
Challenges for Precise Subtyping and Sequencing of a H5N1 Clade 2.3.4.4b Highly Pathogenic Avian Influenza Virus Isolated in Japan in the 2022–2023 Season Using Classical Serological and Molecular Methods
 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Virus Isolation
2.2. RNA Extraction, Real-Time RT-PCR, and Subtype-Specific RT-PCR
2.3. HA Subtyping and Antigenic Characterization by the HI Test
2.4. Amplification and Sanger Sequencing of the HA Gene Using Hoffmann’s Primers
2.5. NGS and Phylogenetic Analysis
3. Results
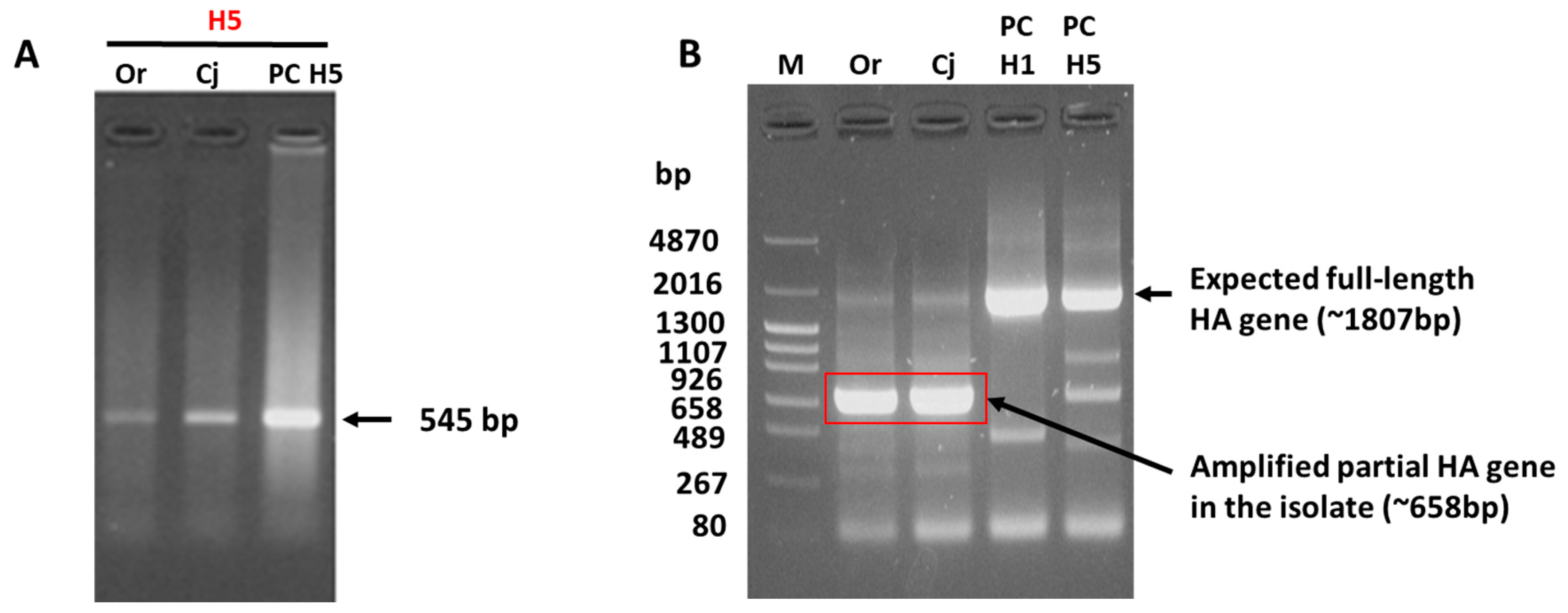
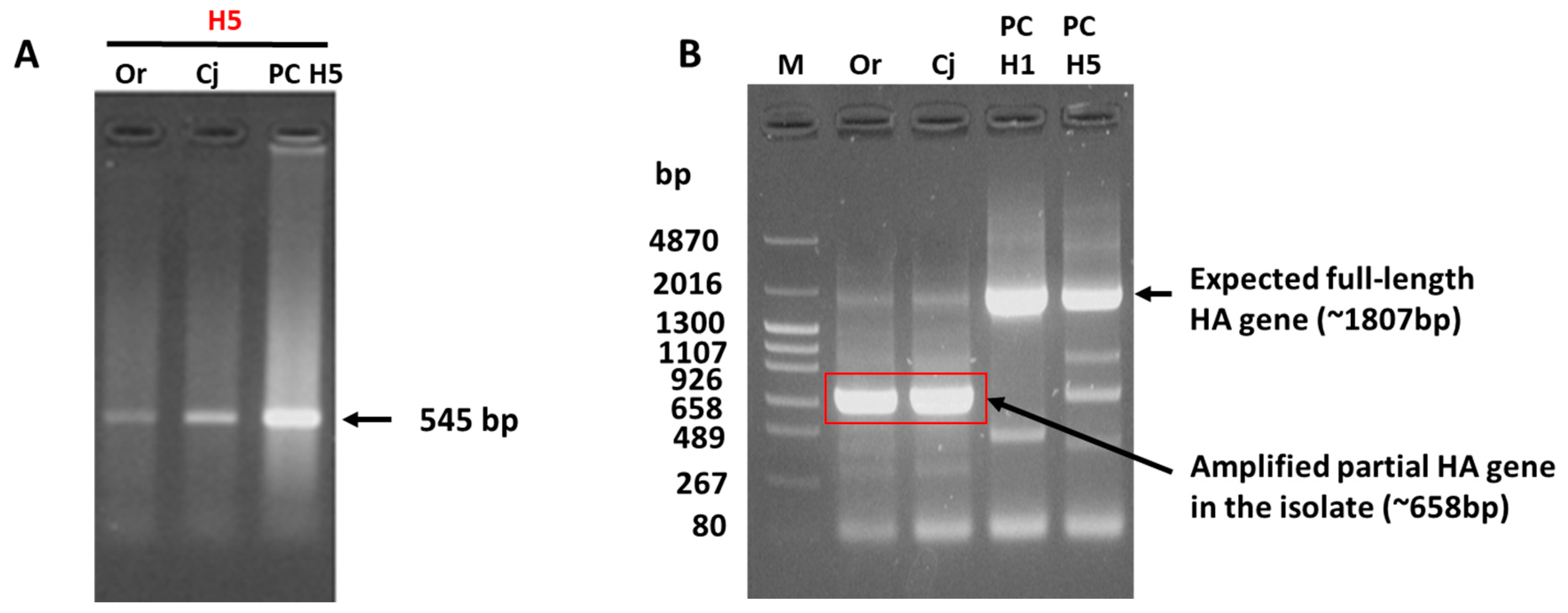
3.1. Failed HA Subtyping and Full-Length HA Gene Amplification