K.R.; Moraes, M.C.B.; Aldrich, J.R. Kairomonal effect of walking traces from Euschistus heros (Heteroptera: Pentatomidae) on two strains of Telenomus podisi (Hymenoptera: Scelionidae). Physiol. Èntomol. 2003, 28, 349–355. [Google Scholar] [CrossRef]
- Parra, J.R.P. Controle Biológico na Agricultura Brasileira. Entomol. Commun. 2019, 1, ec01002. [Google Scholar] [CrossRef]
- Weber, I.D.; Garcia, A.G.; Bueno, A.d.F.; de Oliveira, R.C.; Godoy, W.A.C. Release strategies of Telenomus podisi for control of Euschistus heros: A computational modeling approach. Pest Manag. Sci. 2022, 78, 4544–4556. [Google Scholar] [CrossRef] [PubMed]
- Lüthi, M.N.; Vorburger, C.; Dennis, A.B. A Novel RNA Virus in the Parasitoid Wasp Lysiphlebus fabarum: Genomic Structure, Prevalence, and Transmission. Viruses 2020, 12, 59. [Google Scholar] [CrossRef] [PubMed]
- Izraeli, Y.; Lepetit, D.; Atias, S.; Mozes-Daube, N.; Wodowski, G.; Lachman, O.; Luria, N.; Steinberg, S.; Varaldi, J.; Zchori-Fein, E.; et al. Genomic characterization of viruses associated with the parasitoid Anagyrus vladimiri (Hymenoptera: Encyrtidae). J. Gen. Virol. 2022, 103, 001810. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, F.; Yuan, B.; Yang, L.; Yang, Y.; Fang, Q.; Kuhn, J.H.; Song, Q.; Ye, G. A novel cripavirus of an ectoparasitoid wasp increases pupal duration and fecundity of the wasp’s Drosophila melanogaster host. ISME J. 2021, 15, 3239–3257. [Google Scholar] [CrossRef]
- Wang, F.; Yuan, B.; Xiao, S.; Zhang, J.; Jia, W.; Fang, Q.; Wang, F.; Song, Q.; Ye, G. Diverse RNA Viruses Discovered in Three Parasitoid Wasps of the Rice Weevil Sitophilus oryzae. mSphere 2021, 6, e00331-21. [Google Scholar] [CrossRef]
- Wang, F.; Fang, Q.; Wang, B.; Yan, Z.; Hong, J.; Bao, Y.; Kuhn, J.H.; Werren, J.H.; Song, Q.; Ye, G. A novel negative-stranded RNA virus mediates sex ratio in its parasitoid host. PLoS Pathog. 2017, 13, e1006201. [Google Scholar] [CrossRef]
- Gauthier, J.; Drezen, J.-M.; Herniou, E.A. The recurrent domestication of viruses: Major evolutionary transitions in parasitic wasps. Parasitology 2017, 145, 713–723. [Google Scholar] [CrossRef]
- Drezen, J.-M.; Bézier, A.; Burke, G.R.; Strand, M.R. Bracoviruses, ichnoviruses, and virus-like particles from parasitoid wasps retain many features of their virus ancestors. Curr. Opin. Insect Sci. 2021, 49, 93–100. [Google Scholar] [CrossRef]
- Bézier, A.; Annaheim, M.; Herbinière, J.; Wetterwald, C.; Gyapay, G.; Bernard-Samain, S.; Wincker, P.; Roditi, I.; Heller, M.; Belghazi, M.; et al. Polydnaviruses of Braconid Wasps Derive from an Ancestral Nudivirus. Science 2009, 323, 926–930. [Google Scholar] [CrossRef]
- Burke, G.R.; Hines, H.M.; Sharanowski, B.J. The Presence of Ancient Core Genes Reveals Endogenization from Diverse Viral Ancestors in Parasitoid Wasps. Genome Biol. Evol. 2021, 13, evab105. [Google Scholar] [CrossRef]
