
Molecular Mechanisms of Ferroptosis and Its Role in Viral Pathogenesis


Abstract
:1. Introduction
2. Inducers and Inhibitors of Ferroptosis
3. The Mechanisms of Ferroptosis
3.1. Ferroptosis and Amino Acid Metabolism
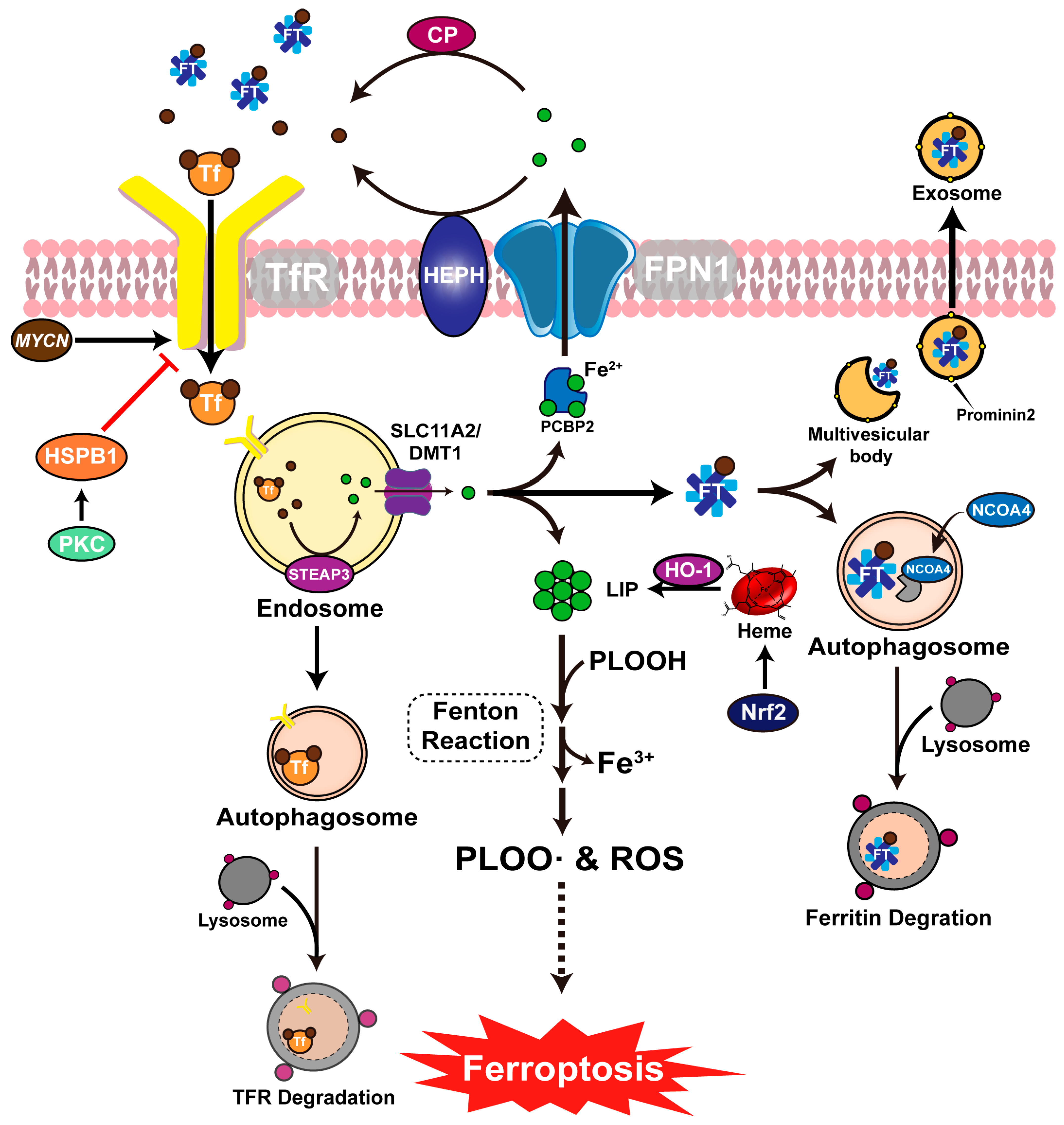
3.2. Ferroptosis and Iron Metabolism
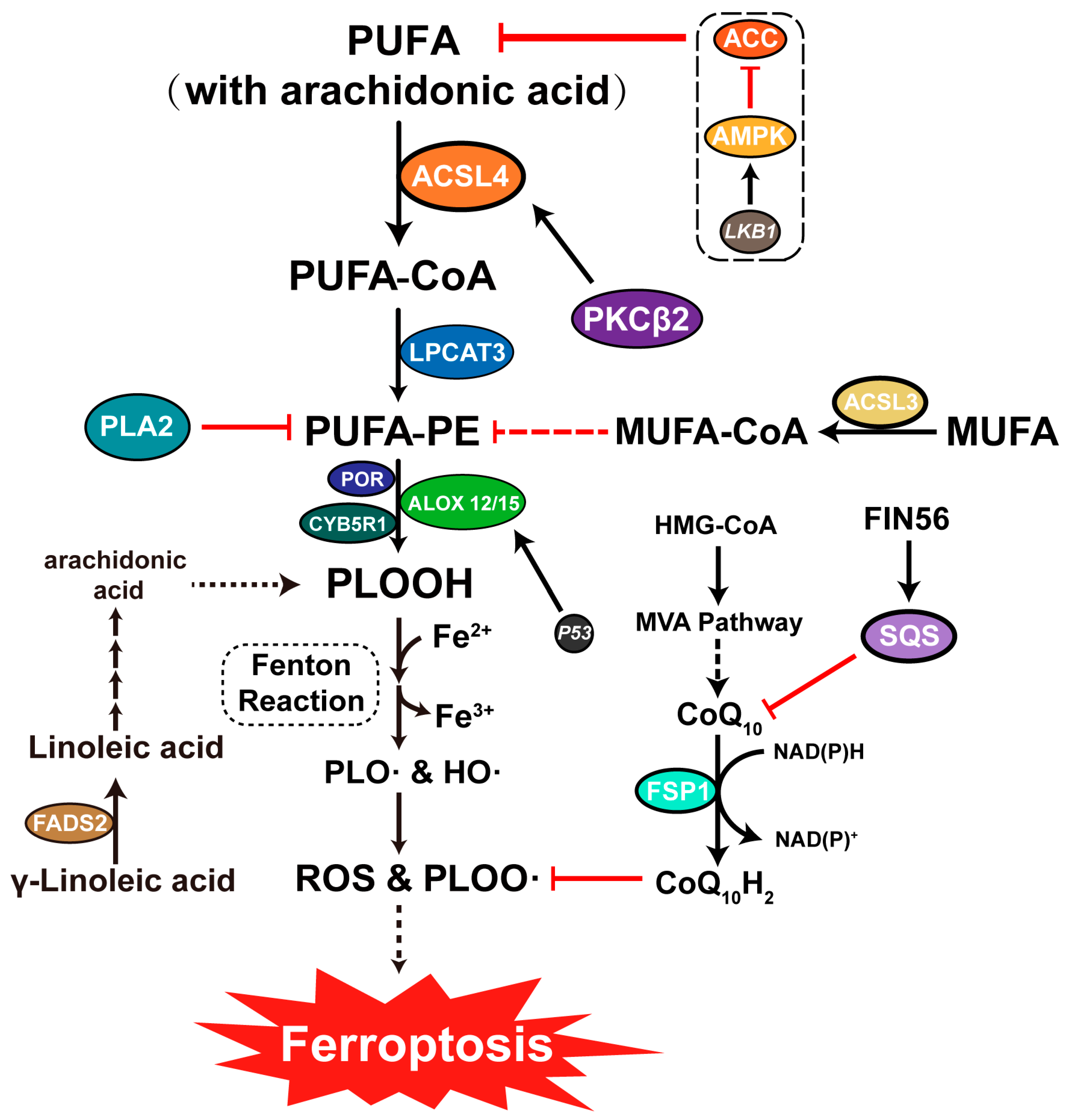
3.3. Ferroptosis and Lipid Metabolism
4. Regulatory Network of Ferroptosis
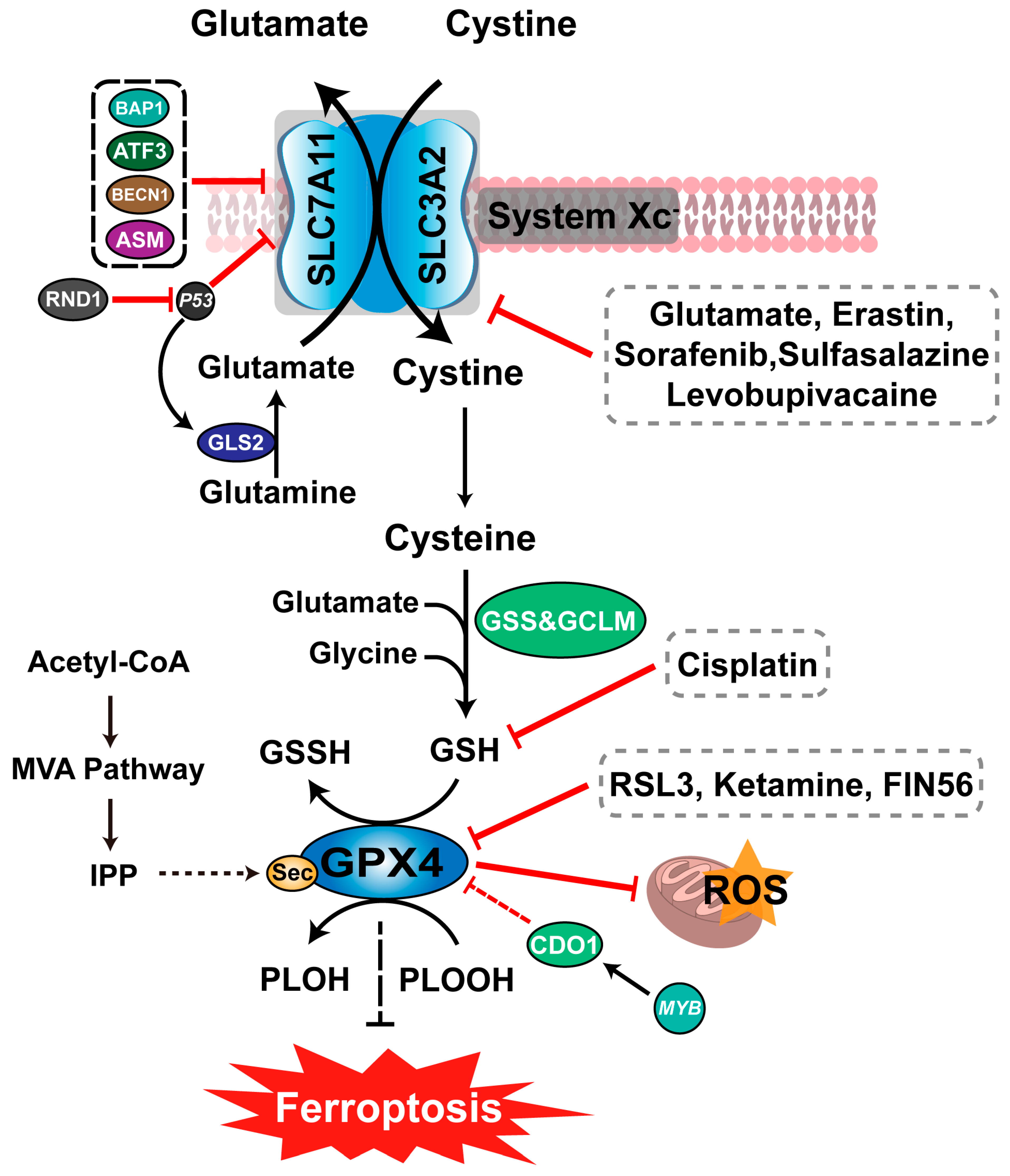
4.1. Inhibition of System Xc− Induces Ferroptosis
4.2. Inhibition of GPX Activity Induces Ferroptosis
4.3. Other Regulatory Networks Associated with Ferroptosis
4.3.1. LKB1-AMPK-ACC-PUFA Pathway
4.3.2. NAD(P)H-FSP1-CoQ10 Pathway
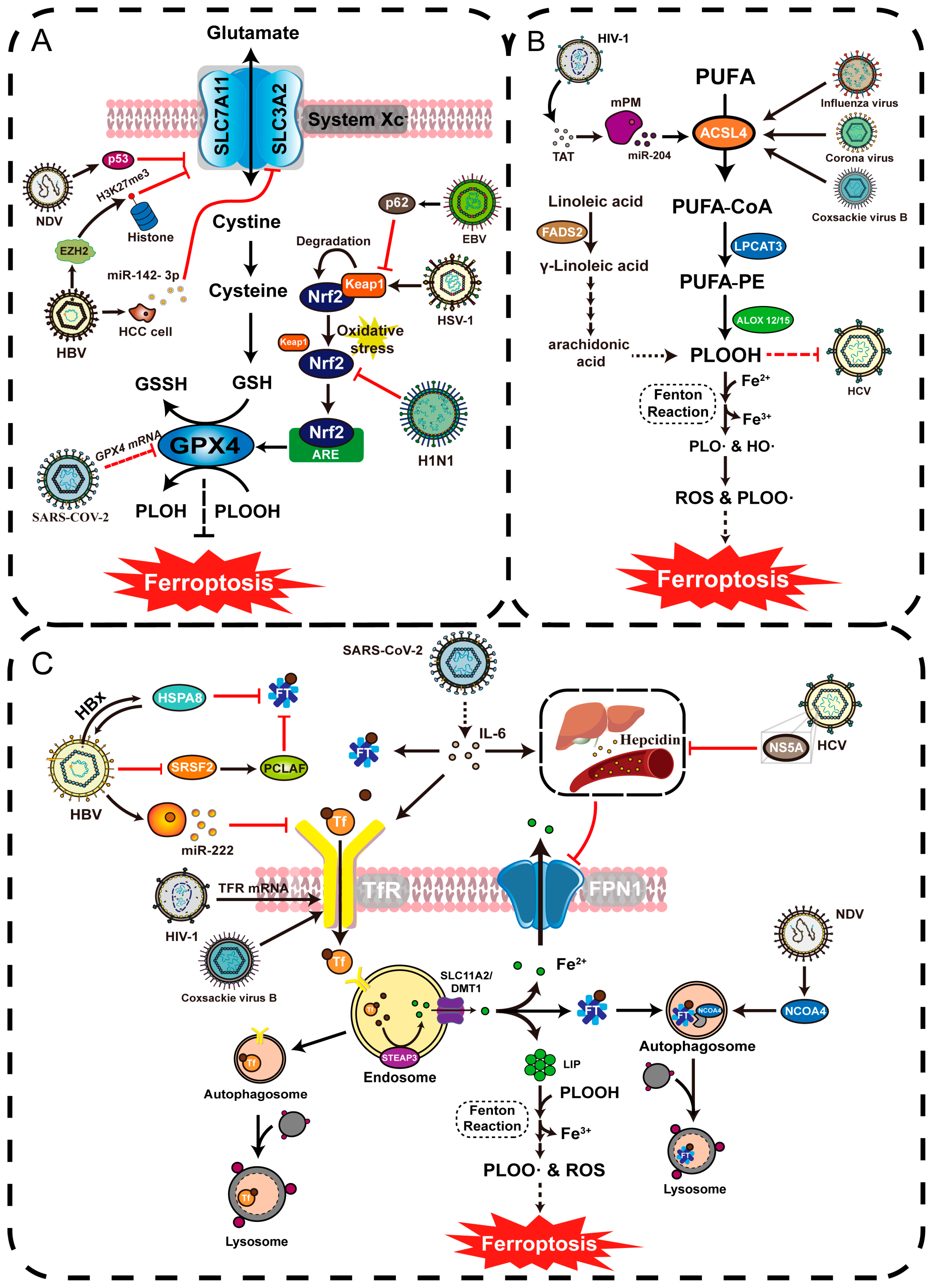
5. The Role of Ferroptosis in the Pathogenesis of Viral Infections
5.1. Transferrin Receptors Mediate Viral Entry Associated with Ferroptosis
5.2. Potential for Virus-Mediated Ferroptosis to Facilitate Viral Release
5.3. Viruses Trigger Ferroptosis to Escape the Immune Response and Promote Viral Proliferation
5.3.1. GPX4 Provides a Stable Oxidation–Reduction State for Immune System
5.3.2. Virus Targeting System Xc−-GSH-GPX4 Axis Induces Ferroptosis in Immune Cells
5.3.3. Viruses Regulate System Xc− and GPX4 to Alter Redox Environment in Infected Cells
5.3.4. Virus Targeting Keap1-Nrf2 Axis Affects Downstream GPX4 Activity
5.4. Viral Infection Alters Host Iron Metabolism
5.4.1. Viral Upregulates Intracellular Iron Levels and Promotes Viral Reproduction
5.4.2. Viral Causes Decreased Intracellular Iron Levels to Promote Disease Progression
5.5. Viral Infection Alters Host Lipid Metabolism Levels
5.5.1. Viruses Mediate Ferroptosis through ACSL Family Upregulation
5.5.2. Virus Causes Persistent Infection through Ferroptosis
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lei, G.; Zhuang, L.; Gan, B. Targeting ferroptosis as a vulnerability in cancer. Nat. Rev. Cancer 2022, 22, 381–396. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.; Minikes, A.M.; Jiang, X. Ferroptosis at the intersection of lipid metabolism and cellular signaling. Mol. Cell 2022, 82, 2215–2227. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Stockwell, B.R. The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 2014, 10, 9–17. [Google Scholar] [CrossRef]
- Golbashirzadeh, M.; Heidari, H.R.; Khosroushahi, A.Y. Molecular mechanisms of reactive oxygen species in regulated cell deaths: Impact of ferroptosis in cancer therapy. Gene Rep. 2022, 27, 101614. [Google Scholar] [CrossRef]
- Jones, D.P.; Sies, H. The Redox Code. Antioxid. Redox Sign 2015, 23, 734–746. [Google Scholar] [CrossRef]
- Scott, J.D.; Kathryn, M.L.; Michael, R.L.; Rachid, S.; Eleina, M.Z.; Caroline, E.G.; Darpan, N.P.; Andras, J.B.; Alexandra, M.C.; Wan, S.Y.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar]
- Yan, H.; Zou, T.; Tuo, Q.; Xu, S.; Li, H.; Belaidi, A.A.; Lei, P. Ferroptosis: Mechanisms and links with diseases. Signal Transduct. Tar. 2021, 6, 49. [Google Scholar] [CrossRef]
- Wang, M.P.; Joshua, B.; Jin, N.Y.; Du, S.W.; Li, C. Ferroptosis in viral infection: The unexplored possibility. Acta Pharmacol. Sin. 2022, 43, 1905–1915. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.L.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef]
- Dolma, S.; Lessnick, S.L.; Hahn, W.C.; Stockwell, B.R. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell 2003, 3, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Gout, P.W.; Buckley, A.R.; Simms, C.R.; Bruchovsky, N. Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the xc− cystine transporter: A new action for an old drug. Leukemia 2001, 15, 1633–1640. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xia, J.; Shao, F.; Zhou, Y.; Yu, J.; Wu, H.; Du, J.; Ren, X. Sorafenib induces mitochondrial dysfunction and exhibits synergistic effect with cysteine depletion by promoting HCC cells ferroptosis. Biochem. Biophys. Res. Commun. 2021, 534, 877–884. [Google Scholar] [CrossRef] [PubMed]
- Mao, S.H.; Zhu, C.H.; Nie, Y.; Yu, J.; Wang, L. Levobupivacaine Induces Ferroptosis by miR-489-3p/SLC7A11 Signaling in Gastric Cancer. Front. Pharmacol. 2021, 12, 681338. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Stockwell, B.R. Synthetic Lethal Screening Identifies Compounds Activating Iron-Dependent, Nonapoptotic Cell Death in Oncogenic-RAS-Harboring Cancer Cells. Chem. Biol. 2008, 15, 234–245. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Skouta, R.; Kaplan, A.; Yang, W.S.; Hayano, M.; Dixon, S.J.; Brown, L.M.; Valenzuela, C.A.; Wolpaw, A.J.; Stockwell, B.R. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat. Chem. Biol. 2016, 12, 497–503. [Google Scholar] [CrossRef]
- Li, H.; Liu, W.; Zhang, X.; Wu, F.; Sun, D.; Wang, Z. Ketamine suppresses proliferation and induces ferroptosis and apoptosis of breast cancer cells by targeting KAT5/GPX4 axis. Biochem. Biophys. Res. Commun. 2021, 585, 111–116. [Google Scholar] [CrossRef]
- He, G.N.; Bao, N.R.; Wang, S.; Xi, M.; Zhang, T.H.; Chen, F.S. Ketamine Induces Ferroptosis of Liver Cancer Cells by Targeting lncRNA PVT1/miR-214-3p/GPX4. Drug Des. Dev. Ther. 2021, 15, 3965–3978. [Google Scholar] [CrossRef]
- Dong, X.Q.; Chu, L.K.; Cao, X.; Xiong, Q.W.; Mao, Y.M.; Chen, C.H.; Bi, Y.L.; Liu, J.; Yan, X.M. Glutathione metabolism rewiring protects renal tubule cells against cisplatin-induced apoptosis and ferroptosis. Redox Rep. 2023, 28, 2152607. [Google Scholar] [CrossRef]
