
Changes in the Murine Microbiome and Bacterial Extracellular Vesicle Production in Response to Antibiotic Treatment and Norovirus Infection
 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Murine Norovirus Production
2.2. Mice Infections and Antibiotic Treatment
2.3. Stool DNA Extraction and Sequencing
2.4. Sequence Processing into Amplicon Sequencing Variants
2.5. Stool Bacterial Extracellular Vesicle Extraction and Removal of Exosomes
2.6. Nanoparticle Tracking Analysis
2.7. Western Blot
2.8. Additional Data Analysis and Statistics
3. Results
3.1. Study Design
3.2. Regionalization of MNV Infection and Increased Bacterial Extracellular Vesicle Production
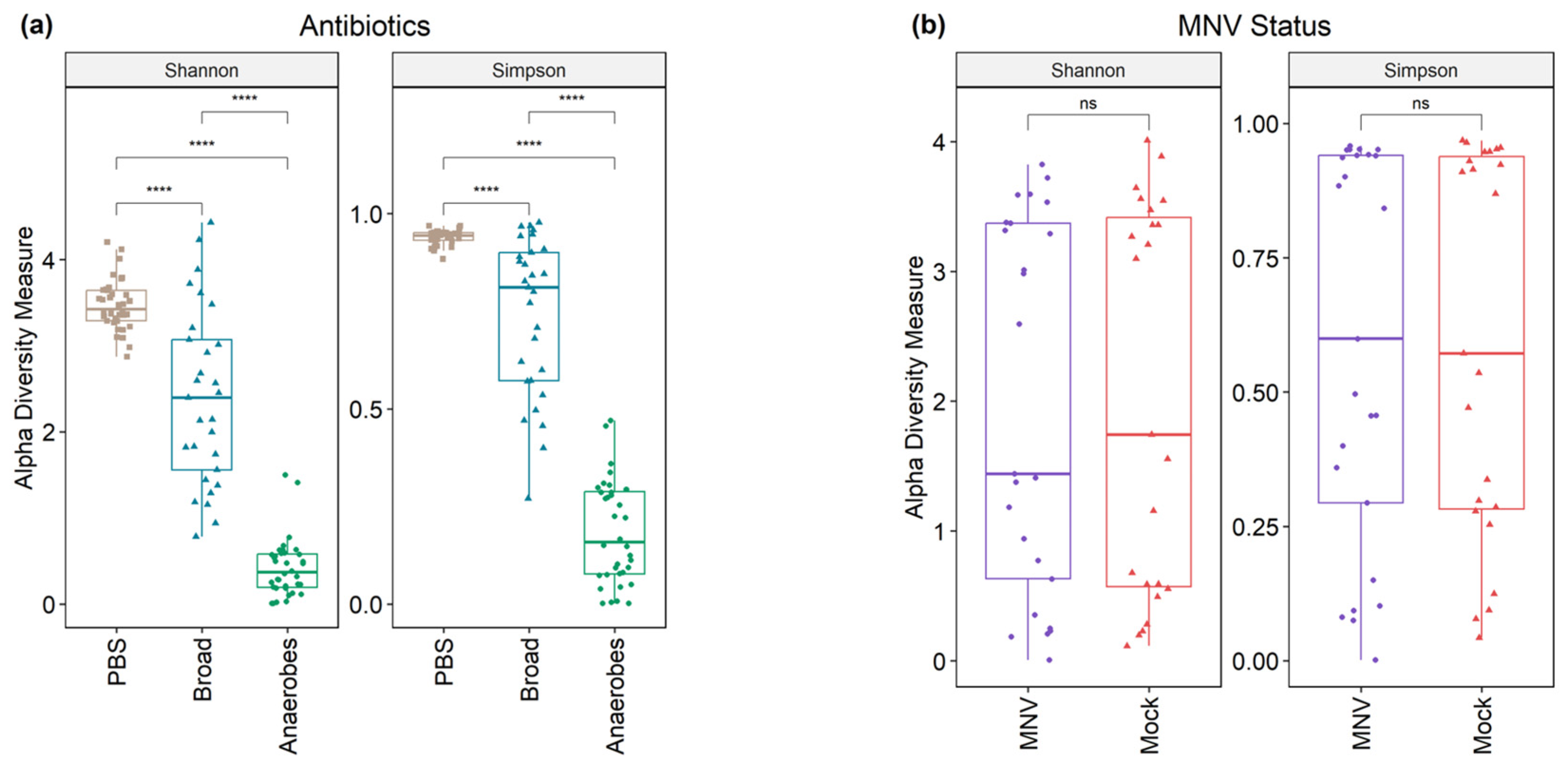
3.3. Differences in Diversity Indices Are Explained by Antibiotic Condition
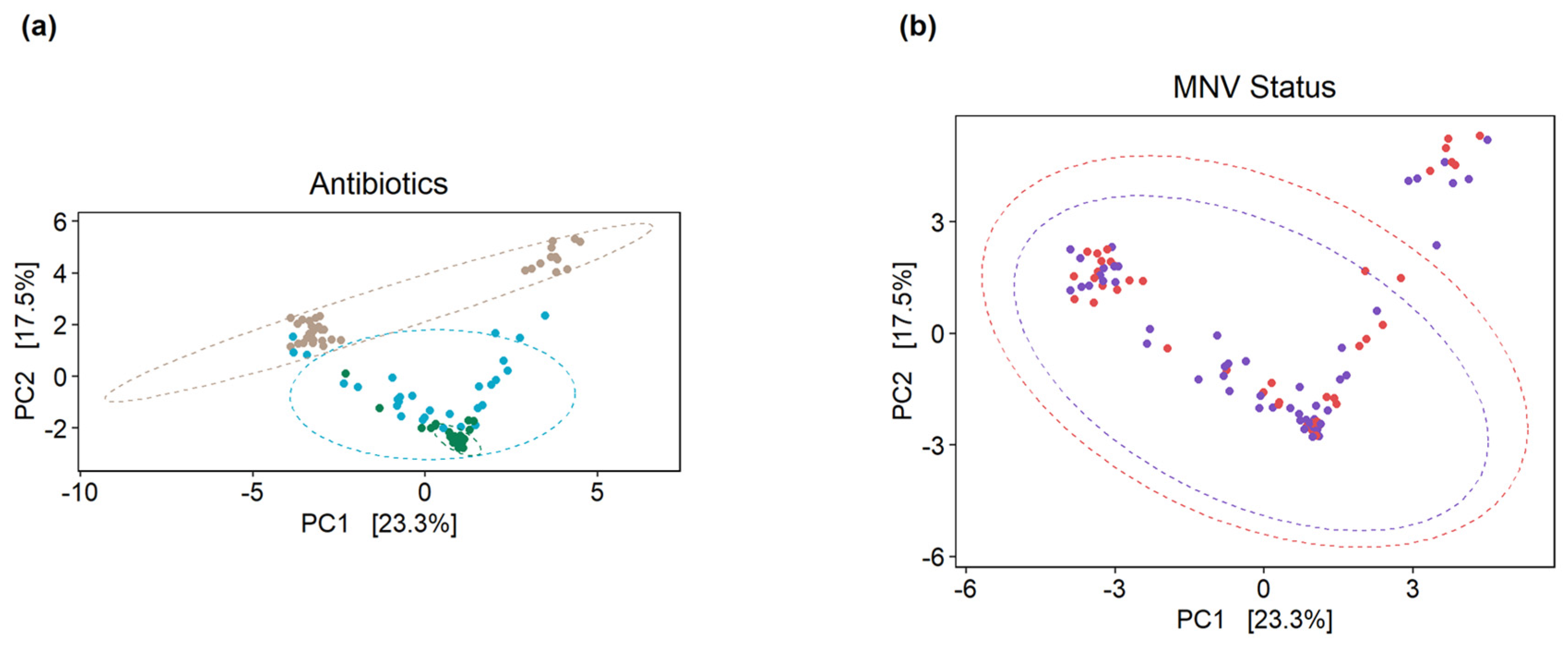
3.4. Use of Antibiotic Cocktails during Norovirus Infection Shows Slight Changes in Overall Gut Microbiota Composition
3.5. Slight Changes in Microbiome Composition When Comparing Pre vs. Post MNV Samples
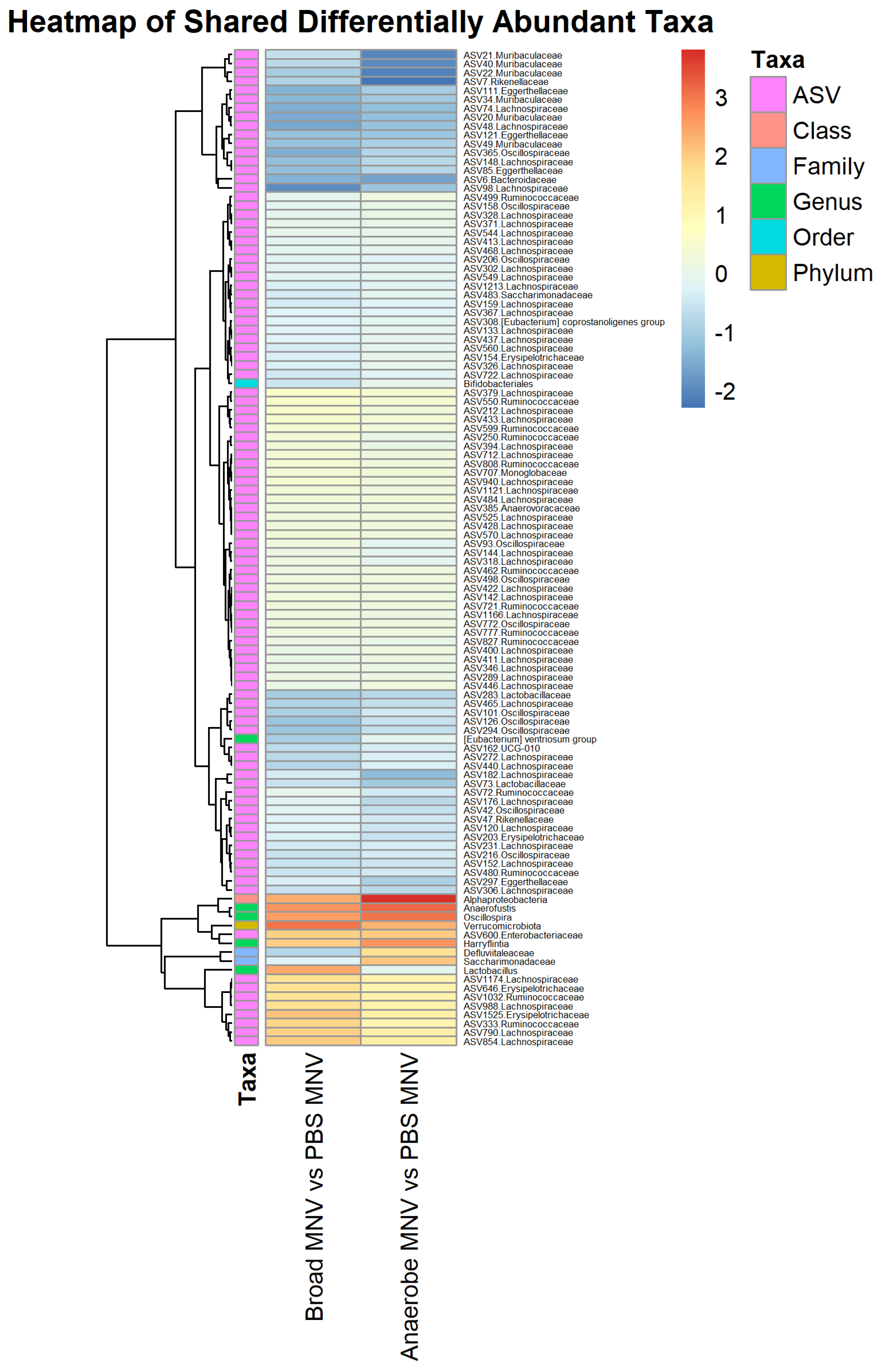
3.6. Comparisons of Post-MNV-Infection Samples Show Antibiotic-Driven Differences in the Microbiome
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Almand, E.A.; Moore, M.D.; Outlaw, J.; Jaykus, L.A. Human norovirus binding to select bacteria representative of the human gut microbiota. PLoS ONE 2017, 12, e0173124. [Google Scholar] [CrossRef]