- Legeai, F.; Santos, B.F.; Robin, S.; Bretaudeau, A.; Dikow, R.B.; Lemaitre, C.; Jouan, V.; Ravallec, M.; Drezen, J.-M.; Tagu, D.; et al. Genomic architecture of endogenous ichnoviruses reveals distinct evolutionary pathways leading to virus domestication in parasitic wasps. BMC Biol. 2020, 18, 89. [Google Scholar] [CrossRef] [PubMed]
- Santos, B.F.; Klopfstein, S.; Whitfield, J.B.; Sharanowski, B.J. Many evolutionary roads led to virus domestication in ichneumonoid parasitoid wasps. Curr. Opin. Insect Sci. 2021, 50, 100861. [Google Scholar] [CrossRef] [PubMed]
- Coffman, K.A.; Harrell, T.C.; Burke, G.R. A Mutualistic Poxvirus Exhibits Convergent Evolution with Other Heritable Viruses in Parasitoid Wasps. J. Virol. 2020, 94, e02059-19. [Google Scholar] [CrossRef]
- Jalili, V.; Afgan, E.; Gu, Q.; Clements, D.; Blankenberg, D.; Goecks, J.; Taylor, J.; Nekrutenko, A. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2020 update. Nucleic Acids Res. 2020, 48, W395–W402. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A Quality Control Tool for High throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraha.ac.uk/projects/fastqc (accessed on 1 October 2022).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2014, 12, 59–60. [Google Scholar] [CrossRef]
- Shen, W.; Ren, H. TaxonKit: A practical and efficient NCBI taxonomy toolkit. J. Genet. Genom. 2021, 48, 844–850. [Google Scholar] [CrossRef]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef]
- Lu, S.; Wang, J.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; Gwadz, M.; Hurwitz, D.I.; Marchler, G.H.; Song, J.S.; et al. CDD/SPARCLE: The conserved domain database in 2020. Nucleic Acids Res. 2020, 48, D265–D268. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed]
- Nurk, S.; Meleshko, D.; Korobeynikov, A.; Pevzner, P.A. metaSPAdes: A new versatile metagenomic assembler. Genome Res. 2017, 27, 824–834. [Google Scholar] [CrossRef] [PubMed]
- Antipov, D.; Korobeynikov, A.; McLean, J.S.; Pevzner, P.A. hybridSPAdes: An algorithm for hybrid assembly of short and long reads. Bioinformatics 2015, 32, 1009–1015. [Google Scholar] [CrossRef] [PubMed]
- Schulz, M.H.; Zerbino, D.R.; Vingron, M.; Birney, E. Oases: Robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics 2012, 28, 1086–1092. [Google Scholar] [CrossRef]
- Li, D.; Liu, C.-M.; Luo, R.; Sadakane, K.; Lam, T.-W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef]
- Huang, X.; Madan, A. CAP3: A DNA Sequence Assembly Program. Genome Res. 1999, 9, 868–877. [Google Scholar] [CrossRef]
- Espinal, R.B.A.; de Santana, S.F.; Santos, V.C.; Lizardo, G.N.R.; Silva, R.J.S.; Corrêa, R.X.; Loguercio, L.L.; Góes-Neto, A.; Pirovani, C.P.; Fonseca, P.L.C.; et al. Uncovering a Complex Virome Associated with the Cacao Pathogens Ceratocystis cacaofunesta and Ceratocystis fimbriata. Pathogens 2023, 12, 287. [Google Scholar] [CrossRef]