- Guo, J.; Xu, B.; Han, Q.; Zhou, H.; Xia, Y.; Gong, C.; Dai, X.; Li, Z.; Wu, G. Ferroptosis: A Novel Anti-tumor Action for Cisplatin. Cancer Res. Treat. 2018, 50, 445–460. [Google Scholar] [CrossRef]
- Cui, Z.; Wang, H.; Li, S.; Qin, T.; Shi, H.; Ma, J.; Li, L.; Yu, G.; Jiang, T.; Li, C. Dihydroartemisinin enhances the inhibitory effect of sorafenib on HepG2 cells by inducing ferroptosis and inhibiting energy metabolism. J. Pharmacol. Sci. 2022, 148, 73–85. [Google Scholar] [CrossRef]
- Zhang, Q.; Yi, H.; Yao, H.; Lu, L.; He, G.; Wu, M.; Zheng, C.; Li, Y.; Chen, S.; Li, L.; et al. Artemisinin Derivatives Inhibit Non-small Cell Lung Cancer Cells Through Induction of ROS-dependent Apoptosis/Ferroptosis. J. Cancer 2021, 12, 4075–4085. [Google Scholar] [CrossRef]
- Chen, G.Q.; Benthani, F.A.; Wu, J.; Liang, D.; Bian, Z.X.; Jiang, X. Artemisinin compounds sensitize cancer cells to ferroptosis by regulating iron homeostasis. Cell Death Differ. 2020, 27, 242–254. [Google Scholar] [CrossRef]
- Kong, Z.; Liu, R.; Cheng, Y. Artesunate alleviates liver fibrosis by regulating ferroptosis signaling pathway. Biomed. Pharmacother. 2019, 109, 2043–2053. [Google Scholar] [CrossRef]
- Eling, N.; Reuter, L.; Hazin, J.; Hamacher-Brady, A.; Brady, N.R. Identification of artesunate as a specific activator of ferroptosis in pancreatic cancer cells. Oncoscience 2015, 2, 517–532. [Google Scholar] [CrossRef]
- Gaschler, M.M.; Andia, A.A.; Liu, H.; Csuka, J.M.; Hurlocker, B.; Vaiana, C.A.; Heindel, D.W.; Zuckerman, D.S.; Bos, P.H.; Reznik, E.; et al. FINO2 initiates ferroptosis through GPX4 inactivation and iron oxidation. Nat. Chem. Biol. 2018, 14, 507–515. [Google Scholar] [CrossRef]
- Wang, X.; Xu, S.; Zhang, L.; Cheng, X.; Yu, H.; Bao, J.; Lu, R. Vitamin C induces ferroptosis in anaplastic thyroid cancer cells by ferritinophagy activation. Biochem. Biophys. Res. Commun. 2021, 551, 46–53. [Google Scholar] [CrossRef]
- Lorenzato, A.; Magri, A.; Matafora, V.; Audrito, V.; Arcella, P.; Lazzari, L.; Montone, M.; Lamba, S.; Deaglio, S.; Siena, S.; et al. Vitamin C Restricts the Emergence of Acquired Resistance to EGFR-Targeted Therapies in Colorectal Cancer. Cancers 2020, 12, 685. [Google Scholar] [CrossRef]
- Wei, S.; Qiu, T.; Yao, X.; Wang, N.; Jiang, L.; Jia, X.; Tao, Y.; Wang, Z.; Pei, P.; Zhang, J.; et al. Arsenic induces pancreatic dysfunction and ferroptosis via mitochondrial ROS-autophagy-lysosomal pathway. J. Hazard. Mater. 2020, 384, 121390. [Google Scholar] [CrossRef] [PubMed]
- Umemura, M.; Kim, J.H.; Aoyama, H.; Hoshino, Y.; Fukumura, H.; Nakakaji, R.; Sato, I.; Ohtake, M.; Akimoto, T.; Narikawa, M.; et al. The iron chelating agent, deferoxamine detoxifies Fe(Salen)-induced cytotoxicity. J. Pharmacol. Sci. 2017, 134, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Angeli, J.P.F.; Shah, R.; Pratt, D.A.; Conrad, M. Ferroptosis Inhibition: Mechanisms and Opportunities. Trends Pharmacol. Sci. 2017, 38, 489–498. [Google Scholar] [CrossRef]
- Sheng, X.; Shan, C.; Liu, J.; Yang, J.; Sun, B.; Chen, D. Theoretical insights into the mechanism of ferroptosis suppression via inactivation of a lipid peroxide radical by liproxstatin-1. Phys. Chem. Chem. Phys. 2017, 19, 13153–13159. [Google Scholar] [CrossRef]
- Zilka, O.; Shah, R.; Li, B.; Friedmann, A.J.; Griesser, M.; Conrad, M.; Pratt, D.A. On the Mechanism of Cytoprotection by Ferrostatin-1 and Liproxstatin-1 and the Role of Lipid Peroxidation in Ferroptotic Cell Death. ACS Cent. Sci. 2017, 3, 232–243. [Google Scholar] [CrossRef]
- Wan, Y.; Shen, K.; Yu, H.; Fan, W. Baicalein limits osteoarthritis development by inhibiting chondrocyte ferroptosis. Free Radic. Biol. Med. 2023, 196, 108–120. [Google Scholar] [CrossRef]
- Xie, Y.; Song, X.; Sun, X.; Huang, J.; Zhong, M.; Lotze, M.T.; Zeh, H.R.; Kang, R.; Tang, D. Identification of baicalein as a ferroptosis inhibitor by natural product library screening. Biochem. Biophys. Res. Commun. 2016, 473, 775–780. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, W.; Li, Y.; Xiao, Y.; Cheng, J.; Jia, J. The 5-Lipoxygenase Inhibitor Zileuton Confers Neuroprotection against Glutamate Oxidative Damage by Inhibiting Ferroptosis. Biol. Pharm. Bull. 2015, 38, 1234–1239. [Google Scholar] [CrossRef]
- Gao, M.; Monian, P.; Quadri, N.; Ramasamy, R.; Jiang, X. Glutaminolysis and Transferrin Regulate Ferroptosis. Mol. Cell 2015, 59, 298–308. [Google Scholar] [CrossRef]
- Lewerenz, J.; Hewett, S.J.; Huang, Y.; Lambros, M.; Gout, P.W.; Kalivas, P.W.; Massie, A.; Smolders, I.; Methner, A.; Pergande, M.; et al. The cystine/glutamate antiporter system xc− in health and disease: From molecular mechanisms to novel therapeutic opportunities. Antioxid. Redox Signal. 2013, 18, 522–555. [Google Scholar] [CrossRef]
- Gao, M.; Monian, P.; Pan, Q.; Zhang, W.; Xiang, J.; Jiang, X. Ferroptosis is an autophagic cell death process. Cell Res. 2016, 26, 1021–1032. [Google Scholar] [CrossRef] [PubMed]
- Ursini, F.; Maiorino, M. Lipid peroxidation and ferroptosis: The role of GSH and GPx4. Free Radic. Biol. Med. 2020, 152, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Ingold, I.; Berndt, C.; Schmitt, S.; Doll, S.; Poschmann, G.; Buday, K.; Roveri, A.; Peng, X.; Porto, F.F.; Seibt, T.; et al. Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis. Cell 2018, 172, 409–422. [Google Scholar] [CrossRef] [PubMed]