- Riddle, M.S.; Walker, R.I. Status of vaccine research and development for norovirus. Vaccine 2016, 34, 2895–2899. [Google Scholar] [CrossRef] [PubMed]
- de Graaf, M.; van Beek, J.; Koopmans, M.P. Human norovirus transmission and evolution in a changing world. Nat. Rev. Microbiol. 2016, 14, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Madrigal, J.L.; Bhar, S.; Hackett, S.; Engelken, H.; Joseph, R.; Keyhani, N.O.; Jones, M.K. Attach Me If You Can: Murine Norovirus Binds to Commensal Bacteria and Fungi. Viruses 2020, 12, 759. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.K.; Watanabe, M.; Zhu, S.; Graves, C.L.; Keyes, L.R.; Grau, K.R.; Gonzalez-Hernandez, M.B.; Iovine, N.M.; Wobus, C.E.; Vinje, J.; et al. Enteric bacteria promote human and mouse norovirus infection of B cells. Science 2014, 346, 755–759. [Google Scholar] [CrossRef] [PubMed]
- Hickman, D.; Jones, M.K.; Zhu, S.; Kirkpatrick, E.; Ostrov, D.A.; Wang, X.; Ukhanova, M.; Sun, Y.; Mai, V.; Salemi, M.; et al. The effect of malnutrition on norovirus infection. MBio 2014, 5, e1003592. [Google Scholar] [CrossRef] [PubMed]
