
Picornavirus 3C Proteins Intervene in Host Cell Processes through Proteolysis and Interactions with RNA



Abstract
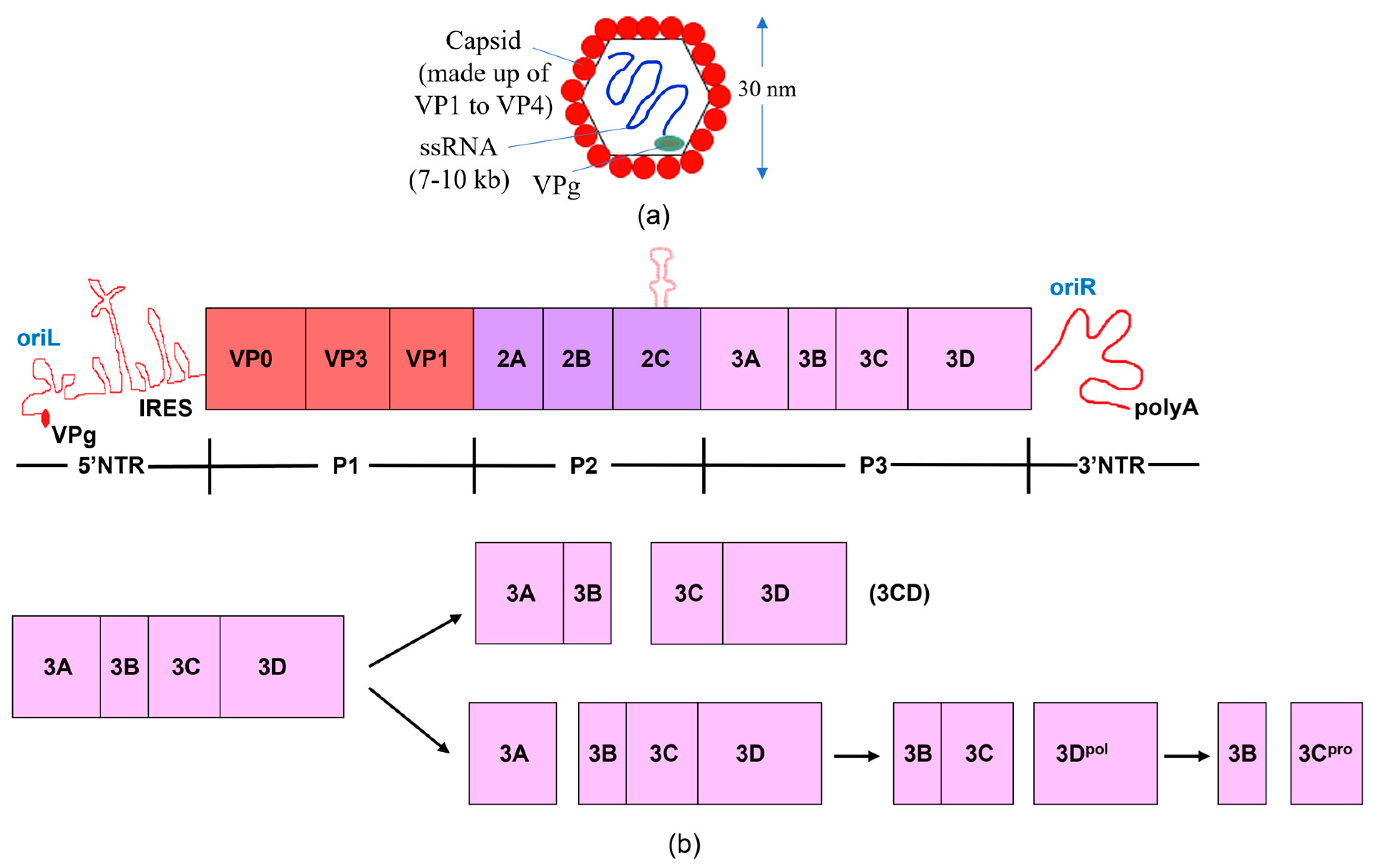
1. Introduction

2. Structure and Functions of 3C
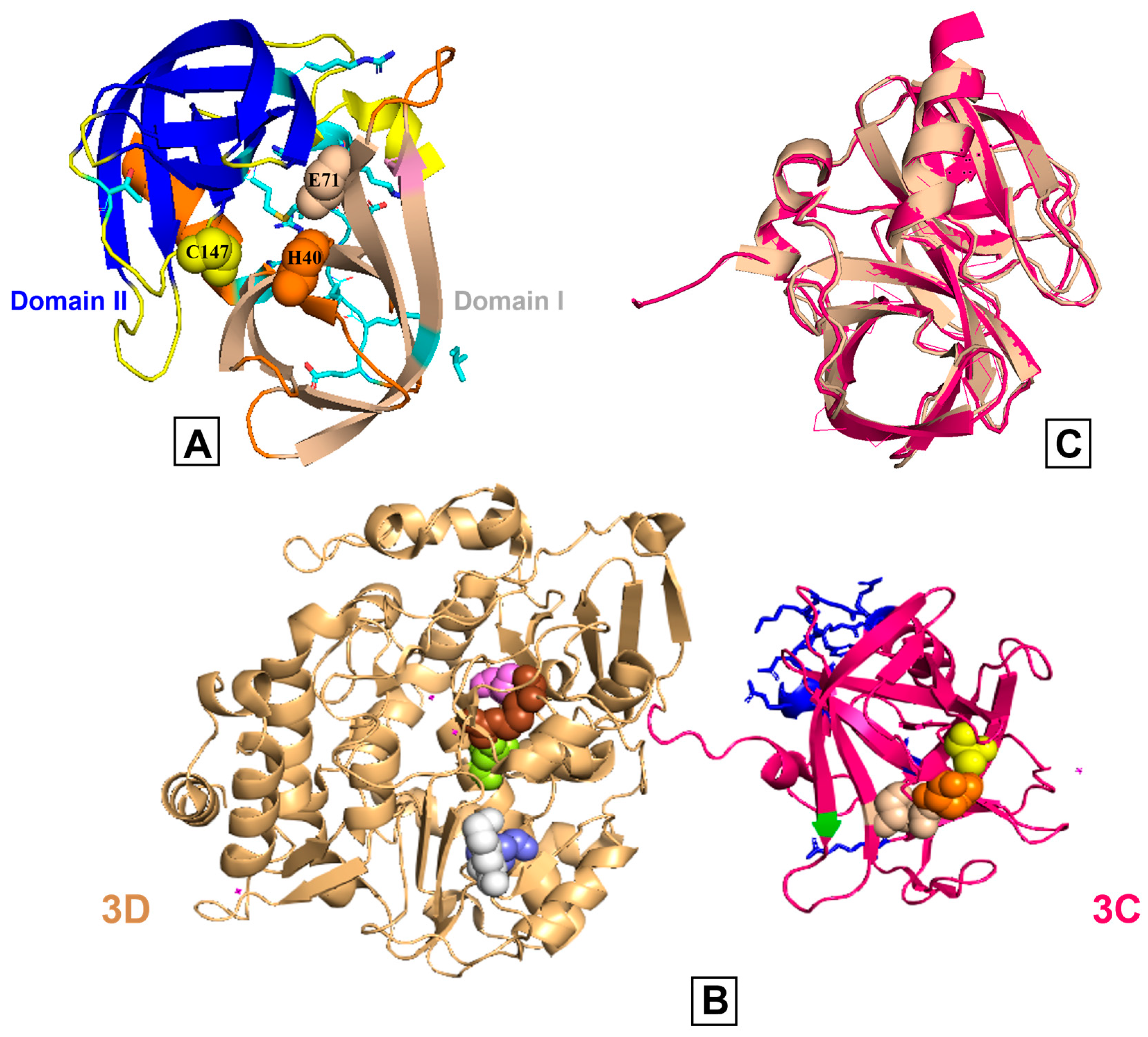
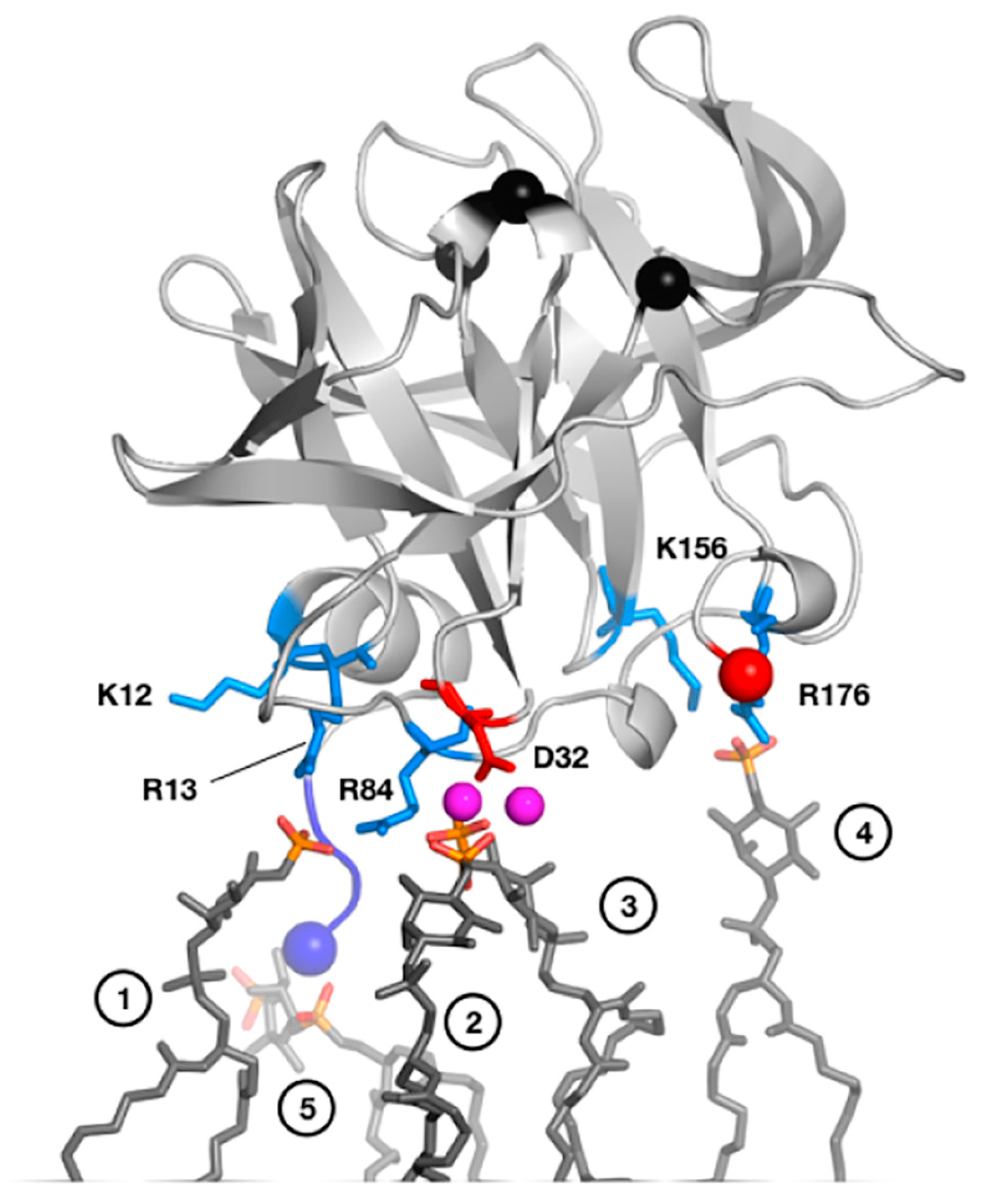
2.1. Catalytic and RNA-Binding Sites
2.2. Viral Polyprotein Processing and Substrate Recognition
2.3. 3C(D) Also Interacts with Virus Replication Membranes
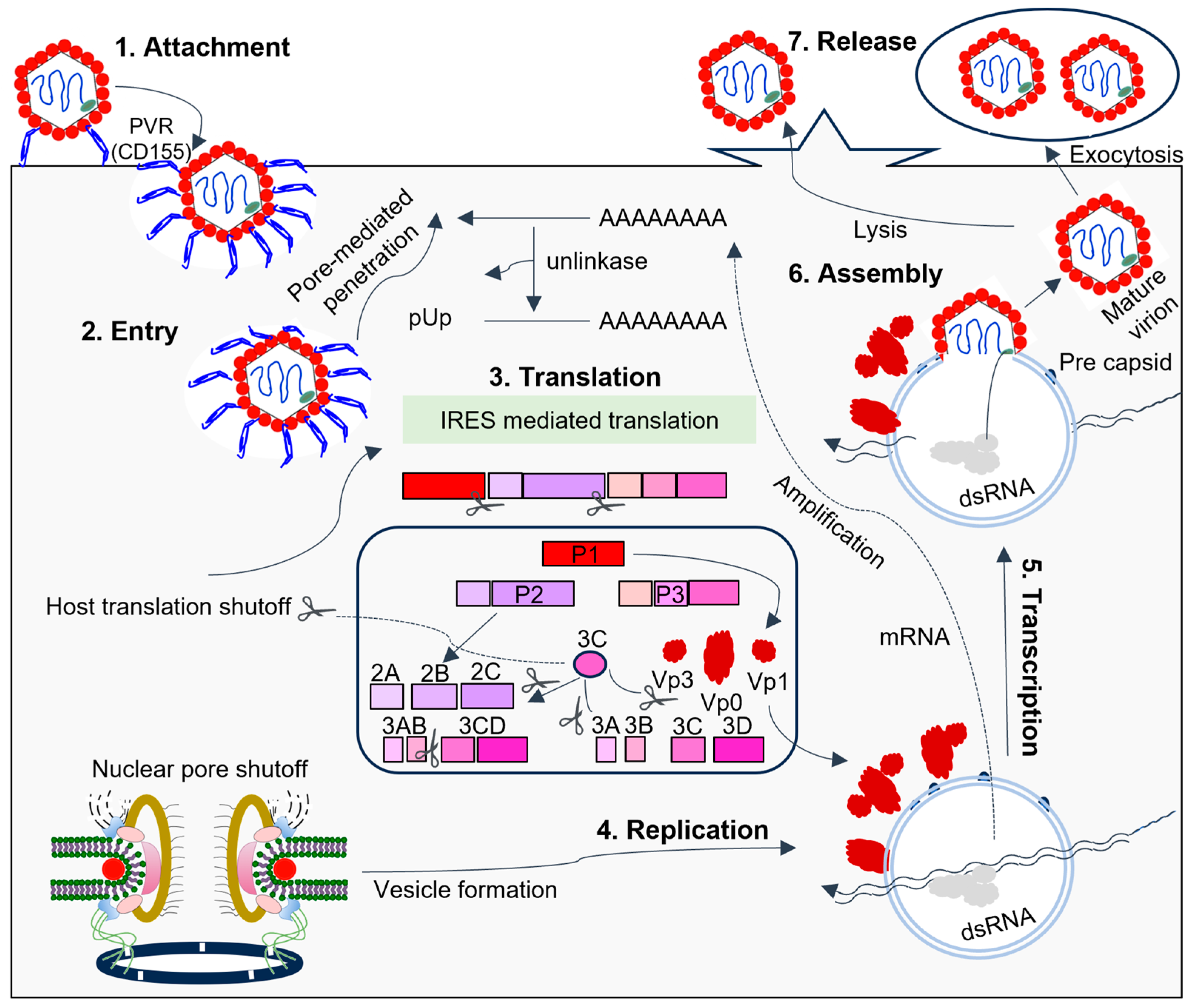
3. The 3C Protease Function Intervenes in Host Cell Processes
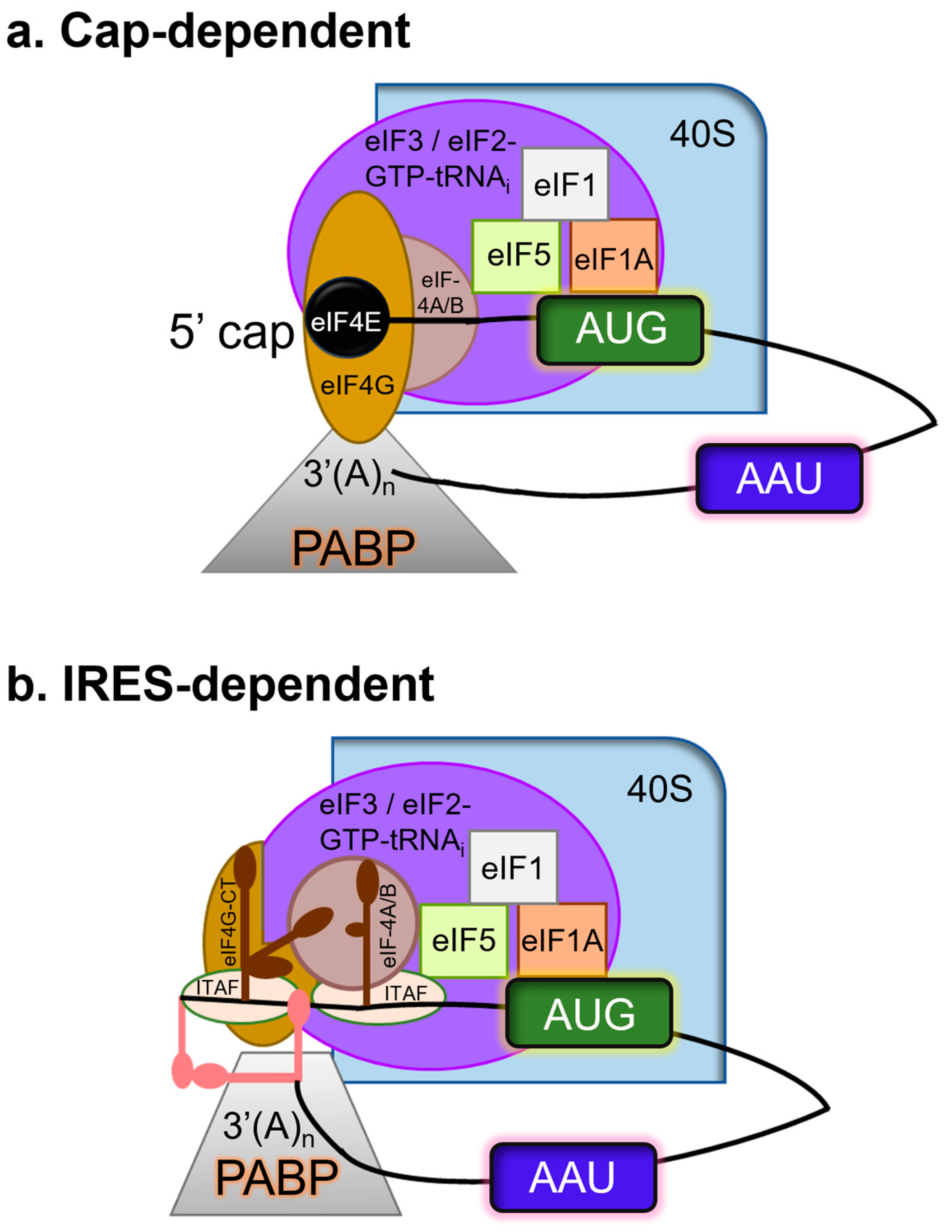
3.1. Proteolysis by 3C Proteases Suppresses the Host Transcription and Translation Mechanisms
3.2. 3C-Promoted Apoptosis for Virus Release
3.3. 3C Interferes with Host Cell Defenses
4. RNA Binding and the Viral RNA Template Transformation
5. Roles of 3C(D) and Other 3C-Containing Polyproteins in Picornavirus RNA Replication
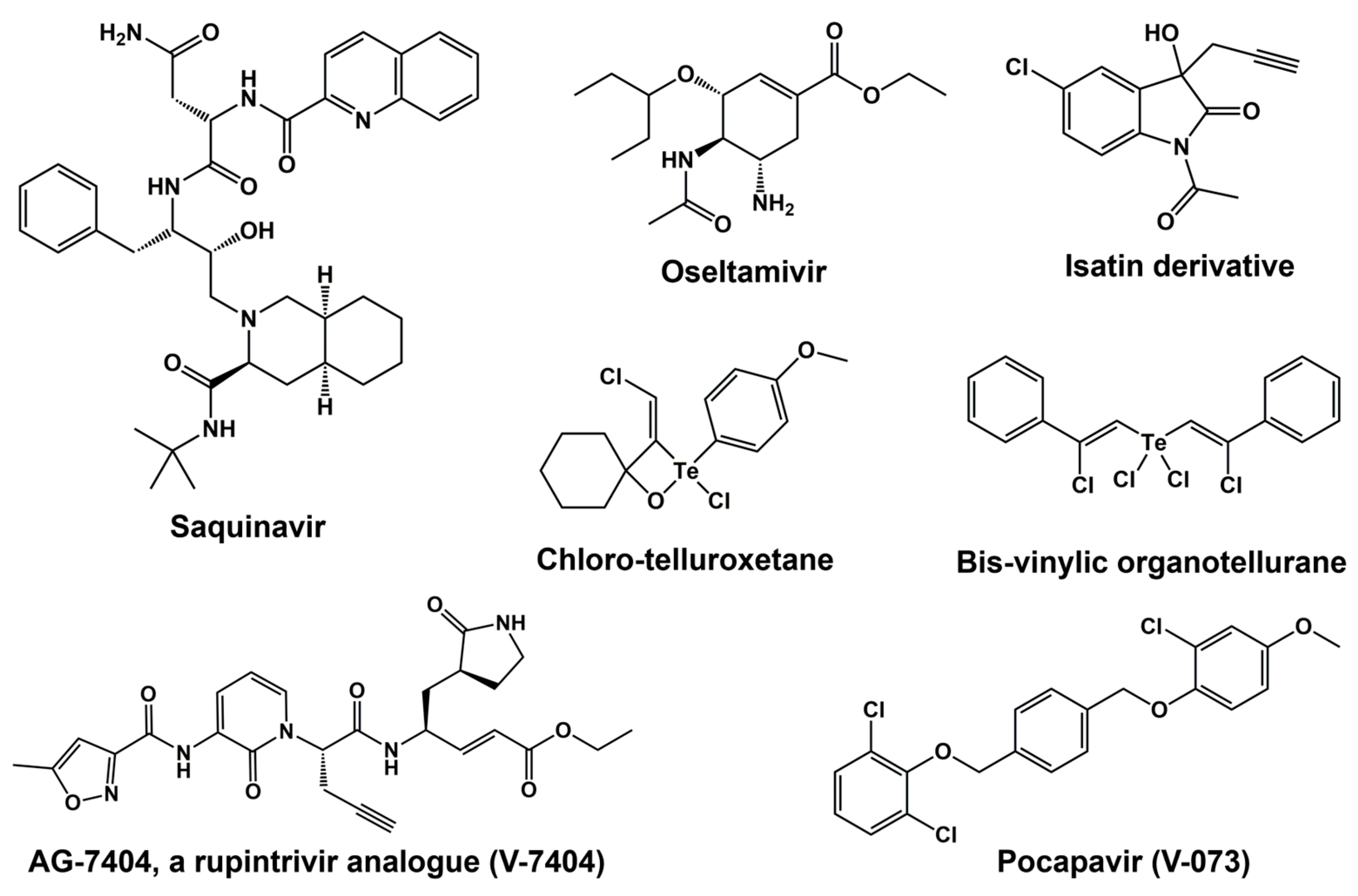
6. 3C Protease Inhibitors
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yi, J.; Peng, J.; Yang, W.; Zhu, G.; Ren, J.; Li, D.; Zheng, H. Picornavirus 3C—A Protease Ensuring Virus Replication and Subverting Host Responses. J. Cell Sci. 2021, 134, jcs253237. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.S.; Stobart, C.C.; Luo, H. Innate Immune Evasion Mediated by Picornaviral 3C Protease: Possible Lessons for Coronaviral 3C-like Protease? Rev. Med. Virol. 2021, 31, 1–22. [Google Scholar] [CrossRef]
- Sun, D.; Chen, S.; Cheng, A.; Wang, M. Roles of the Picornaviral 3C Proteinase in the Viral Life Cycle and Host Cells. Viruses 2016, 8, 82. [Google Scholar] [CrossRef] [PubMed]
- Minor, P.D. Poliovirus Biology. Structure 1996, 4, 775–778. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Weidman, M.K.; Yalamanchili, P.; Ng, B.; Tsai, W.; Dasgupta, A. Poliovirus 3C Protease-Mediated Degradation of Transcriptional Activator P53 Requires a Cellular Activity. Virology 2001, 291, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Gong, P. Structural Basis of Viral RNA-Dependent RNA Polymerase Nucleotide Addition Cycle in Picornaviruses. Enzymes 2021, 49, 215–233. [Google Scholar] [CrossRef]
- Seipelt, J.; Guarné, A.; Bergmann, E.; James, M.; Sommergruber, W.; Fita, I.; Skern, T. The Structures of Picornaviral Proteinases. Virus Res. 1999, 62, 159–168. [Google Scholar] [CrossRef]
- Campagnola, G.; Peersen, O. Co-Folding and RNA Activation of Poliovirus 3Cpro Polyprotein Precursors. J. Biol. Chem. 2023, 299, 105258. [Google Scholar] [CrossRef]
- Takegami, T.; Kuhn, R.J.; Anderson, C.W.; Wimmer, E. Membrane-Dependent Uridylylation of the Genome-Linked Protein VPg of Poliovirus. Proc. Natl. Acad. Sci. USA 1983, 80, 7447–7451. [Google Scholar] [CrossRef]
- Larsen, G.R.; Dorner, A.J.; Harris, T.J.; Wimmer, E. The Structure of Poliovirus Replicative Form. Nucleic Acids Res. 1980, 8, 1217–1229. [Google Scholar] [CrossRef]
- Kitamura, N.; Adler, C.; Wimmer, E. Structure and Expression of the Picornavirus Genome. Ann. N. Y Acad. Sci. 1980, 354, 183–201. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.F.; Nomoto, A.; Detjen, B.M.; Wimmer, E. A Protein Covalently Linked to Poliovirus Genome RNA. Proc. Natl. Acad. Sci. USA 1977, 74, 59–63. [Google Scholar] [CrossRef]
- Erickson, J.W.; Frankenberger, E.A.; Rossmann, M.G.; Fout, G.S.; Medappa, K.C.; Rueckert, R.R. Crystallization of a Common Cold Virus, Human Rhinovirus 14: “Isomorphism” with Poliovirus Crystals. Proc. Natl. Acad. Sci. USA 1983, 80, 931–934. [Google Scholar] [CrossRef] [PubMed]
- Hogle, J.M. Preliminary Studies of Crystals of Poliovirus Type I. J. Mol. Biol. 1982, 160, 663–668. [Google Scholar] [CrossRef]
- Malnou, C.E.; Pöyry, T.A.A.; Jackson, R.J.; Kean, K.M. Poliovirus Internal Ribosome Entry Segment Structure Alterations That Specifically Affect Function in Neuronal Cells: Molecular Genetic Analysis. J. Virol. 2002, 76, 10617–10626. [Google Scholar] [CrossRef]
- Jaafar, Z.A.; Kieft, J.S. Viral RNA Structure-Based Strategies to Manipulate Translation. Nat. Rev. Microbiol. 2019, 17, 110–123. [Google Scholar] [CrossRef] [PubMed]
- Barco, A.; Feduchi, E.; Carrasco, L. Poliovirus Protease 3C(pro) Kills Cells by Apoptosis. Virology 2000, 266, 352–360. [Google Scholar] [CrossRef]
- Francisco-Velilla, R.; Embarc-Buh, A.; Abellan, S.; Martinez-Salas, E. Picornavirus Translation Strategies. FEBS Open Bio 2022, 12, 1125–1141. [Google Scholar] [CrossRef]
- Cohen, L.; Kean, K.M.; Girard, M.; Van der Werf, S. Effects of P2 Cleavage Site Mutations on Poliovirus Polyprotein Processing. Virology 1996, 224, 34–42. [Google Scholar] [CrossRef]
- Hogle, J.M.; Filman, D.J. The Antigenic Structure of Poliovirus. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1989, 323, 467–478. [Google Scholar] [CrossRef]
- Jiang, P.; Liu, Y.; Ma, H.-C.; Paul, A.V.; Wimmer, E. Picornavirus Morphogenesis. Microbiol. Mol. Biol. Rev. 2014, 78, 418–437. [Google Scholar] [CrossRef] [PubMed]
- Hogle, J.M. Poliovirus Cell Entry: Common Structural Themes in Viral Cell Entry Pathways. Annu. Rev. Microbiol. 2002, 56, 677–702. [Google Scholar] [CrossRef] [PubMed]
- Takahara, Y.; Ando, N.; Kohara, M.; Hagino-Yamagishi, K.; Nomoto, A.; Itoh, H.; Numao, N.; Kondo, K. Purification of Enzymatically Active Poliovirus Proteinase 3C Produced in Escherichia Coli. Gene 1989, 79, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Ivanoff, L.A.; Towatari, T.; Ray, J.; Korant, B.D.; Petteway, S.R.J. Expression and Site-Specific Mutagenesis of the Poliovirus 3C Protease in Escherichia Coli. Proc. Natl. Acad. Sci. USA 1986, 83, 5392–5396. [Google Scholar] [CrossRef] [PubMed]
- Winston, D.S.; Boehr, D.D. Dynamics Compared to 3C pro and 3D Pol in Functionally. Viruses 2021, 13, 442. [Google Scholar] [CrossRef]
- Kean, K.M.; Teterina, N.; Girard, M. Cleavage Specificity of the Poliovirus 3C Protease Is Not Restricted to Gln-Gly at the 3C/3D Junction. J. Gen. Virol. 1990, 71 Pt 11, 2553–2563. [Google Scholar] [CrossRef]
- Baltimore, D.; Jacobson, M.F.; Asso, J.; Huang, A.S. The Formation of Poliovirus Proteins. Cold Spring Harb. Symp. Quant. Biol. 1969, 34, 741–746. [Google Scholar] [CrossRef]