- Friedmann, A.J.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef] [PubMed]
- Regina, B.; Matilde, M. Glutathione peroxidases. BBA—Gen. Subj. 2013, 1830, 3289–3303. [Google Scholar]
- Lv, Q.; Niu, H.; Yue, L.; Liu, J.; Yang, L.; Liu, C.; Jiang, H.; Dong, S.; Shao, Z.; Xing, L.; et al. Abnormal Ferroptosis in Myelodysplastic Syndrome. Front. Oncol. 2020, 10, 1656. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef]
- Lei, P.; Bai, T.; Sun, Y. Mechanisms of Ferroptosis and Relations With Regulated Cell Death: A Review. Front. Physiol. 2019, 10, 139. [Google Scholar] [CrossRef]
- Zhu, S.; Zhang, Q.; Sun, X.; Zeh, H.R.; Lotze, M.T.; Kang, R.; Tang, D. HSPA5 Regulates Ferroptotic Cell Death in Cancer Cells. Cancer Res. 2017, 77, 2064–2077. [Google Scholar] [CrossRef]
- Wu, Z.; Geng, Y.; Lu, X.; Shi, Y.; Wu, G.; Zhang, M.; Shan, B.; Pan, H.; Yuan, J. Chaperone-mediated autophagy is involved in the execution of ferroptosis. Proc. Natl. Acad. Sci. USA 2019, 116, 2996–3005. [Google Scholar] [CrossRef]
- Muller, T.; Dewitz, C.; Schmitz, J.; Schroder, A.S.; Brasen, J.H.; Stockwell, B.R.; Murphy, J.M.; Kunzendorf, U.; Krautwald, S. Necroptosis and ferroptosis are alternative cell death pathways that operate in acute kidney failure. Cell. Mol. Life Sci. 2017, 74, 3631–3645. [Google Scholar] [CrossRef]
- Jomova, K.; Valko, M. Advances in metal-induced oxidative stress and human disease. Toxicology 2011, 283, 65–87. [Google Scholar] [CrossRef]
- Ma, S.; Henson, E.S.; Chen, Y.; Gibson, S.B. Ferroptosis is induced following siramesine and lapatinib treatment of breast cancer cells. Cell Death Dis. 2016, 7, e2307. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Li, F.M.; Zhou, Y.F.; Qian, C.; Li, J.; Jiang, L.R.; Qian, Z.M. Effects of alpha-lipoic acid on expression of iron transport and storage proteins in BV-2 microglia cells. Pharmacol. Rep. 2017, 69, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.J.P. Free manganese(II) and iron(II) cations can act as intracellular cell controls. FEBS Lett. 1982, 140, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, M.; Cockrell, A.L.; Park, J.; McCormick, S.P.; Lindahl, L.S.; Lindahl, P.A. Speciation of iron in mouse liver during development, iron deficiency, IRP2 deletion and inflammatory hepatitis. Metallomics 2015, 7, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Liu, T.; Li, Y.; Lau, J.; Yang, Z.; Fan, W.; Zhou, Z.; Shi, C.; Ke, C.; Bregadze, V.I.; et al. Fenton-Reaction-Acceleratable Magnetic Nanoparticles for Ferroptosis Therapy of Orthotopic Brain Tumors. ACS Nano 2018, 12, 11355–11365. [Google Scholar] [CrossRef] [PubMed]
- Ma, P.; Xiao, H.; Yu, C.; Liu, J.; Cheng, Z.; Song, H.; Zhang, X.; Li, C.; Wang, J.; Gu, Z.; et al. Enhanced Cisplatin Chemotherapy by Iron Oxide Nanocarrier-Mediated Generation of Highly Toxic Reactive Oxygen Species. Nano Lett. 2017, 17, 928–937. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.Y.; Dixon, S.J. Mechanisms of ferroptosis. Cell. Mol. Life Sci. 2016, 73, 2195–2209. [Google Scholar] [CrossRef] [PubMed]
- Andrews, N.C.; Schmidt, P.J. Iron homeostasis. Annu. Rev. Physiol. 2007, 69, 69–85. [Google Scholar] [CrossRef]
- Frazer, D.M.; Anderson, G.J. The regulation of iron transport. Biofactors 2014, 40, 206–214. [Google Scholar] [CrossRef]
- Tang, D.; Chen, X.; Kang, R.; Kroemer, G. Ferroptosis: Molecular mechanisms and health implications. Cell Res. 2021, 31, 107–125. [Google Scholar] [CrossRef]
- Ham, J.; Costa, C.; Sano, R.; Lochmann, T.L.; Sennott, E.M.; Patel, N.U.; Dastur, A.; Gomez-Caraballo, M.; Krytska, K.; Hata, A.N.; et al. Exploitation of the Apoptosis-Primed State of MYCN-Amplified Neuroblastoma to Develop a Potent and Specific Targeted Therapy Combination. Cancer Cell 2016, 29, 159–172. [Google Scholar] [CrossRef]
- Ruiz-Perez, M.V.; Henley, A.B.; Arsenian-Henriksson, M. The MYCN Protein in Health and Disease. Genes 2017, 8, 113. [Google Scholar] [CrossRef]
- Lu, Y.; Yang, Q.; Su, Y.; Ji, Y.; Li, G.; Yang, X.; Xu, L.; Lu, Z.; Dong, J.; Wu, Y.; et al. MYCN mediates TFRC-dependent ferroptosis and reveals vulnerabilities in neuroblastoma. Cell Death Dis. 2021, 12, 511. [Google Scholar] [CrossRef]
- Sun, X.; Ou, Z.; Xie, M.; Kang, R.; Fan, Y.; Niu, X.; Wang, H.; Cao, L.; Tang, D. HSPB1 as a novel regulator of ferroptotic cancer cell death. Oncogene 2015, 34, 5617–5625. [Google Scholar] [CrossRef]
- Tang, Z.; Ju, Y.; Dai, X.; Ni, N.; Liu, Y.; Zhang, D.; Gao, H.; Sun, H.; Zhang, J.; Gu, P. HO-1-mediated ferroptosis as a target for protection against retinal pigment epithelium degeneration. Redox Biol. 2021, 43, 101971. [Google Scholar] [CrossRef]
- Hassannia, B.; Wiernicki, B.; Ingold, I.; Qu, F.; Van Herck, S.; Tyurina, Y.Y.; Bayir, H.; Abhari, B.A.; Angeli, J.; Choi, S.M.; et al. Nano-targeted induction of dual ferroptotic mechanisms eradicates high-risk neuroblastoma. J. Clin. Investig. 2018, 128, 3341–3355. [Google Scholar] [CrossRef]
- Sun, X.; Ou, Z.; Chen, R.; Niu, X.; Chen, D.; Kang, R.; Tang, D. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 2016, 63, 173–184. [Google Scholar] [CrossRef]
- Terman, A.; Kurz, T. Lysosomal iron, iron chelation, and cell death. Antioxid. Redox Signal. 2013, 18, 888–898. [Google Scholar] [CrossRef]