- Larsson, A. AliView: A fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 2014, 30, 3276–3278. [Google Scholar] [CrossRef]
- Darriba, D.; Posada, D.; Kozlov, A.M.; Stamatakis, A.; Morel, B.; Flouri, T. ModelTest-NG: A New and Scalable Tool for the Selection of DNA and Protein Evolutionary Models. Mol. Biol. Evol. 2019, 37, 291–294. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In Proceedings of the Gateway Computing Environments Workshop (GCE), New Orleans, LA, USA, 14 November 2010; pp. 1–8. [Google Scholar] [CrossRef]
- Rambaut, A. FigTree v1.4.4: Tree Figure Drawing Tool. GitHub. 2019. Available online: https://github.com/rambaut/figtree/releases (accessed on 1 October 2022).
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed]
- Postler, T.S.; Rubino, L.; Adriaenssens, E.M.; Dutilh, B.E.; Harrach, B.; Junglen, S.; Kropinski, A.M.; Krupovic, M.; Wada, J.; Crane, A.; et al. Guidance for creating individual and batch latinized binomial virus species names. J. Gen. Virol. 2022, 103, 001800. [Google Scholar] [CrossRef]
- Lin, D.A.; Roychoudhury, S.; Palese, P.; Clay, W.C.; Fuller, F.J. Evolutionary relatedness of the predicted gene product of RNA segment 2 of the Tick-Borne Dhori virus and the PB1 polymerase gene of influenza viruses. Virology 1991, 182, 1–7. [Google Scholar] [CrossRef]
- Batts, W.N.; LaPatra, S.E.; Katona, R.; Leis, E.; Ng, T.F.F.; Brieuc, M.S.; Breyta, R.B.; Purcell, M.K.; Conway, C.M.; Waltzek, T.B.; et al. Molecular characterization of a novel orthomyxovirus from rainbow and steelhead trout (Oncorhynchus mykiss). Virus Res. 2017, 230, 38–49. [Google Scholar] [CrossRef] [PubMed]
- Allison, A.B.; Ballard, J.R.; Tesh, R.B.; Brown, J.D.; Ruder, M.G.; Keel, M.K.; Munk, B.A.; Mickley, R.M.; Gibbs, S.E.J.; da Rosa, A.P.A.T.; et al. Cyclic Avian Mass Mortality in the Northeastern United States Is Associated with a Novel Orthomyxovirus. J. Virol. 2015, 89, 1389–1403. [Google Scholar] [CrossRef]
- Shi, M.; Lin, X.-D.; Tian, J.-H.; Chen, L.-J.; Chen, X.; Li, C.-X.; Qin, X.-C.; Li, J.; Cao, J.-P.; Eden, J.-S.; et al. Redefining the invertebrate RNA virosphere. Nature 2016, 540, 539–543. [Google Scholar] [CrossRef]
- Farias, L.R.; Schimmelpfeng, P.H.C.; Togawa, R.C.; Costa, M.M.C.; Grynberg, P.; Martins, N.F.; Borges, M.; Blassioli-Moraes, M.C.; Laumann, R.A.; Báo, S.N.; et al. Transcriptome-Based Identification of Highly Similar Odorant-Binding Proteins among Neotropical Stink Bugs and Their Egg Parasitoid. PLoS ONE 2015, 10, e0132286. [Google Scholar] [CrossRef]
- Sparks, M.E.; Gundersen-Rindal, D.E.; Harrison, R.L. Complete Genome Sequence of a Novel Iflavirus from the Transcriptome of Halyomorpha halys, the Brown Marmorated Stink Bug. Genome Announc. 2013, 1, e00910-13. [Google Scholar] [CrossRef]