- Jacobson, M.F.; Baltimore, D. Polypeptide Cleavages in the Formation of Poliovirus Proteins. Proc. Natl. Acad. Sci. USA 1968, 61, 77–84. [Google Scholar] [CrossRef]
- Jacobson, M.F.; Baltimore, D. Morphogenesis of Poliovirus. I. Association of the Viral RNA with Coat Protein. J. Mol. Biol. 1968, 33, 369–378. [Google Scholar] [CrossRef]
- Holland, J.J.; Kiehn, E.D. Specific Cleavage of Viral Proteins as Steps in the Synthesis and Maturation of Enteroviruses. Proc. Natl. Acad. Sci. USA 1968, 60, 1015–1022. [Google Scholar] [CrossRef]
- Pallansch, M.A.; Kew, O.M.; Palmenberg, A.C.; Golini, F.; Wimmer, E.; Rueckert, R.R. Picornaviral VPg Sequences Are Contained in the Replicase Precursor. J. Virol. 1980, 35, 414–419. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Hu, L.; Huang, X.; Wang, C.; Zhang, Z.; Wang, Y.; Zhang, D.; Ye, W. Potential of Coronavirus 3C-like Protease Inhibitors for the Development of New Anti-SARS-CoV-2 Drugs: Insights from Structures of Protease and Inhibitors. Int. J. Antimicrob. Agents 2020, 56, 106055. [Google Scholar] [CrossRef] [PubMed]
- Lawson, M.A.; Semler, B.L. Poliovirus Thiol Proteinase 3C Can Utilize a Serine Nucleophile within the Putative Catalytic Triad. Proc. Natl. Acad. Sci. USA 1991, 88, 9919–9923. [Google Scholar] [CrossRef] [PubMed]
- Sárkány, Z.; Polgár, L. The Unusual Catalytic Triad of Poliovirus Protease 3C. Biochemistry 2003, 42, 516–522. [Google Scholar] [CrossRef] [PubMed]
- Matthews, D.A.; Smith, W.W.; Ferre, R.A.; Condon, B.; Budahazi, G.; Slsson, W.; Villafranca, J.E.; Janson, C.A.; McElroy, H.E.; Gribskov, C.L.; et al. Structure of Human Rhinovirus 3C Protease Reveals a Trypsin-like Polypeptide Fold, RNA-Binding Site, and Means for Cleaving Precursor Polyprotein. Cell 1994, 77, 761–771. [Google Scholar] [CrossRef]
- Mosimann, S.C.; Cherney, M.M.; Sia, S.; Plotch, S.; James, M.N.G. Refined X-Ray Crystallographic Structure of the Poliovirus 3C Gene Product. J. Mol. Biol. 1997, 273, 1032–1047. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, E.M.; Mosimann, S.C.; Chernaia, M.M.; Malcolm, B.A.; James, M.N. The Refined Crystal Structure of the 3C Gene Product from Hepatitis A Virus: Specific Proteinase Activity and RNA Recognition. J. Virol. 1997, 71, 2436–2448. [Google Scholar] [CrossRef]
- Birtley, J.R.; Knox, S.R.; Jaulent, A.M.; Brick, P.; Leatherbarrow, R.J.; Curry, S. Crystal Structure of Foot-and-Mouth Disease Virus 3C Protease: New Insights into Catalytic Mechanism and Cleavage Specificity. J. Biol. Chem. 2005, 280, 11520–11527. [Google Scholar] [CrossRef]
- Pierce, D.M.; Hayward, C.; Rowlands, D.J.; Stonehouse, N.J.; Herod, M.R. Insights into Polyprotein Processing and RNA-Protein Interactions in Foot-and-Mouth Disease Virus Genome Replication. J. Virol. 2023, 97, e0017123. [Google Scholar] [CrossRef]
- Van Raaij, M.J.; Chouin, E.; Van der Zandt, H.; Bergelson, J.M.; Cusack, S. Erratum: Dimeric Structure of the Coxsackievirus and Adenovirus Receptor D1 Domain at 1.7 Å Resolution (Structure (November 15, 2000) 8 (1147–1155)). Structure 2001, 9, 1147–1155. [Google Scholar] [CrossRef]
- Ohlenschläger, O.; Wöhnert, J.; Bucci, E.; Seitz, S.; Häfner, S.; Ramachandran, R.; Zell, R.; Görlach, M. The Structure of the Stemloop D Subdomain of Coxsackievirus B3 Cloverleaf RNA and Its Interaction with the Proteinase 3C. Structure 2004, 12, 237–248. [Google Scholar] [CrossRef]
- Lee, C.C.; Kuo, C.J.; Ko, T.P.; Hsu, M.F.; Tsui, Y.C.; Chang, S.C.; Yang, S.; Chen, S.J.; Chen, H.C.; Hsu, M.C.; et al. Structural Basis of Inhibition Specificities of 3C and 3C-like Proteases by Zinc-Coordinating and Peptidomimetic Compounds. J. Biol. Chem. 2009, 284, 7646–7655. [Google Scholar] [CrossRef] [PubMed]
- Cui, S.; Wang, J.; Fan, T.; Qin, B.; Guo, L.; Lei, X.; Wang, J.; Wang, M.; Jin, Q. Crystal Structure of Human Enterovirus 71 3C Protease. J. Mol. Biol. 2011, 408, 449–461. [Google Scholar] [CrossRef]
- Sun, D.; Wang, M.; Wen, X.; Mao, S.; Cheng, A.; Jia, R.; Yang, Q.; Wu, Y.; Zhu, D.; Chen, S.; et al. Biochemical Characterization of Recombinant Avihepatovirus 3C Protease and Its Localization. Virol. J. 2019, 16, 54. [Google Scholar] [CrossRef]
- Andino, R.; Kirkegaard, K.; Macadam, A.; Racaniello, V.R.; Rosenfield, A.B. The Picornaviridae Family: Knowledge Gaps, Animal Models, Countermeasures, and Prototype Pathogens. J. Infect. Dis. 2023, 228, S427–S445. [Google Scholar] [CrossRef] [PubMed]
- Nugent, C.I.; Kirkegaard, K. RNA Binding Properties of Poliovirus Subviral Particles. J. Virol. 1995, 69, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.M.; Moustafa, I.M.; Arnold, J.J.; Cameron, C.E.; Boehr, D.D. Long-Range Communication between Different Functional Sites in the Picornaviral 3C Protein. Structure 2016, 24, 509–517. [Google Scholar] [CrossRef][Green Version]
- Tsu, B.V.; Fay, E.J.; Nguyen, K.T.; Corley, M.R.; Hosuru, B.; Dominguez, V.A.; Daugherty, M.D. Running with Scissors: Evolutionary Conflicts between Viral Proteases and the Host Immune System. Front. Immunol. 2021, 12, 769543. [Google Scholar] [CrossRef]
- Shengjuler, D.; Chan, Y.M.; Sun, S.; Moustafa, I.M.; Li, Z.L.; Gohara, D.W.; Buck, M.; Cremer, P.S.; Boehr, D.D.; Cameron, C.E. The RNA-Binding Site of Poliovirus 3C Protein Doubles as a Phosphoinositide-Binding Domain. Structure 2017, 25, 1875–1886.e7. [Google Scholar] [CrossRef]
- Bazan, J.F.; Fletterick, R.J. Viral Cysteine Proteases Are Homologous to the Trypsin-like Family of Serine Proteases: Structural and Functional Implications. Proc. Natl. Acad. Sci. USA 1988, 85, 7872–7876. [Google Scholar] [CrossRef]
- Uma, M.; Hegde, S.R.; Rao, P.P.; Nagalekshmi, K.; Gauthami, S.; Kumar, D.; Hegde, N.R. A Novel Point Mutation (L70P) Inactivates Poliovirus 3C Protease. Acta Virol. 2018, 62, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, T.R.; Roqué-Rosell, N.; Birtley, J.R.; Leatherbarrow, R.J.; Curry, S. Structural and Mutagenic Analysis of Foot-and-Mouth Disease Virus 3C Protease Reveals the Role of the Beta-Ribbon in Proteolysis. J. Virol. 2007, 81, 115–124. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jing, W.; Tingting, F.; Xue, Y.; Zhiqiang, W.; Li, G.; Xiaobo, L.; Jianwei, W.; Meitian, W.; Qi, J.; Sheng, C. Crystal Structures of Enterovirus 71 3C Protease Complexed with Rupintrivir Reveal the Roles of Catalytically Important Residues. J. Virol. 2011, 85, 10021–10030. [Google Scholar] [CrossRef]