- Liang, Y.; Deng, Y.; Zhao, J.; Liu, L.; Wang, J.; Chen, P.; Zhang, Q.; Sun, C.; Wang, Y.; Xiang, Y.; et al. Ferritinophagy is Involved in Experimental Subarachnoid Hemorrhage-Induced Neuronal Ferroptosis. Neurochem. Res. 2021, 47, 692–700. [Google Scholar] [CrossRef] [PubMed]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.F.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef]
- Yuan, H.; Li, X.; Zhang, X.; Kang, R.; Tang, D. Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem. Biophys. Res. Commun. 2016, 478, 1338–1343. [Google Scholar] [CrossRef]
- Shimbara-Matsubayashi, S.; Kuwata, H.; Tanaka, N.; Kato, M.; Hara, S. Analysis on the Substrate Specificity of Recombinant Human Acyl-CoA Synthetase ACSL4 Variants. Biol. Pharm. Bull. 2019, 42, 850–855. [Google Scholar] [CrossRef]
- Zhang, H.; Hu, B.; Li, Z.; Du, T.; Shan, J.; Ye, Z.; Peng, X.; Li, X.; Huang, Y.; Zhu, X.; et al. PKCβII phosphorylates ACSL4 to amplify lipid peroxidation to induce ferroptosis. Nat. Cell Biol. 2022, 24, 88–98. [Google Scholar] [CrossRef]
- Magtanong, L.; Ko, P.J.; To, M.; Cao, J.Y.; Forcina, G.C.; Tarangelo, A.; Ward, C.C.; Cho, K.; Patti, G.J.; Nomura, D.K.; et al. Exogenous Monounsaturated Fatty Acids Promote a Ferroptosis-Resistant Cell State. Cell Chem. Biol. 2019, 26, 420–432. [Google Scholar] [CrossRef]
- Zhou, H.; Li, F.; Niu, J.Y.; Zhong, W.Y.; Tang, M.Y.; Lin, D.; Cui, H.H.; Huang, X.H.; Chen, Y.Y.; Wang, H.Y.; et al. Ferroptosis was involved in the oleic acid-induced acute lung injury in mice. Sheng Li Xue Bao 2019, 71, 689–697. [Google Scholar]
- Galadari, S.; Rahman, A.; Pallichankandy, S.; Thayyullathil, F. Tumor suppressive functions of ceramide: Evidence and mechanisms. Apoptosis 2015, 20, 689–711. [Google Scholar] [CrossRef]
- Novgorodov, S.A.; Voltin, J.R.; Gooz, M.A.; Li, L.; Lemasters, J.J.; Gudz, T.I. Acid sphingomyelinase promotes mitochondrial dysfunction due to glutamate-induced regulated necrosis. J. Lipid Res. 2018, 59, 312–329. [Google Scholar] [CrossRef]
- Thayyullathil, F.; Cheratta, A.R.; Alakkal, A.; Subburayan, K.; Pallichankandy, S.; Hannun, Y.A.; Galadari, S. Acid sphingomyelinase-dependent autophagic degradation of GPX4 is critical for the execution of ferroptosis. Cell Death Dis. 2021, 12, 26. [Google Scholar] [CrossRef]
- Pekárová, M.; Kuhn, H.; Bezáková, L.; Ufer, C.; Heydeck, D. Mutagenesis of triad determinants of rat Alox15 alters the specificity of fatty acid and phospholipid oxygenation. Arch. Biochem. Biophys. 2015, 571, 50–57. [Google Scholar] [CrossRef]
- Kuhn, H.; Belkner, J.; Wiesner, R.; Brash, A.R. Oxygenation of biological membranes by the pure reticulocyte lipoxygenase. J. Biol. Chem. 1990, 265, 18351–18361. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, I.; Kuhn, H.; Heydeck, D. Structural and functional biology of arachidonic acid 15-lipoxygenase-1 (ALOX15). Gene 2015, 573, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Haeggstrom, J.Z.; Funk, C.D. Lipoxygenase and leukotriene pathways: Biochemistry, biology, and roles in disease. Chem. Rev. 2011, 111, 5866–5898. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Ou, Y.; Wang, S.J.; Li, D.; Chu, B.; Gu, W. Activation of SAT1 engages polyamine metabolism with p53-mediated ferroptotic responses. Proc. Natl. Acad. Sci. USA 2016, 113, E6806–E6812. [Google Scholar] [CrossRef] [PubMed]
- Chu, B.; Kon, N.; Chen, D.; Li, T.; Liu, T.; Jiang, L.; Song, S.; Tavana, O.; Gu, W. ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nat. Cell Biol. 2019, 21, 579–591. [Google Scholar] [CrossRef]
- Koppula, P.; Zhuang, L.; Gan, B. Cytochrome P450 reductase (POR) as a ferroptosis fuel. Protein Cell 2021, 12, 675–679. [Google Scholar] [CrossRef] [PubMed]
- Yan, B.; Ai, Y.; Zhang, Z.; Wang, X. Assessing POR and CYB5R1 oxidoreductase-mediated oxidative rupture of PUFA in liposomes. Star. Protoc. 2021, 2, 100360. [Google Scholar] [CrossRef]
- Zou, Y.; Li, H.; Graham, E.T.; Deik, A.A.; Eaton, J.K.; Wang, W.; Sandoval-Gomez, G.; Clish, C.B.; Doench, J.G.; Schreiber, S.L. Cytochrome P450 oxidoreductase contributes tophospholipid peroxidation in ferroptosis. Nat. Chem. Biol. 2020, 16, 302–309. [Google Scholar] [CrossRef]
- Yan, B.; Ai, Y.; Sun, Q.; Ma, Y.; Cao, Y.; Wang, J.; Zhang, Z.; Wang, X. Membrane Damage during Ferroptosis Is Caused by Oxidation of Phospholipids Catalyzed by the Oxidoreductases POR and CYB5R1. Mol. Cell 2021, 81, 355–369. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Patel, D.N.; Welsch, M.; Skouta, R.; Lee, E.D.; Hayano, M.; Thomas, A.G.; Gleason, C.E.; Tatonetti, N.P.; Slusher, B.S.; et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. elife 2014, 3, e2523. [Google Scholar] [CrossRef]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef]
- Liu, Y.; Gu, W. p53 in ferroptosis regulation: The new weapon for the old guardian. Cell Death Differ. 2022, 29, 895–910. [Google Scholar] [CrossRef]
- Huang, C.; Yang, M.; Deng, J.; Li, P.; Su, W.; Jiang, R. Upregulation and activation of p53 by erastininduced reactive oxygen species contribute to cytotoxic and cytostatic effects in A549 lung cancer cells. Oncol. Rep. 2018, 40, 2363–2370. [Google Scholar] [CrossRef]
- Sun, Q.; Xu, Y.; Yuan, F.E.; Qi, Y.; Wang, Y.; Chen, Q.; Liu, B. Rho family GTPase 1 (RND1), a novel regulator of p53, enhances ferroptosis in glioblastoma. Cell Biosci. 2022, 12, 53. [Google Scholar] [CrossRef]
- Hu, W.; Zhang, C.; Wu, R.; Sun, Y.; Levine, A.; Feng, Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc. Natl. Acad. Sci. USA 2010, 107, 7455–7460. [Google Scholar] [CrossRef]