- Polaszek, A.; Vilhemsen, L. Biodiversity of hymenopteran parasitoids. Curr. Opin. Insect Sci. 2023, 56, 101026. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, H.M.; Vanlaerhoven, S.L.; García, M.M.; Hunt, D.W. Food web associations and effect of trophic resources and environmental factors on parasitoids expanding their host range into non-native hosts. Entomol. Exp. Appl. 2018, 166, 277–288. [Google Scholar] [CrossRef]
- Laws, A.N. Climate change effects on predator–prey interactions. Curr. Opin. Insect Sci. 2017, 23, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Manley, R.; Boots, M.; Wilfert, L. REVIEW: Emerging viral disease risk to pollinating insects: Ecological, evolutionary and anthropogenic factors. J. Appl. Ecol. 2015, 52, 331–340. [Google Scholar] [CrossRef]
- Highfield, A.; Kevill, J.; Mordecai, G.; Hunt, J.; Henderson, S.; Sauvard, D.; Feltwell, J.; Martin, S.J.; Sumner, S.; Schroeder, D.C. Detection and Replication of Moku Virus in Honey Bees and Social Wasps. Viruses 2020, 12, 607. [Google Scholar] [CrossRef]
- French, R.K.; Holmes, E.C. An Ecosystems Perspective on Virus Evolution and Emergence. Trends Microbiol. 2019, 28, 165–175. [Google Scholar] [CrossRef]
- Zhang, Y.-Z.; Chen, Y.-M.; Wang, W.; Qin, X.-C.; Holmes, E.C. Expanding the RNA Virosphere by Unbiased Metagenomics. Annu. Rev. Virol. 2019, 6, 119–139. [Google Scholar] [CrossRef]
- Pradeu, T. Mutualistic viruses and the heteronomy of life. Stud. Hist. Philos. Sci. Part C Stud. Hist. Philos. Biol. Biomed. Sci. 2016, 59, 80–88. [Google Scholar] [CrossRef]
- Valles, S.M.; Chen, Y.; Firth, A.E.; Guérin, D.M.A.; Hashimoto, Y.; Herrero, S.; De Miranda, J.R.; Ryabov, E.; ICTV Report Consortium. ICTV Virus Taxonomy Profile: Iflaviridae. J. Gen. Virol. 2017, 98, 527–528. [Google Scholar] [CrossRef]
- Mordecai, G.J.; Brettell, L.E.; Pachori, P.; Villalobos, E.M.; Martin, S.J.; Jones, I.M.; Schroeder, D.C. Moku virus; a new Iflavirus found in wasps, honey bees and Varroa. Sci. Rep. 2016, 6, 34983. [Google Scholar] [CrossRef]
- dos Santos, E.R.; Trentin, L.B.; Ecker, A.; Silva, L.A.; Borges, M.; Mowery, J.D.; Ribeiro, B.M.; Harrison, R.L.; Ardisson-Araújo, D.M. An iflavirus found in stink bugs (Hemiptera: Pentatomidae) of four different species. Virology 2019, 534, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Valverde, R.A.; Khalifa, M.; Okada, R.; Fukuhara, T.; Sabanadzovic, S.; ICTV Report Consortium. ICTV Virus Taxonomy Profile: Endornaviridae. J. Gen. Virol. 2019, 100, 1204–1205. [Google Scholar] [CrossRef] [PubMed]
- Pettersson, J.H.-O.; Shi, M.; Eden, J.-S.; Holmes, E.C.; Hesson, J.C. Meta-Transcriptomic Comparison of the RNA Viromes of the Mosquito Vectors Culex pipiens and Culex torrentium in Northern Europe. Viruses 2019, 11, 1033. [Google Scholar] [CrossRef]