- Yalamanchili, P.; Datta, U.; Dasgupta, A. Inhibition of Host Cell Transcription by Poliovirus: Cleavage of Transcription Factor CREB by Poliovirus-Encoded Protease 3C Pro. J. Virol. 1997, 71, 1220–1226. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Wang, M.; Wen, X.; Cheng, A.; Jia, R.; Sun, K.; Yang, Q.; Wu, Y.; Zhu, D.; Chen, S.; et al. Cleavage of Poly(A)-Binding Protein by Duck Hepatitis A Virus 3C Protease. Sci. Rep. 2017, 7, 16261. [Google Scholar] [CrossRef] [PubMed]
- Roehl, H.H.; Parsley, T.B.; Ho, T.V.; Semler, B.L. Processing of a Cellular Polypeptide by 3CD Proteinase Is Required for Poliovirus Ribonucleoprotein Complex Formation. J. Virol. 1997, 71, 578–585. [Google Scholar] [CrossRef]
- Gamarnik, A.V.; Andino, R. Interactions of Viral Protein 3CD and Poly(RC) Binding Protein with the 5′ Untranslated Region of the Poliovirus Genome. J. Virol. 2000, 74, 2219–2226. [Google Scholar] [CrossRef]
- Wang, Q.; Meng, H.; Ge, D.; Shan, H.; Geri, L.; Liu, F. Structural and Nonstructural Proteins of Senecavirus A: Recent Research Advances, and Lessons Learned from Those of Other Picornaviruses. Virology 2023, 585, 155–163. [Google Scholar] [CrossRef]
- de Breyne, S.; Bonderoff, J.M.; Chumakov, K.M.; Lloyd, R.E.; Hellen, C.U.T. Cleavage of Eukaryotic Initiation Factor EIF5B by Enterovirus 3C Proteases. Virology 2008, 378, 118–122. [Google Scholar] [CrossRef]
- Perera, R.; Daijogo, S.; Walter, B.L.; Nguyen, J.H.C.; Semler, B.L. Cellular Protein Modification by Poliovirus: The Two Faces of Poly(RC)-Binding Protein. J. Virol. 2007, 81, 8919–8932. [Google Scholar] [CrossRef]
- Kuyumcu-Martinez, N.M.; Van Eden, M.E.; Younan, P.; Lloyd, R.E. Cleavage of Poly(A)-Binding Protein by Poliovirus 3C Protease Inhibits Host Cell Translation: A Novel Mechanism for Host Translation Shutoff. Mol. Cell Biol. 2004, 24, 1779–1790. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Ross-Smith, N.; Proud, C.G.; Belsham, G.J. Cleavage of Translation Initiation Factor 4AI (EIF4AI) but Not EIF4AII by Foot-and-Mouth Disease Virus 3C Protease: Identification of the EIF4AI Cleavage Site. FEBS Lett. 2001, 507, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Belsham, G.J.; McInerney, G.M.; Ross-Smith, N. Foot-and-Mouth Disease Virus 3C Protease Induces Cleavage of Translation Initiation Factors EIF4A and EIF4G within Infected Cells. J. Virol. 2000, 74, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, P.; Schafer, E.A.; Rieder, E. The Nuclear Protein Sam68 Is Cleaved by the FMDV 3C Protease Redistributing Sam68 to the Cytoplasm during FMDV Infection of Host Cells. Virology 2012, 425, 40–52. [Google Scholar] [CrossRef] [PubMed]
- Fung, G.; Ng, C.S.; Zhang, J.; Shi, J.; Wong, J.; Piesik, P.; Han, L.; Chu, F.; Jagdeo, J.; Jan, E.; et al. Production of a Dominant-Negative Fragment Due to G3BP1 Cleavage Contributes to the Disruption of Mitochondria-Associated Protective Stress Granules during CVB3 Infection. PLoS ONE 2013, 8, e79546. [Google Scholar] [CrossRef]
- Zhang, B.; Seitz, S.; Kusov, Y.; Zell, R.; Gauss-Müller, V. RNA Interaction and Cleavage of Poly(C)-Binding Protein 2 by Hepatitis A Virus Protease. Biochem. Biophys. Res. Commun. 2007, 364, 725–730. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Morace, G.; Gauss-Müller, V.; Kusov, Y. Poly(A) Binding Protein, C-Terminally Truncated by the Hepatitis A Virus Proteinase 3C, Inhibits Viral Translation. Nucleic Acids Res. 2007, 35, 5975–5984. [Google Scholar] [CrossRef][Green Version]
- Kobayashi, M.; Arias, C.; Garabedian, A.; Palmenberg, A.C.; Mohr, I. Site-Specific Cleavage of the Host Poly(A) Binding Protein by the Encephalomyocarditis Virus 3C Proteinase Stimulates Viral Replication. J. Virol. 2012, 86, 10686–10694. [Google Scholar] [CrossRef]
- Qian, S.; Fan, W.; Liu, T.; Wu, M.; Zhang, H.; Cui, X.; Zhou, Y.; Hu, J.; Wei, S.; Chen, H.; et al. Seneca Valley Virus Suppresses Host Type I Interferon Production by Targeting Adaptor Proteins MAVS, TRIF, and TANK for Cleavage. J. Virol. 2017, 91, e00823-17. [Google Scholar] [CrossRef]
- Zunszain, P.A.; Knox, S.R.; Sweeney, T.R.; Yang, J.; Roqué-Rosell, N.; Belsham, G.J.; Leatherbarrow, R.J.; Curry, S. Insights into Cleavage Specificity from the Crystal Structure of Foot-and-Mouth Disease Virus 3C Protease Complexed with a Peptide Substrate. J. Mol. Biol. 2010, 395, 375–389. [Google Scholar] [CrossRef]
- Cameron, C.E.; Oh, H.S.; Moustafa, I.M. Expanding Knowledge of P3 Proteins in the Poliovirus Lifecycle. Future Microbiol. 2010, 5, 867–881. [Google Scholar] [CrossRef] [PubMed]
- Brandenburg, B.; Lee, L.Y.; Lakadamyali, M.; Rust, M.J.; Zhuang, X.; Hogle, J.M. Imaging Poliovirus Entry in Live Cells. PLoS Biol. 2007, 5, e183. [Google Scholar] [CrossRef] [PubMed]
- Mazzon, M.; Mercer, J. Lipid Interactions during Virus Entry and Infection. Cell Microbiol. 2014, 16, 1493–1502. [Google Scholar] [CrossRef] [PubMed]
- Lenard, J. Viral Membranes. Encycl. Virol. 2008, 308–314. [Google Scholar] [CrossRef]
- Villanueva, R.A.; Rouillé, Y.; Dubuisson, J. Interactions between Virus Proteins and Host Cell Membranes during the Viral Life Cycle. Int. Rev. Cytol. 2005, 245, 171–244. [Google Scholar] [CrossRef]
- Hsu, N.-Y.; Ilnytska, O.; Belov, G.; Santiana, M.; Chen, Y.-H.; Takvorian, P.M.; Pau, C.; van der Schaar, H.; Kaushik-Basu, N.; Balla, T.; et al. Viral Reorganization of the Secretory Pathway Generates Distinct Organelles for RNA Replication. Cell 2010, 141, 799–811. [Google Scholar] [CrossRef]
- Zhang, L.; Hong, Z.; Lin, W.; Shao, R.-X.; Goto, K.; Hsu, V.W.; Chung, R.T. ARF1 and GBF1 Generate a PI4P-Enriched Environment Supportive of Hepatitis C Virus Replication. PLoS ONE 2012, 7, e32135. [Google Scholar] [CrossRef]
- Wen, Y.; Vogt, V.M.; Feigenson, G.W. PI(4,5)P2 Clustering and Its Impact on Biological Functions. Annu. Rev. Biochem. 2021, 90, 681–707. [Google Scholar] [CrossRef]
- Arita, M. Mechanism of Poliovirus Resistance to Host Phosphatidylinositol-4 Kinase III β Inhibitor. ACS Infect. Dis. 2016, 2, 140–148. [Google Scholar] [CrossRef]