- Jennis, M.; Kung, C.P.; Basu, S.; Budina-Kolomets, A.; Leu, J.I.; Khaku, S.; Scott, J.P.; Cai, K.Q.; Campbell, M.R.; Porter, D.K.; et al. An African-specific polymorphism in the TP53 gene impairs p53 tumor suppressor function in a mouse model. Gene Dev. 2016, 30, 918–930. [Google Scholar] [CrossRef]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Gene Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef]
- Sykiotis, G.P.; Bohmann, D. Stress-activated cap‘n’collar transcription factors in aging and human disease. Sci. Signal. 2010, 3, re3. [Google Scholar] [CrossRef]
- Liu, N.; Lin, X.; Huang, C. Activation of the reverse transsulfuration pathway through NRF2/CBS confers erastin-induced ferroptosis resistance. Br. J. Cancer 2020, 122, 279–292. [Google Scholar] [CrossRef]
- Dodson, M.; Castro-Portuguez, R.; Zhang, D.D. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 2019, 23, 101107. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Wirth, A.K.; Chen, D.; Wruck, C.J.; Rauh, M.; Buchfelder, M.; Savaskan, N. Nrf2-Keap1 pathway promotes cell proliferation and diminishes ferroptosis. Oncogenesis 2017, 6, e371. [Google Scholar] [CrossRef] [PubMed]
- Carbone, M.; Yang, H.; Pass, H.I.; Krausz, T.; Testa, J.R.; Gaudino, G. BAP1 and cancer. Nat. Rev. Cancer 2013, 13, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Scheuermann, J.C.; de Ayala, A.A.; Oktaba, K.; Ly-Hartig, N.; McGinty, R.K.; Fraterman, S.; Wilm, M.; Muir, T.W.; Muller, J. Histone H2A deubiquitinase activity of the Polycomb repressive complex PR-DUB. Nature 2010, 465, 243–247. [Google Scholar] [CrossRef]
- Zhang, Y.; Shi, J.; Liu, X.; Feng, L.; Gong, Z.; Koppula, P.; Sirohi, K.; Li, X.; Wei, Y.; Lee, H.; et al. BAP1 links metabolic regulation of ferroptosis to tumour suppression. Nat. Cell Biol. 2018, 20, 1181–1192. [Google Scholar] [CrossRef]
- Wang, L.; Liu, Y.; Du, T.; Yang, H.; Lei, L.; Guo, M.; Ding, H.; Zhang, J.; Wang, H.; Chen, X.; et al. ATF3 promotes erastin-induced ferroptosis by suppressing system Xc–. Cell Death Differ. 2020, 27, 662–675. [Google Scholar] [CrossRef]
- Yan, C.; Lu, D.; Hai, T.; Boyd, D.D. Activating transcription factor 3, a stress sensor, activates p53 by blocking its ubiquitination. EMBO J. 2005, 24, 2425–2435. [Google Scholar] [CrossRef]
- Zhao, J.; Li, X.; Guo, M.; Yu, J.; Yan, C. The common stress responsive transcription factor ATF3 binds genomic sites enriched with p300 and H3K27ac for transcriptional regulation. BMC Genom. 2016, 17, 335. [Google Scholar] [CrossRef]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999, 402, 672–676. [Google Scholar] [CrossRef]
- Song, X.; Zhu, S.; Chen, P.; Hou, W.; Wen, Q.; Liu, J.; Xie, Y.; Liu, J.; Klionsky, D.J.; Kroemer, G.; et al. AMPK-Mediated BECN1 Phosphorylation Promotes Ferroptosis by Directly Blocking System Xc– Activity. Curr. Biol. 2018, 28, 2388–2399. [Google Scholar] [CrossRef] [PubMed]
- Conrad, M.; Schneider, M.; Seiler, A.; Bornkamm, G.W. Physiological role of phospholipid hydroperoxide glutathione peroxidase in mammals. Biol. Chem. 2007, 388, 1019–1025. [Google Scholar] [CrossRef]
- Vindry, C.; Ohlmann, T.; Chavatte, L. Translation regulation of mammalian selenoproteins. BBA—Gen. Subj. 2018, 1862, 2480–2492. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.L.; Brown, M.S. Regulation of the mevalonate pathway. Nature 1990, 343, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Warner, G.J.; Berry, M.J.; Moustafa, M.E.; Carlson, B.A.; Hatfield, D.L.; Faust, J.R. Inhibition of selenoprotein synthesis by selenocysteine tRNA[Ser]Sec lacking isopentenyladenosine. J. Biol. Chem. 2000, 275, 28110–28119. [Google Scholar] [CrossRef] [PubMed]
- Stipanuk, M.H.; Ueki, I.; Dominy, J.J.; Simmons, C.R.; Hirschberger, L.L. Cysteine dioxygenase: A robust system for regulation of cellular cysteine levels. Amino Acids 2009, 37, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Hao, S.; Yu, J.; He, W.; Huang, Q.; Zhao, Y.; Liang, B.; Zhang, S.; Wen, Z.; Dong, S.; Rao, J.; et al. Cysteine Dioxygenase 1 Mediates Erastin-Induced Ferroptosis in Human Gastric Cancer Cells. Neoplasia 2017, 19, 1022–1032. [Google Scholar] [CrossRef] [PubMed]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Zandkarimi, F.; Zhang, Y.; Meena, J.K.; Kim, J.; Zhuang, L.; Tyagi, S.; Ma, L.; Westbrook, T.F.; Steinberg, G.R.; et al. Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat. Cell Biol. 2020, 22, 225–234. [Google Scholar] [CrossRef]
- Hardie, D.G. AMP-activated protein kinase: An energy sensor that regulates all aspects of cell function. Gene Dev. 2011, 25, 1895–1908. [Google Scholar] [CrossRef]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; Da, S.M.; Ingold, I.; Goya, G.A.; Xavier, D.S.T.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, H.; Kawamura, T.; Muroi, M.; Kondoh, Y.; Honda, K.; Kawatani, M.; Aono, H.; Waldmann, H.; Watanabe, N.; Osada, H. Identification of a Small Molecule That Enhances Ferroptosis via Inhibition of Ferroptosis Suppressor Protein 1 (FSP1). ACS Chem. Biol. 2022, 17, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Grove, J.; Marsh, M. The cell biology of receptor-mediated virus entry. J. Cell Biol. 2011, 195, 1071–1082. [Google Scholar] [CrossRef] [PubMed]
- Mayle, K.M.; Le, A.M.; Kamei, D.T. The intracellular trafficking pathway of transferrin. Biochim. Biophys. Acta 2012, 1820, 264–281. [Google Scholar] [CrossRef]