- Nguyen, P.T.T.; Culverwell, C.L.; Suvanto, M.T.; Korhonen, E.M.; Uusitalo, R.; Vapalahti, O.; Smura, T.; Huhtamo, E. Characterisation of the RNA Virome of Nine Ochlerotatus Species in Finland. Viruses 2022, 14, 1489. [Google Scholar] [CrossRef]
- Jacquat, A.G.; Ulla, S.B.; Debat, H.J.; Muñoz-Adalia, E.J.; Theumer, M.G.; Pedrajas, M.D.G.; Dambolena, J.S. An in silico analysis revealed a novel evolutionary lineage of putative mitoviruses. Environ. Microbiol. 2022, 24, 6463–6475. [Google Scholar] [CrossRef] [PubMed]
- Bruenn, J.A.; Warner, B.E.; Yerramsetty, P. Widespread mitovirus sequences in plant genomes. PeerJ 2015, 3, e876. [Google Scholar] [CrossRef]
- Myers, J.M.; Bonds, A.E.; Clemons, R.A.; Thapa, N.A.; Simmons, D.R.; Carter-House, D.; Ortanez, J.; Liu, P.; Miralles-Durán, A.; Desirò, A.; et al. Survey of Early-Diverging Lineages of Fungi Reveals Abundant and Diverse Mycoviruses. mBio 2020, 11, e02027-20. [Google Scholar] [CrossRef]
- Nibert, M.L.; Debat, H.J.; Manny, A.R.; Grigoriev, I.V.; Licht, H.H.D.F. Mitovirus and Mitochondrial Coding Sequences from Basal Fungus Entomophthora muscae. Viruses 2019, 11, 351. [Google Scholar] [CrossRef]
- Manfrino, R.G.; Zumoffen, L.; Salto, C.E.; Lastra, C.C.L. Natural occurrence of entomophthoroid fungi of aphid pests on Medicago sativa L. in Argentina. Rev. Argent. De Microbiol. 2014, 46, 49–52. [Google Scholar] [CrossRef]
- Hillman, B.I.; Cai, G. The Family Narnaviridae. In Advances in Virus Research, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2013; Volume 86, pp. 149–176. [Google Scholar] [CrossRef]
- Li, N.; Huang, Y.; Li, W.; Xu, S. Virome Analysis Reveals Diverse and Divergent RNA Viruses in Wild Insect Pollinators in Beijing, China. Viruses 2022, 14, 227. [Google Scholar] [CrossRef]
- Vainio, E.J.; Chiba, S.; Ghabrial, S.A.; Maiss, E.; Roossinck, M.; Sabanadzovic, S.; Suzuki, N.; Xie, J.; Nibert, M. ICTV Report Consortium. ICTV Virus Taxonomy Profile: Partitiviridae. J. Gen. Virol. 2018, 99, 17–18. [Google Scholar] [CrossRef] [PubMed]
- Cross, S.T.; Maertens, B.L.; Dunham, T.J.; Rodgers, C.P.; Brehm, A.L.; Miller, M.R.; Williams, A.M.; Foy, B.D.; Stenglein, M.D. Partitiviruses Infecting Drosophila melanogaster and Aedes aegypti Exhibit Efficient Biparental Vertical Transmission. J. Virol. 2020, 94, e01070-20. [Google Scholar] [CrossRef] [PubMed]
- Ray, A.M.; Lopez, D.L.; Martinez, J.F.I.; Galbraith, D.A.; Rose, R.; VAN Engelsdorp, D.; Rosa, C.; Evans, J.D.; Grozinger, C.M. Distribution of recently identified bee-infecting viruses in managed honey bee (Apis mellifera) populations in the USA. Apidologie 2020, 51, 736–745. [Google Scholar] [CrossRef]