- Nelson, A.M.; Elftman, M.D.; Pinto, A.K.; Baldridge, M.; Hooper, P.; Kuczynski, J.; Petrosino, J.F.; Young, V.B.; Wobus, C.E. Murine norovirus infection does not cause major disruptions in the murine intestinal microbiota. Microbiome 2013, 1, 7. [Google Scholar] [CrossRef] [PubMed]
- Grau, K.R.; Zhu, S.; Peterson, S.T.; Helm, E.W.; Philip, D.; Phillips, M.; Hernandez, A.; Turula, H.; Frasse, P.; Graziano, V.R.; et al. The intestinal regionalization of acute norovirus infection is regulated by the microbiota via bile acid-mediated priming of type III interferon. Nat. Microbiol. 2020, 5, 84–92. [Google Scholar] [CrossRef]
- Baldridge, M.T.; Nice, T.J.; McCune, B.T.; Yokoyama, C.C.; Kambal, A.; Wheadon, M.; Diamond, M.S.; Ivanova, Y.; Artyomov, M.; Virgin, H.W. Commensal microbes and interferon-lambda determine persistence of enteric murine norovirus infection. Science 2015, 347, 266–269. [Google Scholar] [CrossRef]
- Kuss, S.K.; Best, G.T.; Etheredge, C.A.; Pruijssers, A.J.; Frierson, J.M.; Hooper, L.V.; Dermody, T.S.; Pfeiffer, J.K. Intestinal microbiota promote enteric virus replication and systemic pathogenesis. Science 2011, 334, 249–252. [Google Scholar] [CrossRef]
- Robinson, C.M.; Jesudhasan, P.R.; Pfeiffer, J.K. Bacterial lipopolysaccharide binding enhances virion stability and promotes environmental fitness of an enteric virus. Cell Host Microbe 2014, 15, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Bhar, S.; Zhao, G.; Bartel, J.D.; Sterchele, H.; Del Mazo, A.; Emerson, L.E.; Edelmann, M.J.; Jones, M.K. Bacterial extracellular vesicles control murine norovirus infection through modulation of antiviral immune responses. Front. Immunol. 2022, 13, 909949. [Google Scholar] [CrossRef] [PubMed]
- Mosby, C.A.; Bhar, S.; Phillips, M.B.; Edelmann, M.J.; Jones, M.K. Interaction with mammalian enteric viruses alters outer membrane vesicle production and content by commensal bacteria. J. Extracell. Vesicles 2022, 11, e12172. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Regev, D.; Watanabe, M.; Hickman, D.; Moussatche, N.; Jesus, D.M.; Kahan, S.M.; Napthine, S.; Brierley, I.; Hunter, R.N., III; et al. Identification of immune and viral correlates of norovirus protective immunity through comparative study of intra-cluster norovirus strains. PLoS Pathog. 2013, 9, e1003592. [Google Scholar] [CrossRef] [PubMed]
- Russell, J.T.; Roesch, L.F.W.; Ördberg, M.; Ilonen, J.; Atkinson, M.A.; Schatz, D.A.; Triplett, E.W.; Ludvigsson, J. Genetic risk for autoimmunity is associated with distinct changes in the human gut microbiome. Nat. Commun. 2019, 10, 3621. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FASTQC: A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 21 October 2023).