- Feng, H.; Schorpp, K.; Jin, J.; Yozwiak, C.E.; Hoffstrom, B.G.; Decker, A.M.; Rajbhandari, P.; Stokes, M.E.; Bender, H.G.; Csuka, J.M.; et al. Transferrin Receptor Is a Specific Ferroptosis Marker. Cell Rep. 2020, 30, 3411–3423. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wen, Z.; Cao, H.; Luo, J.; Shuai, L.; Wang, C.; Ge, J.; Wang, X.; Bu, Z.; Wang, J. Transferrin Receptor Protein 1 Is an Entry Factor for Rabies Virus. J. Virol. 2023, 97, e161222. [Google Scholar] [CrossRef] [PubMed]
- Sarute, N.; Ross, S.R. New World Arenavirus Biology. Annu. Rev. Virol. 2017, 4, 141–158. [Google Scholar] [CrossRef] [PubMed]
- Roldan, J.S.; Martinez, M.G.; Forlenza, M.B.; Whittaker, G.R.; Candurra, N.A. Human transferrin receptor triggers an alternative Tacaribe virus internalization pathway. Arch. Virol. 2016, 161, 353–363. [Google Scholar] [CrossRef]
- Martin, V.K.; Droniou-Bonzom, M.E.; Reignier, T.; Oldenburg, J.E.; Cox, A.U.; Cannon, P.M. Investigation of clade B New World arenavirus tropism by using chimeric GP1 proteins. J. Virol. 2010, 84, 1176–1182. [Google Scholar] [CrossRef]
- Sokolov, A.; Isakova-Sivak, I.; Grudinina, N.; Mezhenskaya, D.; Litasova, E.; Kostevich, V.; Stepanova, E.; Rak, A.; Sychev, I.; Kirik, O.; et al. Ferristatin II Efficiently Inhibits SARS-CoV-2 Replication in Vero Cells. Viruses 2022, 14, 317. [Google Scholar] [CrossRef]
- Zhang, S.; Cao, Y.; Yang, Q. Transferrin receptor 1 levels at the cell surface influence the susceptibility of newborn piglets to PEDV infection. PLoS Pathog. 2020, 16, e1008682. [Google Scholar] [CrossRef]
- Mazel-Sanchez, B.; Niu, C.; Williams, N.; Bachmann, M.; Choltus, H.; Silva, F.; Serre-Beinier, V.; Karenovics, W.; Iwaszkiewicz, J.; Zoete, V.; et al. Influenza A virus exploits transferrin receptor recycling to enter host cells. Proc. Natl. Acad. Sci. USA 2023, 120, e2080031176. [Google Scholar] [CrossRef]
- Martin, D.N.; Uprichard, S.L. Identification of transferrin receptor 1 as a hepatitis C virus entry factor. Proc. Natl. Acad. Sci. USA 2013, 110, 10777–10782. [Google Scholar] [CrossRef]
- Wang, G.; Zhang, D.; Orchard, R.; Hancks, D.C.; Reese, T.A. Norovirus MLKL-like pore forming protein initiates programed cell death for viral egress. Biorxiv 2023. [Google Scholar] [CrossRef]
- Matsushita, M.; Freigang, S.; Schneider, C.; Conrad, M.; Bornkamm, G.W.; Kopf, M. T cell lipid peroxidation induces ferroptosis and prevents immunity to infection. J. Exp. Med. 2015, 212, 555–568. [Google Scholar] [CrossRef]
- Jia, M.; Qin, D.; Zhao, C.; Chai, L.; Yu, Z.; Wang, W.; Tong, L.; Lv, L.; Wang, Y.; Rehwinkel, J.; et al. Redox homeostasis maintained by GPX4 facilitates STING activation. Nat. Immunol. 2020, 21, 727–735. [Google Scholar] [CrossRef]
- Hu, Z.; Yin, Y.; Jiang, J.; Yan, C.; Wang, Y.; Wang, D.; Li, L. Exosomal miR-142-3p secreted by hepatitis B virus (HBV)-hepatocellular carcinoma (HCC) cells promotes ferroptosis of M1-type macrophages through SLC3A2 and the mechanism of HCC progression. J. Gastrointest. Oncol. 2022, 13, 754–767. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, J.; Sun, Y.; Stubbs, D.; He, J.; Li, W.; Wang, F.; Liu, Z.; Ruzicka, J.A.; Taylor, E.W.; et al. SARS-CoV-2 suppresses mRNA expression of selenoproteins associated with ferroptosis, endoplasmic reticulum stress and DNA synthesis. Food Chem. Toxicol. 2021, 153, 112286. [Google Scholar] [CrossRef]
- Codo, A.C.; Davanzo, G.G.; Monteiro, L.B.; de Souza, G.F.; Muraro, S.P.; Virgilio-da-Silva, J.V.; Prodonoff, J.S.; Carregari, V.C.; de Biagi, J.C.; Crunfli, F.; et al. Elevated Glucose Levels Favor SARS-CoV-2 Infection and Monocyte Response through a HIF-1alpha/Glycolysis-Dependent Axis. Cell Metab. 2020, 32, 437–446. [Google Scholar] [CrossRef]
- Cheng, J.; Tao, J.; Li, B.; Shi, Y.; Liu, H. Swine influenza virus triggers ferroptosis in A549 cells to enhance virus replication. Virol. J. 2022, 19, 104. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.Z.; Xu, X.W.; Tao, S.H.; Gao, M.J.; Hou, Z.H. HBx facilitates ferroptosis in acute liver failure via EZH2 mediated SLC7A11 suppression. J. Biomed. Sci. 2021, 28, 67. [Google Scholar] [CrossRef] [PubMed]
- Kan, X.; Yin, Y.; Song, C.; Tan, L.; Qiu, X.; Liao, Y.; Liu, W.; Meng, S.; Sun, Y.; Ding, C. Newcastle-disease-virus-induced ferroptosis through nutrient deprivation and ferritinophagy in tumor cells. iScience 2021, 24, 102837. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.; Kim, E.H.; Lee, J.; Roh, J.L. Nrf2 inhibition reverses resistance to GPX4 inhibitor-induced ferroptosis in head and neck cancer. Free Radic. Biol. Med. 2018, 129, 454–462. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.Q.; Xu, T.; Ji, W.; Wang, C.; Ren, Y.; Xiong, X.; Zhou, X.; Lin, S.H.; Xu, Y.; Qiu, Y. Herpes Simplex Virus 1-Induced Ferroptosis Contributes to Viral Encephalitis. mBio 2023, 14, e237022. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Wu, X.; Bing, X.; Qi, W.; Zhu, F.; Guo, N.; Li, C.; Gao, X.; Cao, X.; Zhao, M.; et al. H1N1 influenza virus infection through NRF2-KEAP1-GCLC pathway induces ferroptosis in nasal mucosal epithelial cells. Free Radic. Biol. Med. 2023, 204, 226–242. [Google Scholar] [CrossRef]
- Yuan, L.; Li, S.; Chen, Q.; Xia, T.; Luo, D.; Li, L.; Liu, S.; Guo, S.; Liu, L.; Du, C.; et al. EBV infection-induced GPX4 promotes chemoresistance and tumor progression in nasopharyngeal carcinoma. Cell Death Differ. 2022, 29, 1513–1527. [Google Scholar] [CrossRef]