- Lemmon, M.A. Pleckstrin Homology (PH) Domains and Phosphoinositides. Biochem. Soc. Symp. 2007, 74, 81–93. [Google Scholar] [CrossRef]
- Lemmon, M.A. Pleckstrin Homology (PH) Domains. Handb. Cell Signal. Second. Ed. 2009, 2, 1093–1101. [Google Scholar] [CrossRef]
- Harlan, J.E.; Hajduk, P.J.; Yoon, H.S.; Fesik, S.W. Pleckstrin Homology Domains Bind to Phosphatidylinositol-4,5-Bisphosphate. Nature 1994, 371, 168–170. [Google Scholar] [CrossRef] [PubMed]
- Szentpetery, Z.; Balla, A.; Kim, Y.J.; Lemmon, M.A.; Balla, T. Live Cell Imaging with Protein Domains Capable of Recognizing Phosphatidylinositol 4,5-Bisphosphate; a Comparative Study. BMC Cell Biol. 2009, 10, 67. [Google Scholar] [CrossRef]
- Hammond, G.R.V.; Balla, T. Polyphosphoinositide Binding Domains: Key to Inositol Lipid Biology. Biochim. Biophys. Acta 2015, 1851, 746–758. [Google Scholar] [CrossRef]
- Pemberton, J.G.; Balla, T. Polyphosphoinositide-Binding Domains: Insights from Peripheral Membrane and Lipid-Transfer Proteins. Adv. Exp. Med. Biol. 2019, 1111, 77–137. [Google Scholar] [CrossRef]
- Groux-Degroote, S.; van Dijk, S.M.; Wolthoorn, J.; Neumann, S.; Theos, A.C.; De Mazière, A.M.; Klumperman, J.; van Meer, G.; Sprong, H. Glycolipid-Dependent Sorting of Melanosomal from Lysosomal Membrane Proteins by Lumenal Determinants. Traffic 2008, 9, 951–963. [Google Scholar] [CrossRef]
- Wang, Y.; Ma, L.; Stipkovits, L.; Szathmary, S.; Li, X.; Liu, Y. The Strategy of Picornavirus Evading Host Antiviral Responses: Nonstructural Proteins Suppress the Production of IFNs. Front. Microbiol. 2018, 9, 2943. [Google Scholar] [CrossRef]
- Flather, D.; Semler, B.L. Picornaviruses and Nuclear Functions: Targeting a Cellular Compartment Distinct from the Replication Site of a Positive-Strand RNA Virus. Front. Microbiol. 2015, 6, 594. [Google Scholar] [CrossRef]
- Lin, J.-Y.; Chen, T.-C.; Weng, K.-F.; Chang, S.-C.; Chen, L.-L.; Shih, S.-R. Viral and Host Proteins Involved in Picornavirus Life Cycle. J. Biomed. Sci. 2009, 16, 103. [Google Scholar] [CrossRef]
- Martínez-Salas, E.; Francisco-Velilla, R.; Fernandez-Chamorro, J.; Lozano, G.; Diaz-Toledano, R. Picornavirus IRES Elements: RNA Structure and Host Protein Interactions. Virus Res. 2015, 206, 62–73. [Google Scholar] [CrossRef]
- Kim, H.; Aponte-Diaz, D.; Sotoudegan, M.S.; Shengjuler, D.; Arnold, J.J.; Cameron, C.E. The Enterovirus Genome Can Be Translated in an IRES-Independent Manner That Requires the Initiation Factors EIF2A/EIF2D. PLoS Biol. 2023, 21, e3001693. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Guo, Y.; Wang, D.; Quan, R.; Wang, J.; Liu, J. Seneca Valley Virus 3C pro Antagonizes Type I Interferon Response by Targeting STAT1-STAT2-IRF9 and KPNA1 Signals. J. Virol. 2023, 97, e0072723. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Quan, R.; Wang, D.; Liu, J. Seneca Valley Virus 3Cpro Cleaves Heterogeneous Nuclear Ribonucleoprotein K to Facilitate Viral Replication. Front. Microbiol. 2022, 13, 945443. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, J.D.; Tsai, W.-C.; Lloyd, R.E. Multiple Poliovirus Proteins Repress Cytoplasmic RNA Granules. Viruses 2015, 7, 6127–6140. [Google Scholar] [CrossRef]
- Fitzgerald, K.D.; Chase, A.J.; Cathcart, A.L.; Tran, G.P.; Semler, B.L. Viral Proteinase Requirements for the Nucleocytoplasmic Relocalization of Cellular Splicing Factor SRp20 during Picornavirus Infections. J. Virol. 2013, 87, 2390–2400. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jackson, T.; Belsham, G.J. Picornaviruses: A View from 3A. Viruses 2021, 13, 456. [Google Scholar] [CrossRef]
- White, J.P.; Cardenas, A.M.; Marissen, W.E.; Lloyd, R.E. Inhibition of Cytoplasmic MRNA Stress Granule Formation by a Viral Proteinase. Cell Host Microbe 2007, 2, 295–305. [Google Scholar] [CrossRef]
- Yi, S.-J.; Kim, K. Histone Tail Cleavage as a Novel Epigenetic Regulatory Mechanism for Gene Expression. BMB Rep. 2018, 51, 211–218. [Google Scholar] [CrossRef]
- Das, C.; Tyler, J.K. Histone Exchange and Histone Modifications during Transcription and Aging. Biochim. Biophys. Acta 2013, 1819, 332–342. [Google Scholar] [CrossRef]
- Werbeck, N.D.; Shukla, V.K.; Kunze, M.B.A.; Yalinca, H.; Pritchard, R.B.; Siemons, L.; Mondal, S.; Greenwood, S.O.R.; Kirkpatrick, J.; Marson, C.M.; et al. A Distal Regulatory Region of a Class I Human Histone Deacetylase. Nat. Commun. 2020, 11, 3841. [Google Scholar] [CrossRef]
- Zhou, P.; Wu, E.; Alam, H.B.; Li, Y. Histone Cleavage as a Mechanism for Epigenetic Regulation: Current Insights and Perspectives. Curr. Mol. Med. 2014, 14, 1164–1172. [Google Scholar] [CrossRef]
- Shi, J.; Perryman, J.M.; Yang, X.; Liu, X.; Musser, D.M.; Boehr, A.K.; Moustafa, I.M.; Arnold, J.J.; Cameron, C.E.; Boehr, D.D. Rational Control of Poliovirus RNA-Dependent RNA Polymerase Fidelity by Modulating Motif-D Loop Conformational Dynamics. Biochemistry 2019, 58, 3735–3743. [Google Scholar] [CrossRef] [PubMed]
- Boehr, D.D.; Liu, X.; Yang, X. Targeting Structural Dynamics of the RNA-Dependent RNA Polymerase for Anti-Viral Strategies. Curr. Opin. Virol. 2014, 9, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Gohara, D.W.; Crotty, S.; Arnold, J.J.; Yoder, J.D.; Andino, R.; Cameron, C.E. Poliovirus RNA-Dependent RNA Polymerase (3Dpol): Structural, Biochemical, and Biological Analysis of Conserved Structural Motifs A and B. J. Biol. Chem. 2000, 275, 25523–25532. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yang, X.; Lee, C.A.; Moustafa, I.M.; Smidansky, E.D.; Lum, D.; Arnold, J.J.; Cameron, C.E.; Boehr, D.D. Vaccine-Derived Mutation in Motif D of Poliovirus RNA-Dependent RNA Polymerase Lowers Nucleotide Incorporation Fidelity*. J. Biol. Chem. 2013, 288, 32753–32765. [Google Scholar] [CrossRef]
- Sholders, A.J.; Peersen, O.B. Distinct Conformations of a Putative Translocation Element in Poliovirus Polymerase. J. Mol. Biol. 2014, 426, 1407–1419. [Google Scholar] [CrossRef] [PubMed]
- Kundu, P.; Raychaudhuri, S.; Tsai, W.; Dasgupta, A. Shutoff of RNA Polymerase II Transcription by Poliovirus Involves 3C Protease-Mediated Cleavage of the TATA-Binding Protein at an Alternative Site: Incomplete Shutoff of Transcription Interferes with Efficient Viral Replication. J. Virol. 2005, 79, 9702–9713. [Google Scholar] [CrossRef]