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 3. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Yilmaz, P.; Parfrey, L.W.; Yarza, P.; Gerken, J.; Pruesse, E.; Quast, C.; Schweer, T.; Peplies, J.; Ludwig, W.; Glöckner, F.O. The SILVA and “All-species Living Tree Project (LTP)” taxonomic frameworks. Nucleic Acids Res. 2014, 42, D643–D648. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef]
- Zhou, H.; He, K.; Chen, J.; Zhang, X. LinDA: Linear models for differential abundance analysis of microbiome compositional data. Genome Biol. 2022, 23, 95. [Google Scholar] [CrossRef] [PubMed]
- Team, R. RStudio: Integrated Development Environment for R; RStudio, PBC.: Boston, MA, USA, 2020. [Google Scholar]
- Hadadi-Fishani, M.; Najar-Peerayeh, S.; Siadat, S.D.; Sekhavati, M.; Mobarez, A.M. Isolation and immunogenicity of extracted outer membrane vesicles from Pseudomonas aeruginosa under antibiotics treatment conditions. Iran. J. Microbiol. 2021, 13, 824–831. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro de Freitas, M.C.; Elaine de Almeida, P.; Vieira, W.V.; Ferreira-Machado, A.B.; Resende, J.A.; Lúcia da Silva, V.; Diniz, C.G. Inflammatory modulation and outer membrane vesicles (OMV) production associated to Bacteroides fragilis response to subinhibitory concentrations of metronidazole during experimental infection. Anaerobe 2022, 73, 102504. [Google Scholar] [CrossRef] [PubMed]
- Bauwens, A.; Kunsmann, L.; Karch, H.; Mellmann, A.; Bielaszewska, M. Antibiotic-Mediated Modulations of Outer Membrane Vesicles in Enterohemorrhagic Escherichia coli O104:H4 and O157:H7. Antimicrob. Agents Chemother. 2017, 61, e00937-17. [Google Scholar] [CrossRef]
- Siqueira, V.L.; Cardoso, R.F.; Caleffi-Ferracioli, K.R.; Scodro, R.B.; Fernandez, M.A.; Fiorini, A.; Ueda-Nakamura, T.; Dias-Filho, B.P.; Nakamura, C.V. Structural changes and differentially expressed genes in Pseudomonas aeruginosa exposed to meropenem-ciprofloxacin combination. Antimicrob. Agents Chemother. 2014, 58, 3957–3967. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kadurugamuwa, J.L.; Beveridge, T.J. Natural release of virulence factors in membrane vesicles by Pseudomonas aeruginosa and the effect of aminoglycoside antibiotics on their release. J. Antimicrob. Chemother. 1997, 40, 615–621. [Google Scholar] [CrossRef] [PubMed]
- Odamaki, T.; Kato, K.; Sugahara, H.; Hashikura, N.; Takahashi, S.; Xiao, J.-z.; Abe, F.; Osawa, R. Age-related changes in gut microbiota composition from newborn to centenarian: A cross-sectional study. BMC Microbiol. 2016, 16, 90. [Google Scholar] [CrossRef]
- Cong, X.; Judge, M.; Xu, W.; Diallo, A.; Janton, S.; Brownell, E.A.; Maas, K.; Graf, J. Influence of Feeding Type on Gut Microbiome Development in Hospitalized Preterm Infants. Nurs. Res. 2017, 66, 123–133. [Google Scholar] [CrossRef]
- Devillard, E.; McIntosh Freda, M.; Duncan Sylvia, H.; Wallace, R.J. Metabolism of Linoleic Acid by Human Gut Bacteria: Different Routes for Biosynthesis of Conjugated Linoleic Acid. J. Bacteriol. 2007, 189, 2566–2570. [Google Scholar] [CrossRef]