- Adams, M.J.; Adkins, S.; Bragard, C.; Gilmer, D.; Li, D.; MacFarlane, S.A.; Wong, S.-M.; Melcher, U.; Ratti, C.; Ryu, K.H.; et al. ICTV Virus Taxonomy Profile: Virgaviridae. J. Gen. Virol. 2017, 98, 1999–2000. [Google Scholar] [CrossRef]
- Kondo, H.; Chiba, S.; Maruyama, K.; Andika, I.B.; Suzuki, N. A novel insect-infecting virga/nege-like virus group and its pervasive endogenization into insect genomes. Virus Res. 2019, 262, 37–47. [Google Scholar] [CrossRef]
- Mailleux, A.-C.; Roques, A.; Molenberg, J.-M.; Grégoire, J.-C. A North American invasive seed pest, Megastigmus spermotrophus (Wachtl) (Hymenoptera: Torymidae): Its populations and parasitoids in a European introduction zone. Biol. Control 2008, 44, 137–141. [Google Scholar] [CrossRef]
- Walker, P.J.; Freitas-Astúa, J.; Bejerman, N.; Blasdell, K.R.; Breyta, R.; Dietzgen, R.G.; Fooks, A.R.; Kondo, H.; Kurath, G.; Kuzmin, I.V.; et al. ICTV Virus Taxonomy Profile: Rhabdoviridae 2022. J. Gen. Virol. 2022, 103, 001689. [Google Scholar] [CrossRef]
- Kleanthous, E.; Olendraite, I.; Lukhovitskaya, N.I.; Firth, A.E. Discovery of three RNA viruses using ant transcriptomic datasets. Arch. Virol. 2018, 164, 643–647. [Google Scholar] [CrossRef]
- Shrestha, G.; Skovgård, H.; Reddy, G.V.P.; Steenberg, T.; Enkegaard, A. Role of the aphid species and their feeding locations in parasitization behavior of Aphelinus abdominalis, a parasitoid of the lettuce aphid Nasonovia ribisnigri. PLoS ONE 2017, 12, e0184080. [Google Scholar] [CrossRef]
- Thekke-Veetil, T.; Lagos-Kutz, D.; McCoppin, N.K.; Hartman, G.L.; Ju, H.-K.; Lim, H.-S.; Domier, L.L. Soybean Thrips (Thysanoptera: Thripidae) Harbor Highly Diverse Populations of Arthropod, Fungal and Plant Viruses. Viruses 2020, 12, 1376. [Google Scholar] [CrossRef]
- Wu, H.; Pang, R.; Cheng, T.; Xue, L.; Zeng, H.; Lei, T.; Chen, M.; Wu, S.; Ding, Y.; Zhang, J.; et al. Abundant and Diverse RNA Viruses in Insects Revealed by RNA-Seq Analysis: Ecological and Evolutionary Implications. mSystems 2020, 5, e00039-20. [Google Scholar] [CrossRef] [PubMed]
- Kwon, M.; Jung, C.; Kil, E.-J. Metagenomic analysis of viromes in honey bee colonies (Apis mellifera; Hymenoptera: Apidae) after mass disappearance in Korea. Front. Cell. Infect. Microbiol. 2023, 13, 1124596. [Google Scholar] [CrossRef] [PubMed]
- Mahar, J.E.; Shi, M.; Hall, R.N.; Strive, T.; Holmes, E.C. Comparative Analysis of RNA Virome Composition in Rabbits and Associated Ectoparasites. J. Virol. 2020, 94, e02119-19. [Google Scholar] [CrossRef] [PubMed]
- An, X.; Zhang, W.; Ye, C.; Smagghe, G.; Wang, J.; Niu, J. Discovery of a widespread presence bunyavirus that may have symbiont-like relationships with different species of aphids. Insect Sci. 2021, 29, 1120–1134. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Wasp/Contigs | Virus | Segment | Query Size (nt) | Type | Query Coverage (%) | Identity (%) | E-Value | Closest Sequence in GenBank | Accession |