- Weng, K.F.; Li, M.L.; Hung, C.T.; Shih, S.R. Enterovirus 71 3C Protease Cleaves a Novel Target CstF-64 and Inhibits Cellular Polyadenylation. PLoS Pathog. 2009, 5, e1000593. [Google Scholar] [CrossRef]
- Xue, Q.; Liu, H.; Zhu, Z.; Yang, F.; Ma, L.; Cai, X.; Xue, Q.; Zheng, H. Seneca Valley Virus 3Cpro Abrogates the IRF3- and IRF7-Mediated Innate Immune Response by Degrading IRF3 and IRF7. Virology 2018, 518, 1–7. [Google Scholar] [CrossRef]
- Wen, W.; Yin, M.; Zhang, H.; Liu, T.; Chen, H.; Qian, P.; Hu, J.; Li, X. Seneca Valley Virus 2C and 3C Inhibit Type I Interferon Production by Inducing the Degradation of RIG-I. Virology 2019, 535, 122–129. [Google Scholar] [CrossRef]
- Martínez-Salas, E. The Impact of RNA Structure on Picornavirus IRES Activity. Trends Microbiol. 2008, 16, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Yalamanchili, P.; Weidman, K.; Dasgupta, A. Cleavage of Transcriptional Activator Oct-1 by Poliovirus Encoded Protease 3C Pro. Virology 1997, 239, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Maggioli, M.F.; Fernandes, M.H.V.; Joshi, L.R.; Sharma, B.; Tweet, M.M.; Noll, J.C.G.; Bauermann, F.V.; Diel, D.G. Persistent Infection and Transmission of Senecavirus A from Carrier Sows to Contact Piglets. J. Virol. 2019, 93, e00819-19. [Google Scholar] [CrossRef] [PubMed]
- Galicia-Vázquez, G.; Cencic, R.; Robert, F.; Agenor, A.Q.; Pelletier, J. A Cellular Response Linking EIF4AI Activity to EIF4AII Transcription. RNA 2012, 18, 1373–1384. [Google Scholar] [CrossRef]
- Kim, Y.; Lovell, S.; Tiew, K.-C.; Mandadapu, S.R.; Alliston, K.R.; Battaile, K.P.; Groutas, W.C.; Chang, K.-O. Broad-Spectrum Antivirals against 3C or 3C-Like Proteases of Picornaviruses, Noroviruses, and Coronaviruses. J. Virol. 2012, 86, 11754–11762. [Google Scholar] [CrossRef]
- Novoa, I.; Carrasco, L. Cleavage of Eukaryotic Translation Initiation Factor 4G by Exogenously Added Hybrid Proteins Containing Poliovirus 2Apro in HeLa Cells: Effects on Gene Expression. Mol. Cell Biol. 1999, 19, 2445–2454. [Google Scholar] [CrossRef]
- Kafasla, P.; Lin, H.; Curry, S.; Jackson, R.J. Activation of Picornaviral IRESs by PTB Shows Differential Dependence on Each PTB RNA-Binding Domain. RNA 2011, 17, 1120–1131. [Google Scholar] [CrossRef]
- Gradi, A.; Foeger, N.; Strong, R.; Svitkin, Y.V.; Sonenberg, N.; Skern, T.; Belsham, G.J. Cleavage of Eukaryotic Translation Initiation Factor 4GII within Foot-and-Mouth Disease Virus-Infected Cells: Identification of the L-Protease Cleavage Site In Vitro. J. Virol. 2004, 78, 3271–3278. [Google Scholar] [CrossRef]
- Huot, M.-É.; Brown, C.M.; Lamarche-Vane, N.; Richard, S. An Adaptor Role for Cytoplasmic Sam68 in Modulating Src Activity during Cell Polarization. Mol. Cell Biol. 2009, 29, 1933–1943. [Google Scholar] [CrossRef]
- Ullmer, W.; Semler, B.L. Diverse Strategies Used by Picornaviruses to Escape Host RNA Decay Pathways. Viruses 2016, 8, 335. [Google Scholar] [CrossRef]
- De Jesús-González, L.A.; Palacios-Rápalo, S.; Reyes-Ruiz, J.M.; Osuna-Ramos, J.F.; Cordero-Rivera, C.D.; Farfan-Morales, C.N.; Gutiérrez-Escolano, A.L.; Del Ángel, R.M. The Nuclear Pore Complex Is a Key Target of Viral Proteases to Promote Viral Replication. Viruses 2021, 13, 706. [Google Scholar] [CrossRef]
- Alber, F.; Dokudovskaya, S.; Veenhoff, L.M.; Zhang, W.; Kipper, J.; Devos, D.; Suprapto, A.; Karni-Schmidt, O.; Williams, R.; Chait, B.T.; et al. The Molecular Architecture of the Nuclear Pore Complex. Nature 2007, 450, 695–701. [Google Scholar] [CrossRef]
- Grossman, E.; Medalia, O.; Zwerger, M. Functional Architecture of the Nuclear Pore Complex. Annu. Rev. Biophys. 2012, 41, 557–584. [Google Scholar] [CrossRef]
- Lizcano-Perret, B.; Michiels, T. Nucleocytoplasmic Trafficking Perturbation Induced by Picornaviruses. Viruses 2021, 13, 1210. [Google Scholar] [CrossRef]
- Sun, D.; Wen, X.; Wang, M.; Mao, S.; Cheng, A.; Yang, X.; Jia, R.; Chen, S.; Yang, Q.; Wu, Y.; et al. Apoptosis and Autophagy in Picornavirus Infection. Front. Microbiol. 2019, 10, 2032. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, Y.; Wang, S.; Meng, X.; Song, F.; Huo, W.; Zhang, S.; Chang, J.; Li, J.; Zheng, B.; et al. Coxsackievirus A6 Induces Cell Cycle Arrest in G0/G1 Phase for Viral Production. Front. Cell Infect. Microbiol. 2018, 8, 279. [Google Scholar] [CrossRef] [PubMed]
- Shubin, A.V.; Demidyuk, I.V.; Lunina, N.A.; Komissarov, A.A.; Roschina, M.P.; Leonova, O.G.; Kostrov, S. V Protease 3C of Hepatitis A Virus Induces Vacuolization of Lysosomal/Endosomal Organelles and Caspase-Independent Cell Death. BMC Cell Biol. 2015, 16, 4. [Google Scholar] [CrossRef] [PubMed]
- Calandria, C.; Irurzun, A.; Barco, Á.; Carrasco, L. Individual Expression of Poliovirus 2Apro and 3Cpro Induces Activation of Caspase-3 and PARP Cleavage in HeLa Cells. Virus Res. 2004, 104, 39–49. [Google Scholar] [CrossRef]
- Odell, A.F.; Mannion, A.J.; Jones, P.F.; Cook, G.P. Negative Regulation of P53 by the Poliovirus Receptor PVR Is a Target of a Human Cytomegalovirus Immune Evasion Molecule. bioRxiv 2022. [Google Scholar] [CrossRef]
- Autret, A.; Martin-Latil, S.; Mousson, L.; Wirotius, A.; Petit, F.; Arnoult, D.; Colbère-Garapin, F.; Estaquier, J.; Blondel, B. Poliovirus Induces Bax-Dependent Cell Death Mediated by c-Jun NH2-Terminal Kinase. J. Virol. 2007, 81, 7504–7516. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pampin, M.; Simonin, Y.; Blondel, B.; Percherancier, Y.; Chelbi-Alix, M.K. Cross Talk between PML and P53 during Poliovirus Infection: Implications for Antiviral Defense. J. Virol. 2006, 80, 8582–8592. [Google Scholar] [CrossRef]
- Belov, G.A.; Romanova, L.I.; Tolskaya, E.A.; Kolesnikova, M.S.; Lazebnik, Y.A.; Agol, V.I. The Major Apoptotic Pathway Activated and Suppressed by Poliovirus. J. Virol. 2003, 77, 45–56. [Google Scholar] [CrossRef]
- Li, M.-L.; Lin, J.-Y.; Chen, B.-S.; Weng, K.-F.; Shih, S.-R.; Calderon, J.D.; Tolbert, B.S.; Brewer, G. EV71 3C Protease Induces Apoptosis by Cleavage of HnRNP A1 to Promote Apaf-1 Translation. PLoS ONE 2019, 14, e0221048. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Jiang, Y.; Wu, C.; Zhou, D.; Gong, J.; Zhao, T.; Jin, Z. Development of FRET and Stress Granule Dual-Based System to Screen for Viral 3C Protease Inhibitors. Molecules 2023, 28, 3020. [Google Scholar] [CrossRef] [PubMed]