- Biddle, A.; Stewart, L.; Blanchard, J.; Leschine, S. Untangling the Genetic Basis of Fibrolytic Specialization by Lachnospiraceae and Ruminococcaceae in Diverse Gut Communities. Diversity 2013, 5, 627–640. [Google Scholar] [CrossRef]
- Steinmeyer, S.; Lee, K.; Jayaraman, A.; Alaniz, R.C. Microbiota Metabolite Regulation of Host Immune Homeostasis: A Mechanistic Missing Link. Curr. Allergy Asthma Rep. 2015, 15, 24. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibiotic Cocktail | Targeted Bacterial Population (s) | Antibiotic | Administration Amount | Drinking Water Amount |
|---|---|---|---|---|
| Broad Abx | Gram-negative, Gram-positive, aerobes, anaerobes | Ampicillin | 10 mg | 1 g/L |
| Neomycin | 10 mg | 1 g/L | ||
| Metronidazole | 10 mg | 1 g/L | ||
| Vancomycin | 10 mg | 500 mg/L | ||
| Anaerobic Abx | Anaerobes | Metronidazole | 10 mg | 1 g/L |
| Clindamycin | 2 mg | 1 g/L |
| Taxa ID | Broad vs. PBS | Anaerobe vs. PBS |
|---|---|---|
| Increased Abundance | ||
| Verrucomicrobiota | 4.195023523 | 5.278166274 |
| Anaerofustis | 3.402296713 | 8.084034485 |
| Oscillospira | 3.121227438 | 7.798614837 |
| Lactobacillus | 2.837273213 | −2.939306235 |
| Alphaproteobacteria | 2.796732207 | 10.3676823 |
| Harryflintia | 1.818983454 | 6.593577373 |
| Decreased Abundance | ||
| ASV21.Muribaculaceae | −4.065631202 | −9.578255593 |
| ASV40.Muribaculaceae | −4.353977958 | −9.269787528 |
| ASV7.Rikenellaceae | −4.565836714 | −10.45958671 |
| ASV22.Muribaculaceae | −4.94867859 | −9.72542094 |
| ASV6.Bacteroidaceae | −5.794439552 | −8.174547192 |
| ASV74.Lachnospiraceae | −6.858943809 | |
| ASV365.Oscillospiraceae | −6.021839134 | −5.256030931 |
| ASV20.Muribaculaceae | −6.202646373 | −6.744189982 |
| ASV48.Lachnospiraceae | −6.271731816 | −6.547431892 |
| ASV98.Lachnospiraceae | −7.061084208 | −6.343099048 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mosby, C.A.; Long, K.J.; Phillips, M.B.; Bartel, J.; Jones, M.K. Changes in the Murine Microbiome and Bacterial Extracellular Vesicle Production in Response to Antibiotic Treatment and Norovirus Infection. Viruses 2023, 15, 2443. https://doi.org/10.3390/v15122443
Mosby CA, Long KJ, Phillips MB, Bartel J, Jones MK. Changes in the Murine Microbiome and Bacterial Extracellular Vesicle Production in Response to Antibiotic Treatment and Norovirus Infection. Viruses. 2023; 15(12):2443. https://doi.org/10.3390/v15122443
Chicago/Turabian StyleMosby, Chanel A., Kendall J. Long, Matthew B. Phillips, Julia Bartel, and Melissa K. Jones. 2023. "Changes in the Murine Microbiome and Bacterial Extracellular Vesicle Production in Response to Antibiotic Treatment and Norovirus Infection" Viruses 15, no. 12: 2443. https://doi.org/10.3390/v15122443
APA StyleMosby, C. A., Long, K. J., Phillips, M. B., Bartel, J., & Jones, M. K. (2023). Changes in the Murine Microbiome and Bacterial Extracellular Vesicle Production in Response to Antibiotic Treatment and Norovirus Infection. Viruses, 15(12), 2443. https://doi.org/10.3390/v15122443