|---|---|---|---|---|---|---|---|---|---|
| A. compressa | Ampulexvirus narnaviri | single | 3732 | nt | 22 | 67 | 3.00 × 10−5 | Xiangshan narna-like virus | OK491482.1 |
| Ac_Contig1 | aa | 68 | 51 | 0.0 | Xiangshan narna-like virus | UDL13948.1 | |||
| C. vestalis | Cotesiavirus virgavi | single | 9075 | nt | 1 | 76 | 4.00 × 10−9 | Abisko virus | KY662294.1 |
| Cv_Contig1 | aa | 31 | 40 | 0.0 | Sanya virga-like virus 1 | UHM27517.1 | |||
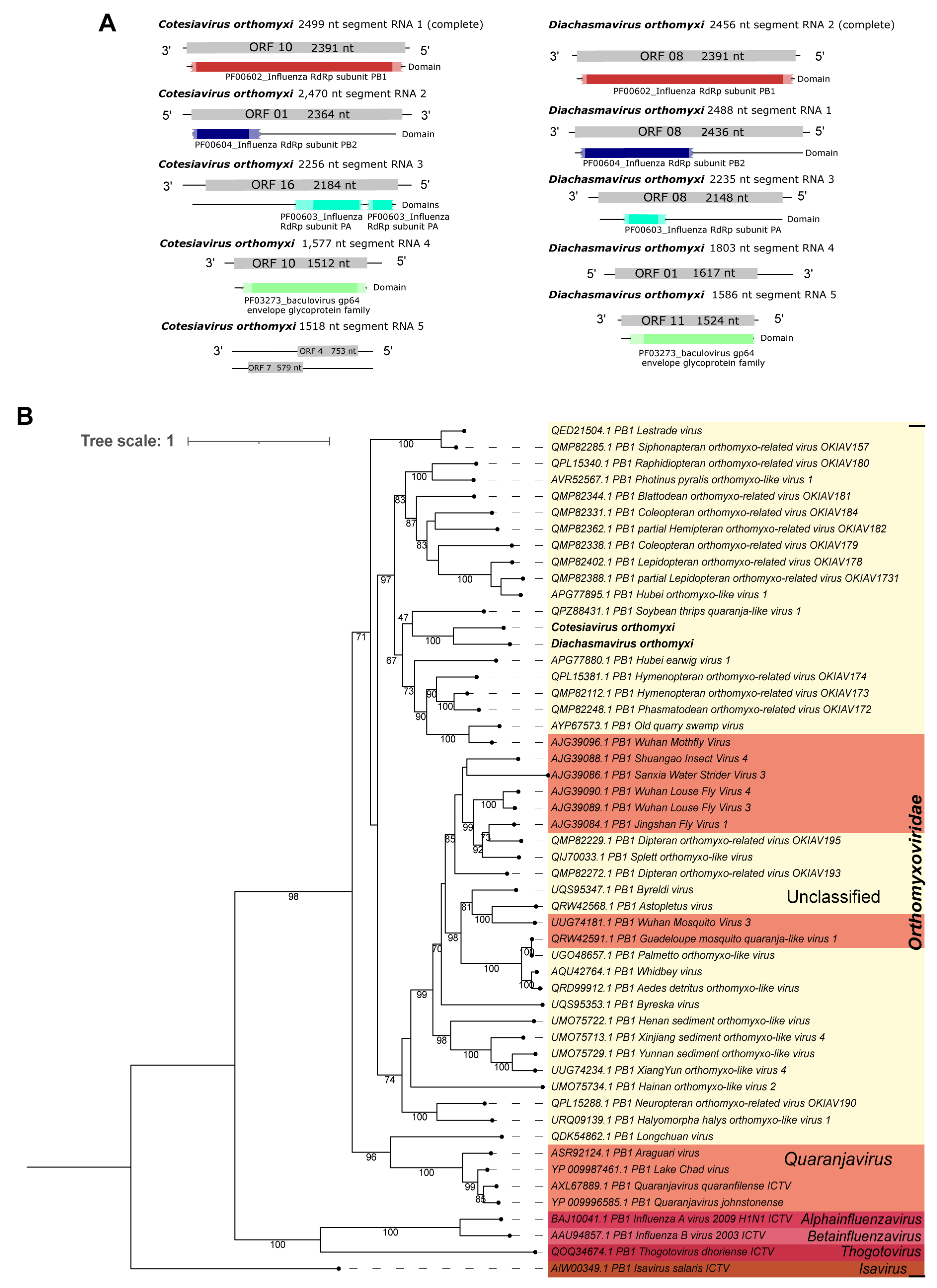
| Cv_RNA_ segment_1 | Cotesiavirus orthomyxi | 1 | 2499 | aa | 94 | 48 | 0.0 | Phasmatodean orthomyxo-related virus OKIAV172 | QMP82248.1 |
| Cv_RNA_ segment_2 | 2 | 2470 | aa | 92 | 35 | 3.00 × 10−132 | Phasmatodean orthomyxo-related virus OKIAV172 | QMP82297.1 | |
| Cv_RNA_ segment_3 | 3 | 2256 | aa | 93 | 36 | 5.00 × 10−148 | Hymenopteran orthomyxo-related virus OKIAV171 | QPL15315.1 | |
| Cv_RNA_ segment_4 | 4 | 1577 | aa | 83 | 27 | 9.00 × 10−45 | Hymenopteran orthomyxo-related virus OKIAV173 | QMP82117.1 | |
| Cv_RNA_ segment_5 | 5 | 1518 | aa | 66 | 34 | 1.00 × 10−21 | Blattodean orthomyxo-related virus OKIAV181 | QMP82185.1 | |
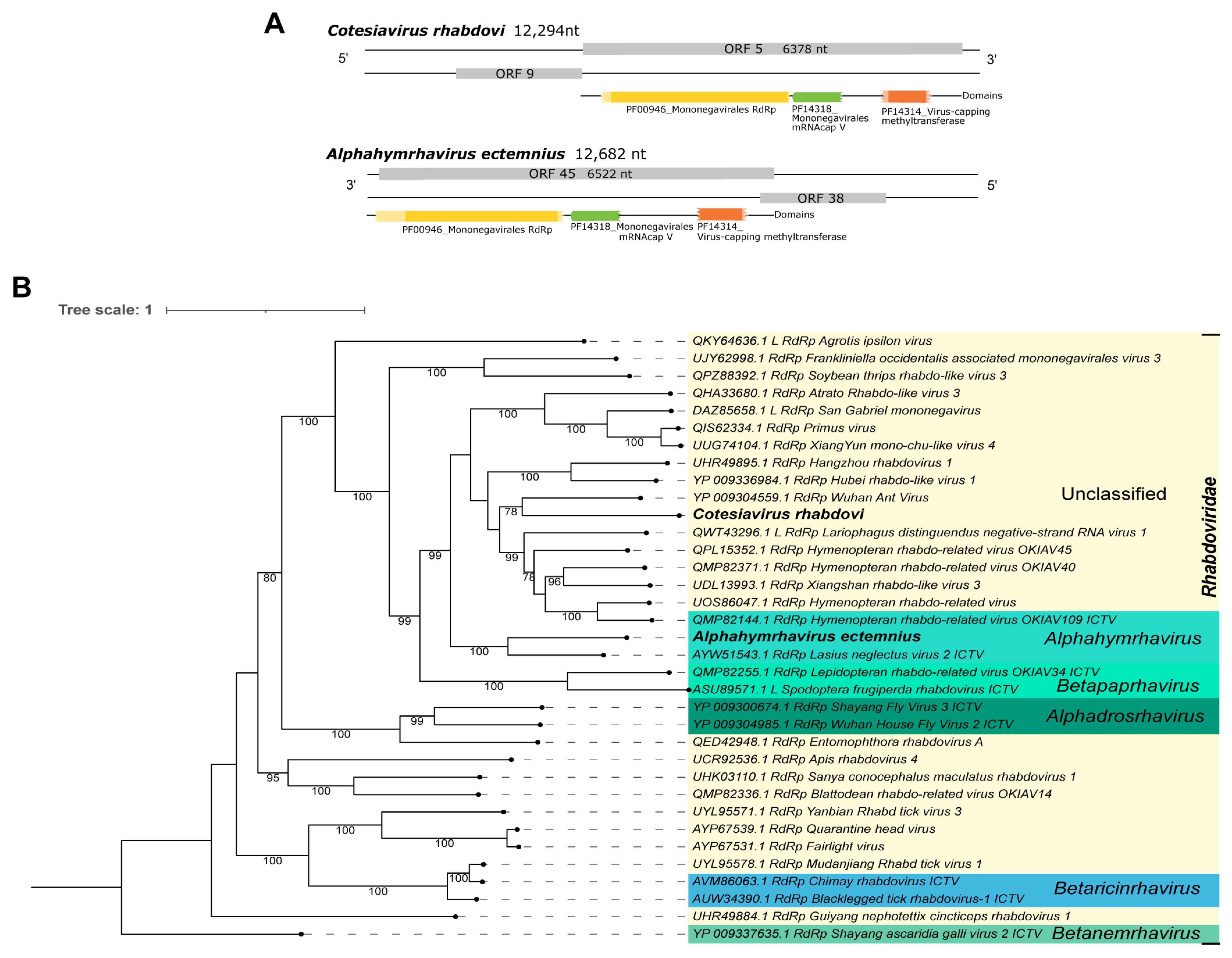
| Cv_Contig2 | Cotesiavirus rhabdovi | single | 12,294 | nt | 4 | 70 | 5.00 × 10−42 | San Gabriel mononegavirus | BK059423.1 |
| aa | 51 | 43 | 0.0 | Hymenopteran rhabdo-related virus | UOS86047.1 | ||||
| aa | 51 | 42 | 0.0 | Wuhan Ant Virus | YP_009304559.1 | ||||
| Cv_Contig3 | Cotesiavirus chinense | 1 | 6636 | nt | 10 | 69 | 3.00 × 10−16 | Wuhan insect virus 16 | KX884733.1 |
| aa | 92 | 41 | 0.0 | Wuhan insect virus 16 | APG79216.1 | ||||
| 2 | 1579 | aa | 67 | 29 | 1.00 × 10−39 | Hymenopteran phenui-related virus OKIAV282 | QMP82201.1 | ||
| 3 | 868 | aa | 72 | 33 | 4.00 × 10−26 | Hymenopteran phenui-related virus OKIAV275 | QPL15371.1 | ||
| D. alloeum | Diachasmavirus michiganense | single | 11,634 | nt | 28 | 67 | 0.0 | Gudgenby Calliphora mononega-like virus | MT129693.1 |
| Da_Contig1 | aa | 51 | 51 | 0.0 | Gudgenby Calliphora mononega-like virus | QIJ70030.1 | |||
| Da_RNA_ segment_1 | Diachasmavirus orthomyxi | 1 | 2488 | aa | 85 | 32 | 5.00 × 10−115 | Phasmatodean orthomyxo-related virus OKIAV172 | QMP82297.1 |
| Da_RNA_ segment_2 | 2 | 2456 | aa | 91 | 49 | 0.0 | Hymenopteran orthomyxo-related virus OKIAV173 | QMP82112.1 | |
| Da_RNA_ segment_3 | 3 | 2235 | aa | 95 | 37 | 1.00 × 10−150 | Hymenopteran orthomyxo-related virus OKIAV173 | QMP82372.1 | |
| Da_RNA_ segment_4 | 4 | 1803 | aa | 88 | 33 | 3.00 × 10−82 | Old quarry swamp virus | AYP67576.1 | |
| Da_RNA_ segment_5 | 5 | 1586 | aa | 85 | 29 | 2.00 × 10−40 | Hymenopteran orthomyxo-related virus OKIAV173 | QMP82117.1 | |