- Rui, Y.; Su, J.; Wang, H.; Chang, J.; Wang, S.; Zheng, W.; Cai, Y.; Wei, W.; Gordy, J.T.; Markham, R.; et al. Disruption of MDA5-Mediated Innate Immune Responses by the 3C Proteins of Coxsackievirus A16, Coxsackievirus A6, and Enterovirus D68. J. Virol. 2017, 91, 10-1128. [Google Scholar] [CrossRef]
- Song, J.; Quan, R.; Wang, D.; Liu, J. Seneca Valley Virus 3C pro Mediates Cleavage and Redistribution of Nucleolin To Facilitate Viral Replication. Microbiol. Spectr. 2022, 10, e0030422. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Liu, Q.; Zhang, L.; Zhang, Q.; Hu, L.; Li, C.; Wang, S.; Li, J.; Zhang, Y.; Yu, H.; et al. Encephalomyocarditis Virus 3C Protease Relieves TRAF Family Member-Associated NF-KB Activator (TANK) Inhibitory Effect on TRAF6-Mediated NF-KB Signaling through Cleavage of TANK. J. Biol. Chem. 2015, 290, 27618–27632. [Google Scholar] [CrossRef]
- Hung, H.-C.; Wang, H.-C.; Shih, S.-R.; Teng, I.-F.; Tseng, C.-P.; Hsu, J.T.-A. Synergistic Inhibition of Enterovirus 71 Replication by Interferon and Rupintrivir. J. Infect. Dis. 2011, 203, 1784–1790. [Google Scholar] [CrossRef]
- Lei, X.; Liu, X.; Ma, Y.; Sun, Z.; Yang, Y.; Jin, Q.; He, B.; Wang, J. The 3C Protein of Enterovirus 71 Inhibits Retinoid Acid-Inducible Gene I-Mediated Interferon Regulatory Factor 3 Activation and Type I Interferon Responses. J. Virol. 2010, 84, 8051–8061. [Google Scholar] [CrossRef]
- Mukherjee, A.; Morosky, S.A.; Delorme-Axford, E.; Dybdahl-Sissoko, N.; Oberste, M.S.; Wang, T.; Coyne, C.B. The Coxsackievirus B 3Cpro Protease Cleaves MAVS and TRIF to Attenuate Host Type I Interferon and Apoptotic Signaling. PLoS Pathog. 2011, 7, e1001311. [Google Scholar] [CrossRef]
- Zhao, T.; Yang, L.; Sun, Q.; Arguello, M.; Ballard, D.W.; Hiscott, J.; Lin, R. The NEMO Adaptor Bridges the Nuclear Factor-ΚB and Interferon Regulatory Factor Signaling Pathways. Nat. Immunol. 2007, 8, 592–600. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liang, Y.; Qu, L.; Chen, Z.; Yi, M.; Li, K.; Lemon, S.M. Disruption of Innate Immunity Due to Mitochondrial Targeting of a Picornaviral Protease Precursor. Proc. Natl. Acad. Sci. USA 2007, 104, 7253–7258. [Google Scholar] [CrossRef]
- Bruurs, L.J.M.; Müller, M.; Schipper, J.G.; Rabouw, H.H.; Boersma, S.; van Kuppeveld, F.J.M.; Tanenbaum, M.E. Antiviral Responses Are Shaped by Heterogeneity in Viral Replication Dynamics. Nat. Microbiol. 2023, 8, 2115–2129. [Google Scholar] [CrossRef] [PubMed]
- Dias Junior, A.G.; Sampaio, N.G.; Rehwinkel, J. A Balancing Act: MDA5 in Antiviral Immunity and Autoinflammation. Trends Microbiol. 2019, 27, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Andreas, P.; Oliver, S.; Choon-Ping, T.; Jan, R.; Hiroki, K.; Osamu, T.; Shizuo, A.; Michael, W.; Giampietro, S.; Caetano, R.e.S. Activation of MDA5 Requires Higher-Order RNA Structures Generated during Virus Infection. J. Virol. 2009, 83, 10761–10769. [Google Scholar] [CrossRef]
- Rehwinkel, J.; Gack, M.U. RIG-I-like Receptors: Their Regulation and Roles in RNA Sensing. Nat. Rev. Immunol. 2020, 20, 537–551. [Google Scholar] [CrossRef]
- Savitsky, D.; Tamura, T.; Yanai, H.; Taniguchi, T. Regulation of Immunity and Oncogenesis by the IRF Transcription Factor Family. Cancer Immunol. Immunother. 2010, 59, 489–510. [Google Scholar] [CrossRef]
- Andersen, J.; VanScoy, S.; Cheng, T.-F.; Gomez, D.; Reich, N.C. IRF-3-Dependent and Augmented Target Genes during Viral Infection. Genes. Immun. 2008, 9, 168–175. [Google Scholar] [CrossRef]
- Gamarnik, A.V.; Andino, R. Switch from Translation to RNA Replication in a Positive-Stranded RNA Virus. Minerva Anestesiol. 1998, 12, 2293–2304. [Google Scholar] [CrossRef]
- Lloyd, R.E. Translational Control by Viral Proteinases. Virus Res. 2006, 119, 76–88. [Google Scholar] [CrossRef]
- Amero, C.D.; Arnold, J.J.; Moustafa, I.M.; Cameron, C.E.; Foster, M.P. Identification of the OriI-Binding Site of Poliovirus 3C Protein by Nuclear Magnetic Resonance Spectroscopy. J. Virol. 2008, 82, 4363–4370. [Google Scholar] [CrossRef] [PubMed]
- Kanda, T.; Gauss-Müller, V.; Cordes, S.; Tamura, R.; Okitsu, K.; Shuang, W.; Nakamoto, S.; Fujiwara, K.; Imazeki, F.; Yokosuka, O. Hepatitis A Virus (HAV) Proteinase 3C Inhibits HAV IRES-Dependent Translation and Cleaves the Polypyrimidine Tract-Binding Protein. J. Viral Hepat. 2010, 17, 618–623. [Google Scholar] [CrossRef] [PubMed]
- Hoon, B.S.; Ki, K.Y.; Jae, K.W.; Sungchan, C.; Rang, O.H.; Jung-Eun, K.; Key, J.S. Translation of Polioviral MRNA Is Inhibited by Cleavage of Polypyrimidine Tract-Binding Proteins Executed by Polioviral 3Cpro. J. Virol. 2002, 76, 2529–2542. [Google Scholar] [CrossRef]
- Valesano, A.L.; Taniuchi, M.; Fitzsimmons, W.J.; Islam, M.O.; Ahmed, T.; Zaman, K.; Haque, R.; Wong, W.; Famulare, M.; Lauring, A.S. The Early Evolution of Oral Poliovirus Vaccine Is Shaped by Strong Positive Selection and Tight Transmission Bottlenecks. Cell Host Microbe 2021, 29, 32–43.e4. [Google Scholar] [CrossRef] [PubMed]
- Reineke, L.C.; Lloyd, R.E. The Stress Granule Protein G3BP1 Recruits Protein Kinase R to Promote Multiple Innate Immune Antiviral Responses. J. Virol. 2015, 89, 2575–2589. [Google Scholar] [CrossRef] [PubMed]
- Hyun, J.; Kanagavelu, S.; Fukata, M. A Unique Host Defense Pathway: TRIF Mediates Both Antiviral and Antibacterial Immune Responses. Microbes Infect. 2013, 15, 1–10. [Google Scholar] [CrossRef][Green Version]
- Lei, X.; Xiao, X.; Xue, Q.; Jin, Q.; He, B.; Wang, J. Cleavage of Interferon Regulatory Factor 7 by Enterovirus 71 3C Suppresses Cellular Responses. J. Virol. 2013, 87, 1690–1698. [Google Scholar] [CrossRef]
- Andino, R.; Rieckhof, G.E.; Achacoso, P.L.; Baltimore, D. Poliovirus RNA Synthesis Utilizes an RNP Complex Formed around the 5’-End of Viral RNA. EMBO J. 1993, 12, 3587–3598. [Google Scholar] [CrossRef]
- Fass, D.M.; Reis, S.A.; Ghosh, B.; Hennig, K.M.; Joseph, N.F.; Zhao, W.N.; Nieland, T.J.; Guan, J.S.; Kuhnle, C.E.G.; Tang, W.; et al. Crebinostat: A novel cognitive enhancer that inhibits histone deacetylase activity and modulates chromatin-mediated neuroplasticity. Neuropharmacology 2013, 64, 81–96. [Google Scholar] [CrossRef]