- He, F.; Zhang, P.; Liu, J.; Wang, R.; Kaufman, R.J.; Yaden, B.C.; Karin, M. ATF4 suppresses hepatocarcinogenesis by inducing SLC7A11 (xCT) to block stress-related ferroptosis. J. Hepatol. 2023, 79, 362–377. [Google Scholar] [CrossRef]
- Gao, R.; Kalathur, R.; Coto-Llerena, M.; Ercan, C.; Buechel, D.; Shuang, S.; Piscuoglio, S.; Dill, M.T.; Camargo, F.D.; Christofori, G.; et al. YAP/TAZ and ATF4 drive resistance to Sorafenib in hepatocellular carcinoma by preventing ferroptosis. EMBO Mol. Med. 2021, 13, e14351. [Google Scholar] [CrossRef]
- Mu, Q.; Chen, L.; Gao, X.; Shen, S.; Sheng, W.; Min, J.; Wang, F. The role of iron homeostasis in remodeling immune function and regulating inflammatory disease. Sci. Bull. 2021, 66, 1806–1816. [Google Scholar] [CrossRef]
- Marx, J.J. Iron and infection: Competition between host and microbes for a precious element. Best Pract. Res. Clin. Haematol. 2002, 15, 411–426. [Google Scholar] [CrossRef]
- Drakesmith, H.; Prentice, A. Viral infection and iron metabolism. Nat. Rev. Microbiol. 2008, 6, 541–552. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.B.; Donovan, A.; Ward, D.M.; Ganz, T.; Kaplan, J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004, 306, 2090–2093. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Li, R.; Cheng, G.; Li, Z.; Zhang, Z.; Li, J.; Zhang, G.; Bi, C.; Hu, C.; Yang, L.; et al. Role of hepcidin and iron metabolism in the onset of prostate cancer. Oncol. Lett. 2018, 15, 9953–9958. [Google Scholar] [CrossRef]
- Chang, H.C.; Bayeva, M.; Taiwo, B.; Palella, F.J.; Hope, T.J.; Ardehali, H. Short communication: High cellular iron levels are associated with increased HIV infection and replication. Aids Res. Hum. Retrov. 2015, 31, 305–312. [Google Scholar] [CrossRef]
- Halcrow, P.W.; Lakpa, K.L.; Khan, N.; Afghah, Z.; Miller, N.; Datta, G.; Chen, X.; Geiger, J.D. HIV-1 gp120-Induced Endolysosome de-Acidification Leads to Efflux of Endolysosome Iron, and Increases in Mitochondrial Iron and Reactive Oxygen Species. J. Neuroimmune Pharm. 2022, 17, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.; Halcrow, P.W.; Afghah, Z.; Baral, A.; Geiger, J.D.; Chen, X. HIV-1 Tat endocytosis and retention in endolysosomes affects HIV-1 Tat-induced LTR transactivation in astrocytes. FASEB J. 2022, 36, e22184. [Google Scholar] [CrossRef] [PubMed]
- Yi, L.; Hu, Y.; Wu, Z.; Li, Y.; Kong, M.; Kang, Z.; Zuoyuan, B.; Yang, Z. TFRC upregulation promotes ferroptosis in CVB3 infection via nucleus recruitment of Sp1. Cell Death Dis. 2022, 13, 592. [Google Scholar] [CrossRef]
- Majidpoor, J.; Mortezaee, K. Interleukin-6 in SARS-CoV-2 induced disease: Interactions and therapeutic applications. Biomed. Pharmacother. 2022, 145, 112419. [Google Scholar] [CrossRef]
- Kobune, M.; Kohgo, Y.; Kato, J.; Miyazaki, E.; Niitsu, Y. Interleukin-6 enhances hepatic transferrin uptake and ferritin expression in rats. Hepatology 1994, 19, 1468–1475. [Google Scholar] [CrossRef]
- Nemeth, E.; Rivera, S.; Gabayan, V.; Keller, C.; Taudorf, S.; Pedersen, B.K.; Ganz, T. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J. Clin. Investig. 2004, 113, 1271–1276. [Google Scholar] [CrossRef]
- Jia, F.; Liu, H.; Kang, S. NCOA4-Mediated Ferritinophagy: A Vicious Culprit in COVID-19 Pathogenesis? Front. Mol. Biosci. 2021, 8, 761793. [Google Scholar] [CrossRef] [PubMed]
- Silva, I.S.; Perez, R.M.; Oliveira, P.V.; Cantagalo, M.I.; Dantas, E.; Sisti, C.; Figueiredo-Mendes, C.; Lanzoni, V.P.; Silva, A.; Ferraz, M.L. Iron overload in patients with chronic hepatitis C virus infection: Clinical and histological study. J. Gastroen. Hepatol. 2005, 20, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Kanda, T.; Goto, T.; Hirotsu, Y.; Moriyama, M.; Omata, M. Molecular Mechanisms Driving Progression of Liver Cirrhosis towards Hepatocellular Carcinoma in Chronic Hepatitis B and C Infections: A Review. Int. J. Mol. Sci. 2019, 20, 1358. [Google Scholar] [CrossRef] [PubMed]
- Foka, P.; Dimitriadis, A.; Karamichali, E.; Kyratzopoulou, E.; Giannimaras, D.; Koskinas, J.; Varaklioti, A.; Mamalaki, A.; Georgopoulou, U. Alterations in the iron homeostasis network: A driving force for macrophage-mediated hepatitis C virus persistency. Virulence 2016, 7, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.Z.; Xiao, X.Q.; Cheng, D.; Jiang, Y.F.; Gong, G.Z. HCV NS5A protein down-regulates hepcidin gene expression and increases hepatic intracellular iron storage. Zhonghua Gan Zang Bing Za Zhi 2011, 19, 894–897. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; An, P.; Xie, E.; Wu, Q.; Fang, X.; Gao, H.; Zhang, Z.; Li, Y.; Wang, X.; Zhang, J.; et al. Characterization of ferroptosis in murine models of hemochromatosis. Hepatology 2017, 66, 449–465. [Google Scholar] [CrossRef]
- Liu, L.; Lv, Z.; Wang, M.; Zhang, D.; Liu, D.; Zhu, F. HBV Enhances Sorafenib Resistance in Hepatocellular Carcinoma by Reducing Ferroptosis via SRSF2-Mediated Abnormal PCLAF Splicing. Int. J. Mol. Sci. 2023, 24, 3263. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, M.; Zhao, L.; Geng, Y.; Li, G.; Chen, L.; Yu, J.; Yuan, H.; Zhang, H.; Yun, H.; et al. HBx-Induced HSPA8 Stimulates HBV Replication and Suppresses Ferroptosis to Support Liver Cancer Progression. Cancer Res. 2023, 83, 1048–1061. [Google Scholar] [CrossRef]