| Da_Contig2 | Pterovirus diachasmae | single | 4108 | aa | 96 | 31 | 0.0 | Hymenopteran chu-related virus OKIAV147 | QPB73971.1 |
| E. lituratus | Alphahymrhavirus ectemnius | single | 12,682 | nt | 12 | 69 | 5.00 × 10−111 | Hymenopteran rhabdo-related virus OKIAV38 | MT153454.1 |
| El_Contig1 | aa | 49 | 49 | 0.0 | Lasius neglectus virus 2 | AYW51543.1 | |||
| P. lugubris | |||||||||
| Pl_Contig1 | Pemphredonvirus anglici | single | 5813 | aa | 50 | 43 | 0.0 | Megastigmus ssRNA virus | QDZ71189.1 |
| Pl_Contig2 | Betapartitivirus pemphredoni | 1 | 2397 | nt | 97 | 79 | 0.0 | Dill cryptic virus 2segment 1 | JX971984.1 |
| Pl_Contig2.2 | 2 | 2271 | nt | 99 | 75 | 0.0 | Dill cryptic virus 2segment 2 | JX971985.1 | |
| Pl_Contig3 | Alphaendornavirus pemphredoni 1 | single | 3462 | nt | 5 | 73 | 2.00 × 10−21 | Phaseolus lunatus alphaendornavirus | MT792849.1 |
| aa | 96 | 36 | 0.0 | Lily alphaendornavirus | UPO25292.1 | ||||
| Pl_Contig4 | Alphaendornavirus pemphredoni 2 | single | 11,115 | aa | 75 | 30 | 0.0 | Geranium carolinianum endornavirus | QBB21108.1 |
| Pl_Contig5 | Pemphredonvirus endornavi | single | 14,232 | aa | 80 | 33 | 0.0 | Hallsjon virus | UYL94274.1 |
| Pl_Contig6 | Mitovirus pemphredoni | single | 2141 | aa | 85 | 34 | 9.00 × 10−107 | Hangzhou altica cyanea mitovirus 1 | UHK03009.1 |
| Pl_Contig7 | Unuamitovirus pemphredoni | single | 1056 | aa | 86 | 38 | 2.00 × 10−82 | Entomophthora muscae mitovirus 2 | QCF24453.1 |
| Pl_Contig10 | Hubei narna-like virus 25 isolate P. lugubris | single | 1143 | nt | 100 | 92 | 0.0 | Hubei narna-like virus 25 strain SCM51430 | KX883546.1 |
| T. podisi Tp_Contig1 | Halyomorpha halys virus isolate T. podisi | single | 8285 | nt | 100 | 98 | 0.0 | Halyomorpha halys virus isolate Beltsville | KF699344.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caldas-Garcia, G.B.; Santos, V.C.; Fonseca, P.L.C.; de Almeida, J.P.P.; Costa, M.A.; Aguiar, E.R.G.R. The Viromes of Six Ecosystem Service Provider Parasitoid Wasps. Viruses 2023, 15, 2448. https://doi.org/10.3390/v15122448
Caldas-Garcia GB, Santos VC, Fonseca PLC, de Almeida JPP, Costa MA, Aguiar ERGR. The Viromes of Six Ecosystem Service Provider Parasitoid Wasps. Viruses. 2023; 15(12):2448. https://doi.org/10.3390/v15122448
Chicago/Turabian StyleCaldas-Garcia, Gabriela B., Vinícius Castro Santos, Paula Luize Camargos Fonseca, João Paulo Pereira de Almeida, Marco Antônio Costa, and Eric Roberto Guimarães Rocha Aguiar. 2023. "The Viromes of Six Ecosystem Service Provider Parasitoid Wasps" Viruses 15, no. 12: 2448. https://doi.org/10.3390/v15122448
APA StyleCaldas-Garcia, G. B., Santos, V. C., Fonseca, P. L. C., de Almeida, J. P. P., Costa, M. A., & Aguiar, E. R. G. R. (2023). The Viromes of Six Ecosystem Service Provider Parasitoid Wasps. Viruses, 15(12), 2448. https://doi.org/10.3390/v15122448