- Paul, A.V.; Wimmer, E. Initiation of Protein-Primed Picornavirus RNA Synthesis. Virus Res. 2015, 206, 12–26. [Google Scholar] [CrossRef]
- Herold, J.; Andino, R. Poliovirus RNA Replication Requires Genome Circularization through a Protein–Protein Bridge. Mol. Cell 2001, 7, 581–591. [Google Scholar] [CrossRef] [PubMed]
- Parsley, T.B.; Towner, J.S.; Blyn, L.B.; Ehrenfeld, E.; Semler, B.L. Poly (RC) Binding Protein 2 Forms a Ternary Complex with the 5’-Terminal Sequences of Poliovirus RNA and the Viral 3CD Proteinase. RNA 1997, 3, 1124–1134. [Google Scholar] [PubMed]
- Harris, K.S.; Xiang, W.; Alexander, L.; Lane, W.S.; Paul, A.V.; Wimmer, E. Interaction of Poliovirus Polypeptide 3CDpro with the 5′ and 3′ Termini of the Poliovirus Genome. Identification of Viral and Cellular Cofactors Needed for Efficient Binding. J. Biol. Chem. 1994, 269, 27004–27014. [Google Scholar] [CrossRef] [PubMed]
- Xiang, W.; Harris, K.S.; Alexander, L.; Wimmer, E. Interaction between the 5’-Terminal Cloverleaf and 3AB/3CDpro of Poliovirus Is Essential for RNA Replication. J. Virol. 1995, 69, 3658–3667. [Google Scholar] [CrossRef]
- Yang, Y.; Rijnbrand, R.; Watowich, S.; Lemon, S.M. Genetic Evidence for an Interaction between a Picornaviral Cis-Acting RNA Replication Element and 3CD Protein*. J. Biol. Chem. 2004, 279, 12659–12667. [Google Scholar] [CrossRef]
- Shen, M.; Wang, Q.; Yang, Y.; Pathak, H.B.; Arnold, J.J.; Castro, C.; Lemon, S.M.; Cameron, C.E. Human Rhinovirus Type 14 Gain-of-Function Mutants for OriI Utilization Define Residues of 3C(D) and 3Dpol That Contribute to Assembly and Stability of the Picornavirus VPg Uridylylation Complex. J. Virol. 2007, 81, 12485–12495. [Google Scholar] [CrossRef]
- Kusov, Y.Y.; Gauss-Müller, V. In Vitro RNA Binding of the Hepatitis A Virus Proteinase 3C (HAV 3Cpro) to Secondary Structure Elements within the 5’ Terminus of the HAV Genome. RNA 1997, 3, 291–302. [Google Scholar]
- Peters, H.; Kusov, Y.Y.; Meyer, S.; Benie, A.J.; Bäuml, E.; Wolff, M.; Rademacher, C.; Peters, T.; Gauss-Müller, V. Hepatitis A Virus Proteinase 3C Binding to Viral RNA: Correlation with Substrate Binding and Enzyme Dimerization. Biochem. J. 2005, 385, 363–370. [Google Scholar] [CrossRef]
- Kusov, Y.Y.; Morace, G.; Probst, C.; Gauss-Müller, V. Interaction of Hepatitis A Virus (HAV) Precursor Proteins 3AB and 3ABC with the 5’ and 3’ Termini of the HAV RNA. Virus Res. 1997, 51, 151–157. [Google Scholar] [CrossRef]
- Walker, P.A.; Leong, L.E.-C.; Porter, A.G. Sequence and Structural Determinants of the Interaction between the 5′-Noncoding Region of Picornavirus RNA and Rhinovirus Protease 3C*. J. Biol. Chem. 1995, 270, 14510–14516. [Google Scholar] [CrossRef]
- Leong, L.E.; Walker, P.A.; Porter, A.G. Human Rhinovirus-14 Protease 3C (3Cpro) Binds Specifically to the 5′-Noncoding Region of the Viral RNA. Evidence That 3Cpro Has Different Domains for the RNA Binding and Proteolytic Activities. J. Biol. Chem. 1993, 268, 25735–25739. [Google Scholar] [CrossRef] [PubMed]
- Nayak, A.; Goodfellow, I.G.; Woolaway, K.E.; Birtley, J.; Curry, S.; Belsham, G.J. Role of RNA Structure and RNA Binding Activity of Foot-and-Mouth Disease Virus 3C Protein in VPg Uridylylation and Virus Replication. J. Virol. 2006, 80, 9865–9875. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Molla, A.; Harris, K.S.; Paul, A.V.; Shin, S.H.; Mugavero, J.; Wimmer, E. Stimulation of Poliovirus Proteinase 3Cpro-Related Proteolysis by the Genome-Linked Protein VPg and Its Precursor 3AB. J. Biol. Chem. 1994, 269, 27015–27020. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Welch, J.L.; Arnold, J.J.; Boehr, D.D. Long-Range Interaction Networks in the Function and Fidelity of Poliovirus RNA-Dependent RNA Polymerase Studied by Nuclear Magnetic Resonance. Biochemistry 2010, 49, 9361–9371. [Google Scholar] [CrossRef] [PubMed]
- Plotch, S.J.; Palant, O. Poliovirus Protein 3AB Forms a Complex with and Stimulates the Activity of the Viral RNA Polymerase, 3Dpol. J. Virol. 1995, 69, 7169–7179. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, K.; Bhat, R.; Rao, V.U.B.; Nain, A.; Rallapalli, K.L.; Gangopadhyay, S.; Singh, R.P.; Banerjee, M.; Jayaram, B. Toward Development of Generic Inhibitors against the 3C Proteases of Picornaviruses. FEBS J. 2019, 286, 765–787. [Google Scholar] [CrossRef] [PubMed]
- Gouvea, I.E.; Santos, J.A.N.; Burlandy, F.M.; Tersariol, I.L.S.; da Silva, E.E.; Juliano, M.A.; Juliano, L.; Cunha, R.L.O.R. Poliovirus 3C Proteinase Inhibition by Organotelluranes. Biol. Chem. 2011, 392, 587–591. [Google Scholar] [CrossRef]
- McKinlay, M.A.; Collett, M.S.; Hincks, J.R.; Oberste, M.S.; Pallansch, M.A.; Okayasu, H.; Sutter, R.W.; Modlin, J.F.; Dowdle, W.R. Progress in the Development of Poliovirus Antiviral Agents and Their Essential Role in Reducing Risks That Threaten Eradication. J. Infect. Dis. 2014, 210, S447–S453. [Google Scholar] [CrossRef]
- Rhoden, E.; Liu, H.-M.; Wang-Chern, S.; Oberste, M.S. Anti-Poliovirus Activity of Protease Inhibitor AG-7404, and Assessment of In Vitro Activity in Combination with Antiviral Capsid Inhibitor Compounds. Antivir. Res. 2013, 98, 186–191. [Google Scholar] [CrossRef]
- Patick, A.K.; Brothers, M.A.; Maldonado, F.; Binford, S.; Maldonado, O.; Fuhrman, S.; Petersen, A.; Smith, G.J.; Zalman, L.S.; Burns-Naas, L.A.; et al. In Vitro Antiviral Activity and Single-Dose Pharmacokinetics in Humans of a Novel, Orally Bioavailable Inhibitor of Human Rhinovirus 3C Protease. Antimicrob. Agents Chemother. 2005, 49, 2267–2275. [Google Scholar] [CrossRef]
- Groaz, E.; De Clercq, E.; Herdewijn, P. Anno 2021: Which Antivirals for the Coming Decade? Annu. Rep. Med. Chem. 2021, 57, 49–107. [Google Scholar] [CrossRef] [PubMed]
- Jagdeo, J.M.; Dufour, A.; Klein, T.; Solis, N.; Kleifeld, O.; Kizhakkedathu, J.; Luo, H.; Overall, C.M.; Jan, E. N-Terminomics TAILS Identifies Host Cell Substrates of Poliovirus and Coxsackievirus B3 3C Proteinases That Modulate Virus Infection. J. Virol. 2018, 92, e02211-17. [Google Scholar] [CrossRef] [PubMed]