- Zhang, Q.; Qu, Y.; Zhang, Q.; Li, F.; Li, B.; Li, Z.; Dong, Y.; Lu, L.; Cai, X. Exosomes derived from hepatitis B virus-infected hepatocytes promote liver fibrosis via miR-222/TFRC axis. Cell Biol. Toxicol. 2023, 39, 467–481. [Google Scholar] [CrossRef]
- Kung, Y.A.; Chiang, H.J.; Li, M.L.; Gong, Y.N.; Chiu, H.P.; Hung, C.T.; Huang, P.N.; Huang, S.Y.; Wang, P.Y.; Hsu, T.A.; et al. Acyl-Coenzyme A Synthetase Long-Chain Family Member 4 Is Involved in Viral Replication Organelle Formation and Facilitates Virus Replication via Ferroptosis. Mbio 2022, 13, e02717-21. [Google Scholar] [CrossRef]
- Kannan, M.; Sil, S.; Oladapo, A.; Thangaraj, A.; Periyasamy, P.; Buch, S. HIV-1 Tat-mediated microglial ferroptosis involves the miR-204-ACSL4 signaling axis. Redox Biol. 2023, 62, 102689. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Zhang, Z.; You, F. Inhibiting ACSL1-Related Ferroptosis Restrains Murine Coronavirus Infection. Viruses 2021, 13, 2383. [Google Scholar] [CrossRef] [PubMed]
- Yamane, D.; Hayashi, Y.; Matsumoto, M.; Nakanishi, H.; Imagawa, H.; Kohara, M.; Lemon, S.M.; Ichi, I. FADS2-dependent fatty acid desaturation dictates cellular sensitivity to ferroptosis and permissiveness for hepatitis C virus replication. Cell Chem. Biol. 2022, 29, 799–810. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fuction | Target | Molecule | Chemical Structure | Action Site |
|---|---|---|---|---|
| Inducer | System Xc− | Erastin [11] | 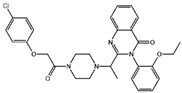 | Erastin prevents inhibition of microtubule proteins on VDAC, causing an increase in mitochondrial metabolism and promoting ROS. Erastin reduces cystine input by inhibiting System Xc−. |
| Sulfasalazine (SAS) [12] | 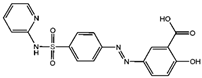 | SSZ reduces cystine input by inhibiting System Xc−. | ||
| Sorafenib [13] | 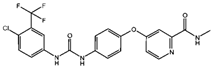 | Sorafenib binds to System Xc− and inhibits its activity. | ||
| Glutamate [7] | 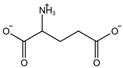 | High extracellular concentrations of Glutamate affect glutamate and cystine transport and thus System Xc−. | ||
| Levobupivacaine [14] | 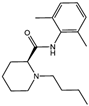 | Levobupivacaine inhibits SLC7A11 or GPX4 by upregulating miR-489-3p to induced ferroptosis. | ||
| GPX4 | (1S,3R)-RSL3 [15] | 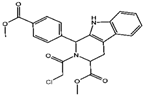 | RSL3 directly inactivates GPX4 by covalently binding to Sec46 of GPX4. | |
| FIN56 [16] | 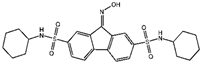 | FIN56 promotes the degradation of GPX4 and leads to CoQ10 depletion by binding to squalene synthase. | ||
| Ketamine [17,18] | 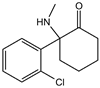 | Ketamine inhibits GPX4 expression via lncRNA PVT1 or GPX4 transcript levels. | ||
| GSH | Cisplatin (Cis) [19,20] | 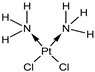 | Cis binds to GSH forming a complex thereby reducing GSH concentration. | |
| ROS or iron | Dihydroartemisinin (DHA) [21,22,23] |  | DHA induces ferritin degradation, upregulates TfR to increase free iron levels promoting ROS deposition and downregulates System Xc−. | |
| Inducer | ROS or iron | Artesunate (ART) [24,25] |  | ART acts similarly to DHA in promoting ferritin degradation and ROS Accumulation. |
| FINO2 [26] |  | FINO2 undergoes the Fenton reaction with Fe2+ and oxidizes it to Fe3+ leading to the production of free radicals. | ||
| Vitamin C [27,28] | 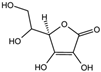 | Vitamin C induces ferritin degradation, leading to free iron release. | ||
| Sodium arsenite (NaAsO2) [29] | 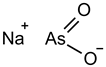 | NaAsO2 induces ROS release by inducing mitochondrial damage and enhances ferritin degradation thereby promoting Fe2+ release. | ||
| Inhibitor | ROS or iron | DFO (Deferoxamine) [30] | 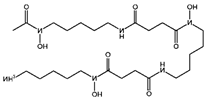 | DFO binds to intracellular iron and prevents its participation in the Fenton reaction that generates ROS. |
| Fer-1 (Ferrostatin-1) [7] | 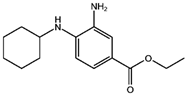 | Fer-1 inhibits lipid peroxidation by trapping free radicals and stabilizing lipid peroxides. And it can it binds to free Fe2+ to reduce mitochondrial Fe2+ content. | ||
| VE (Vitamin E) [31] | 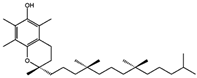 | Vitamin E forms hydroquinone substances and subsequently interact with iron in the active center of the LOXs enzyme. | ||
| liproxstatin-1 (Lip-1) [32,33] | 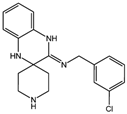 | Lip-1 stays inside the lipid bilayer and removes ROS and also activates Nrf2 to restore GPX4 levels. | ||
| LOX | Baicalein [34,35] |  | Baicalein chelates iron and inhibits 12/15-LOX activity. | |
| zileuton [36] | 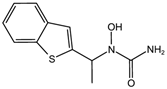 | Zileuton inhibits 5-LOX activity. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, R.; Wu, J.; Ma, Y.; Kang, K. Molecular Mechanisms of Ferroptosis and Its Role in Viral Pathogenesis. Viruses 2023, 15, 2373. https://doi.org/10.3390/v15122373
Huang R, Wu J, Ma Y, Kang K. Molecular Mechanisms of Ferroptosis and Its Role in Viral Pathogenesis. Viruses. 2023; 15(12):2373. https://doi.org/10.3390/v15122373
Chicago/Turabian StyleHuang, Riwei, Jiang Wu, Yaodan Ma, and Kai Kang. 2023. "Molecular Mechanisms of Ferroptosis and Its Role in Viral Pathogenesis" Viruses 15, no. 12: 2373. https://doi.org/10.3390/v15122373
APA StyleHuang, R., Wu, J., Ma, Y., & Kang, K. (2023). Molecular Mechanisms of Ferroptosis and Its Role in Viral Pathogenesis. Viruses, 15(12), 2373. https://doi.org/10.3390/v15122373