- Nomoto, A. Molecular Aspects of Poliovirus Pathogenesis. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2007, 83, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Kafasla, P.; Morgner, N.; Robinson, C.V.; Jackson, R.J. Polypyrimidine Tract-Binding Protein Stimulates the Poliovirus IRES by Modulating EIF4G Binding. EMBO J. 2010, 29, 3710–3722. [Google Scholar] [CrossRef]
- Chase, A.J.; Daijogo, S.; Semler, B.L. Inhibition of Poliovirus-Induced Cleavage of Cellular Protein PCBP2 Reduces the Levels of Viral RNA Replication. J. Virol. 2014, 88, 3192–3201. [Google Scholar] [CrossRef] [PubMed]
- Kuyumcu-Martinez, N.M.; Joachims, M.; Lloyd, R.E. Efficient Cleavage of Ribosome-Associated Poly(A)-Binding Protein by Enterovirus 3C Protease. J. Virol. 2002, 76, 2062–2074. [Google Scholar] [CrossRef] [PubMed]
- Kuyumcu-Martinez, M.; Belliot, G.; Sosnovtsev, S.V.; Chang, K.-O.; Green, K.Y.; Lloyd, R.E. Calicivirus 3C-like Proteinase Inhibits Cellular Translation by Cleavage of Poly(A)-Binding Protein. J. Virol. 2004, 78, 8172–8182. [Google Scholar] [CrossRef]
- Wan, Q.; Song, D.; Li, H.; He, M. Stress Proteins: The Biological Functions in Virus Infection, Present and Challenges for Target-Based Antiviral Drug Development. Signal Transduct. Target. Ther. 2020, 5, 125. [Google Scholar] [CrossRef]
- Gorman, S.D.; D’Amico, R.N.; Winston, D.S.; Boehr, D.D. Engineering Allostery into Proteins BT—Protein Allostery in Drug Discovery; Zhang, J., Nussinov, R., Eds.; Springer: Singapore, 2019; pp. 359–384. ISBN 978-981-13-8719-7. [Google Scholar]
- Ma, Y.; Li, L.; He, S.; Shang, C.; Sun, Y.; Liu, N.; Meek, T.D.; Wang, Y.; Shang, L. Application of Dually Activated Michael Acceptor to the Rational Design of Reversible Covalent Inhibitor for Enterovirus 71 3C Protease. J. Med. Chem. 2019, 62, 6146–6162. [Google Scholar] [CrossRef]
- Xu, Y.; Ma, S.; Huang, Y.; Chen, F.; Chen, L.; Ding, D.; Zheng, Y.; Li, H.; Xiao, J.; Feng, J.; et al. Virus-like Particle Vaccines for Poliovirus Types 1, 2, and 3 with Enhanced Thermostability Expressed in Insect Cells. Vaccine 2019, 37, 2340–2347. [Google Scholar] [CrossRef]
- Kassem, A.F.; Batran, R.Z.; Abbas, E.M.H.; Elseginy, S.A.; Shaheen, M.N.F.; Elmahdy, E.M. New 4-Phenylcoumarin Derivatives as Potent 3C Protease Inhibitors: Design, Synthesis, Anti-HAV Effect and Molecular Modeling. Eur. J. Med. Chem. 2019, 168, 447–460. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus | Protein from the Host Cell | Activity | Cleaving Site | Reference(s) |
|---|---|---|---|---|
| Poliovirus (PV) | eIF5B | Inserts the initiation tRNA for methionine on the start codon of mRNA | LeuCysAlaAlaValGluValMetGluGln478GlyValProGluLysGluGluThr | De Bryne S et al. (2008) [59] |
| PCBP2 | Regulation of gene expression through translational activation | AlaMetGlnGln253SerHisPhePro… IleGlyArgGln306GlyAlaLys | Perera R et al. (2007) [60] | |
| PABP | Involved in the start of the translation and the shortening of poly (A) | Not certain | Kuyumcu-Martinez NM et al. (2004) [61] | |
| Foot-and-mouth disease virus (FMDV) | eIF4A I | Unwinds double-stranded RNA and allows the ribosomal subunit 40S to bind capped mRNA | CysIleGlyGlyThrAsnValArgAlaGlu143ValGlnLysLeuGlnMetGluAla | Li W et al. (2001) [62] |
| eIF4G I | Transports mRNA to the 40S ribosome to initiate translation | ArgArgSerGlnGlnGlyProArgLysGlu712ProArgLysIleIleAlaThrValLeu | Belsham GJ et al. (2000) [63] | |
| Sam68 | Responsible for cell growth and division | C-terminal region | Lawrence P et al. (2012) [64] | |
| Coxsackievirus (CVB) | eIF5B | Inserts the initiation tRNA for methionine on the start codon of mRNA | LeuCysAlaAlaValGluValMetGluGln478GlyValProGluLysGluGluThr | De Bryne S et al. (2008) [59] |
| G3BP1 | Ras–GAP-interacting RNA-binding protein | GluAlaGlyGluGln325GlyAspIleGluPro | Fung G et al. (2013) [65] | |
| Human rhinovirus (HRV) | eIF5B | Inserts the initiation tRNA for methionine on the start codon of mRNA | LeuCysAlaAlaValGluValMetGluGln478GlyValProGluLysGluGluThr | De Bryne S et al. (2008) [59] |
| Hepatitis A virus (HAV) | PCBP2 | Regulation of gene expression through translational activation | IleGlyArgGln306GlyAlaLysIle (Postulated) | Zhang B et al. (2007) [66] |
| PABP | Involved in the start of the translation and the shortening of poly (A) | Not certain | Zhang B et al. (2007) [67] | |
| Encephalomyocarditis virus (EMCV) | PABP | Involved in the start of the translation and the shortening of poly (A) | ValArgProProAlaAlaIleGln437GlyValGlnAlaGlyAla | Mariko K et al. (2012) [68] |
| Seneca valley virus (SVV) | TRIP MAVS TANK | Suppresses host type-I interferon production | Glu-Gln or, Gln-Gly | Qian S et al. (2017) [69] |
| Virus | Signaling Pathways | IFN Type | References |
|---|---|---|---|
| EV71 | Inhibits IRF7 and IRF9 | Type I IFNs | Lei X et al. 2010 [139] |
| EMCV | Cleaves TANK and inhibits TRAF6-mediated NF-κB signaling | Type I IFNs | Huang L et al. 2015 [137] |
| CVB3 | Cleaves MAVS and TRIF | Type I IFNs | Mukherjee A et al. 2011 [140] |
| CV–A6, EV–D68 | Cleaves TAK1 to inhibit NF-κB signalling | Not clear | Yajuan R et al. 2017 [135] |
| FMDV | Cleaving TANK and NEMO | Not clear | Zhao T et al. 2007 [141] |
| HAV | Cleaving MAVS and NEMO | Type I IFNs | Yang Y et al. 2007 [142] |
| SVV | Cleaving MAVS, TRIF, and TANK | Type I IFNs | Qian S et al. 2017 [69] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mondal, S.; Sarvari, G.; Boehr, D.D. Picornavirus 3C Proteins Intervene in Host Cell Processes through Proteolysis and Interactions with RNA. Viruses 2023, 15, 2413. https://doi.org/10.3390/v15122413
Mondal S, Sarvari G, Boehr DD. Picornavirus 3C Proteins Intervene in Host Cell Processes through Proteolysis and Interactions with RNA. Viruses. 2023; 15(12):2413. https://doi.org/10.3390/v15122413
Chicago/Turabian StyleMondal, Somnath, Gisoo Sarvari, and David D. Boehr. 2023. "Picornavirus 3C Proteins Intervene in Host Cell Processes through Proteolysis and Interactions with RNA" Viruses 15, no. 12: 2413. https://doi.org/10.3390/v15122413
APA StyleMondal, S., Sarvari, G., & Boehr, D. D. (2023). Picornavirus 3C Proteins Intervene in Host Cell Processes through Proteolysis and Interactions with RNA. Viruses, 15(12), 2413. https://doi.org/10.3390/v15122413