of the H5N1 Isolate by Classical Methods
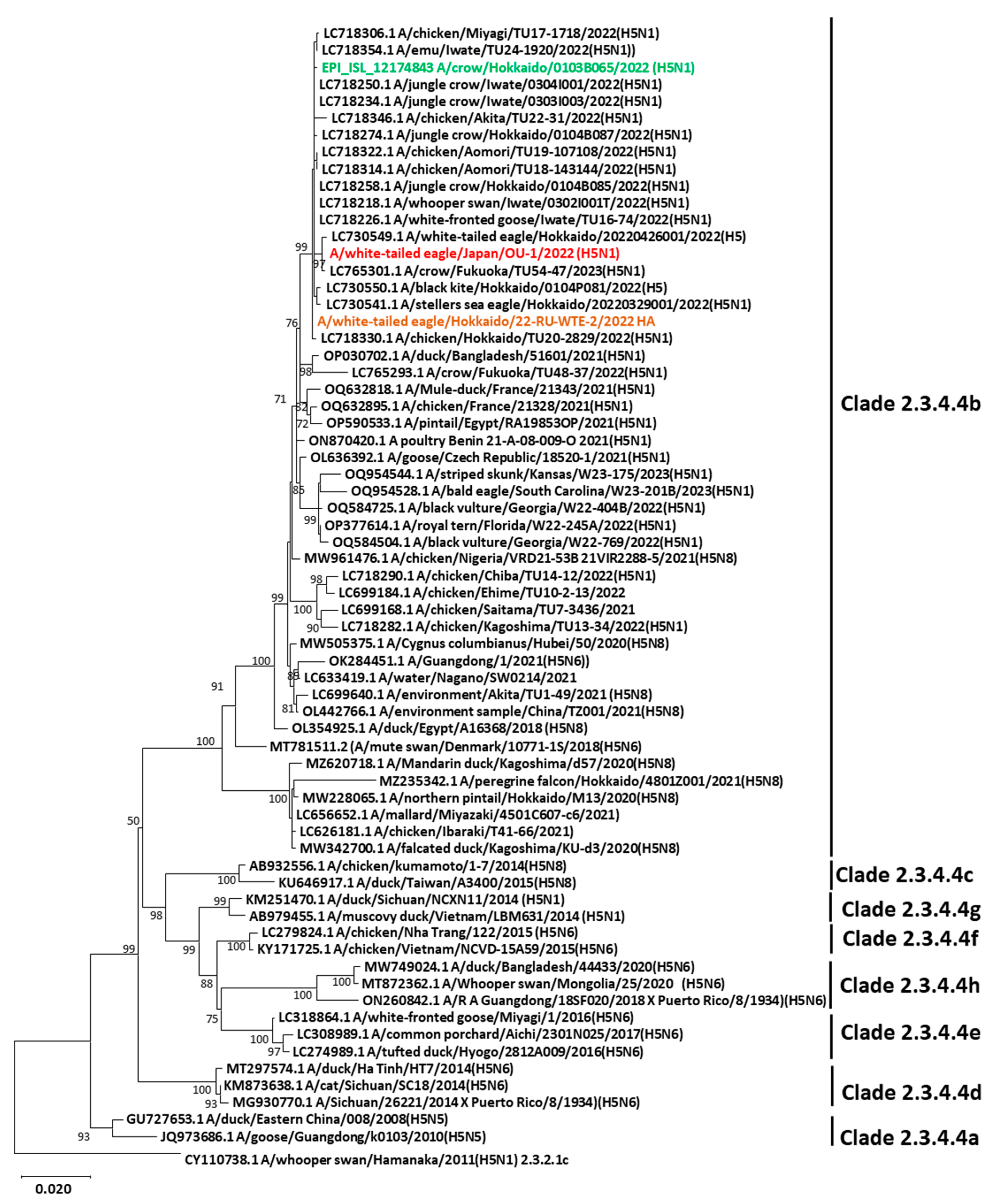
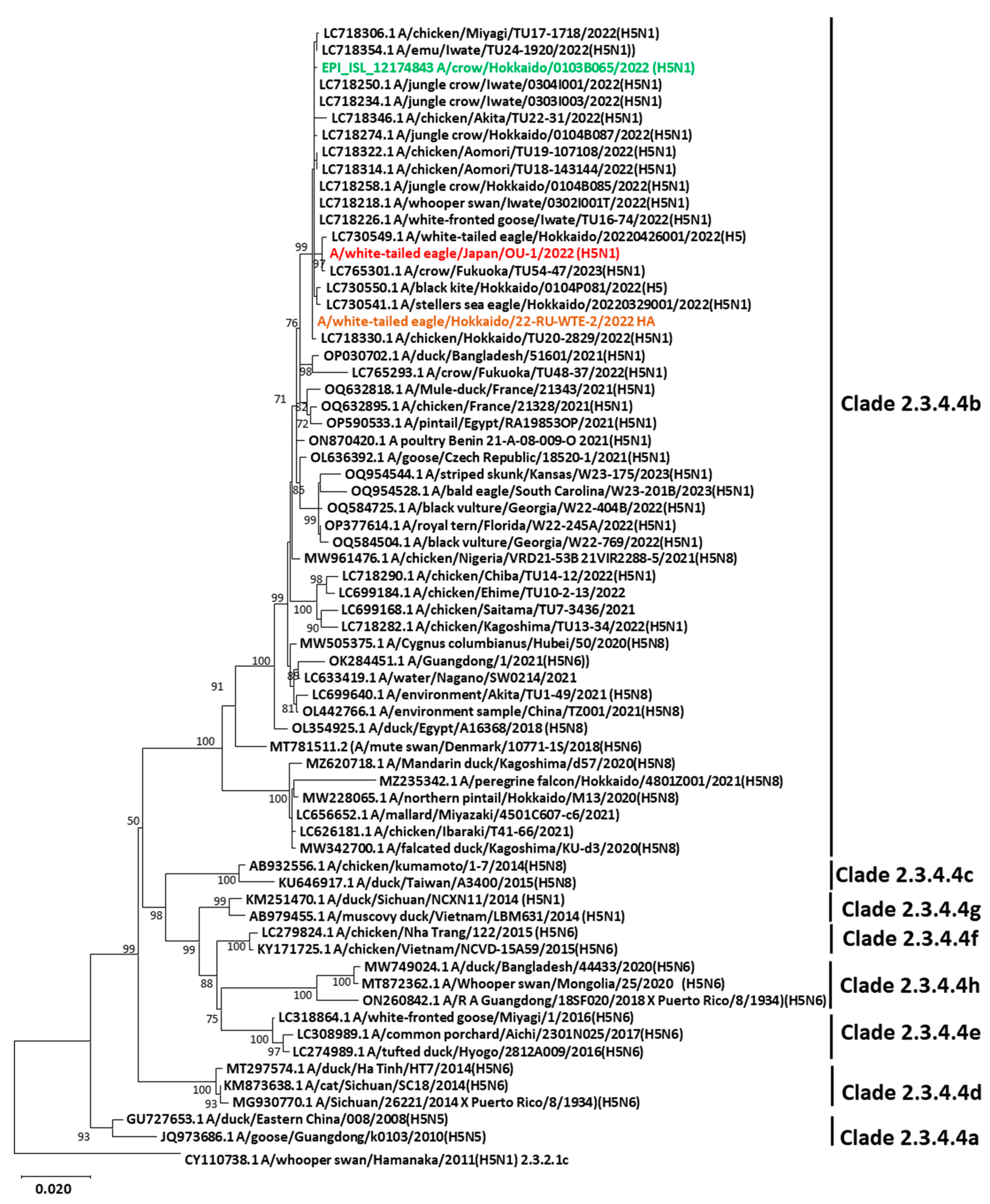
3.2. Genetic Characterization and Phylogenetic Analysis
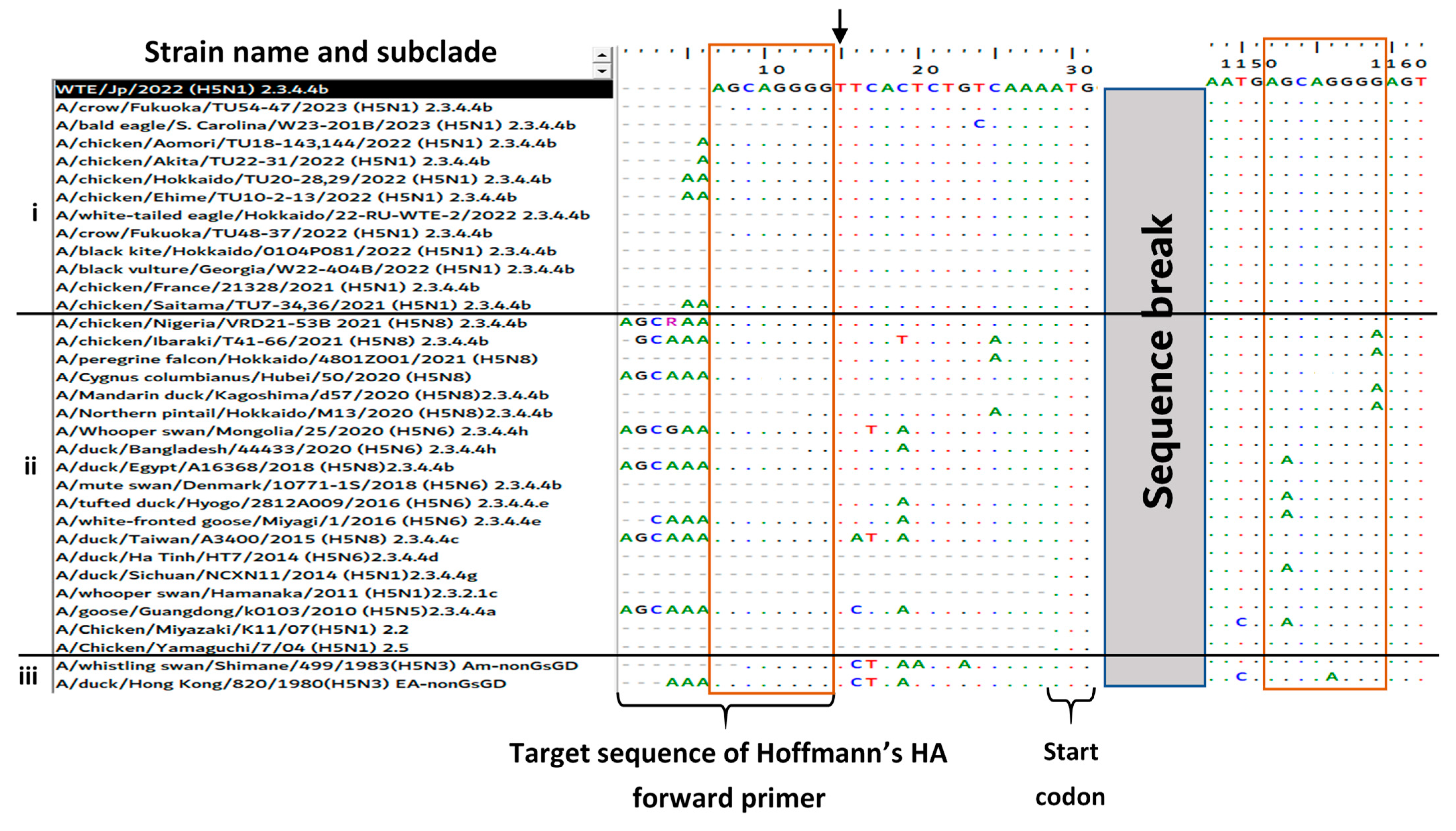
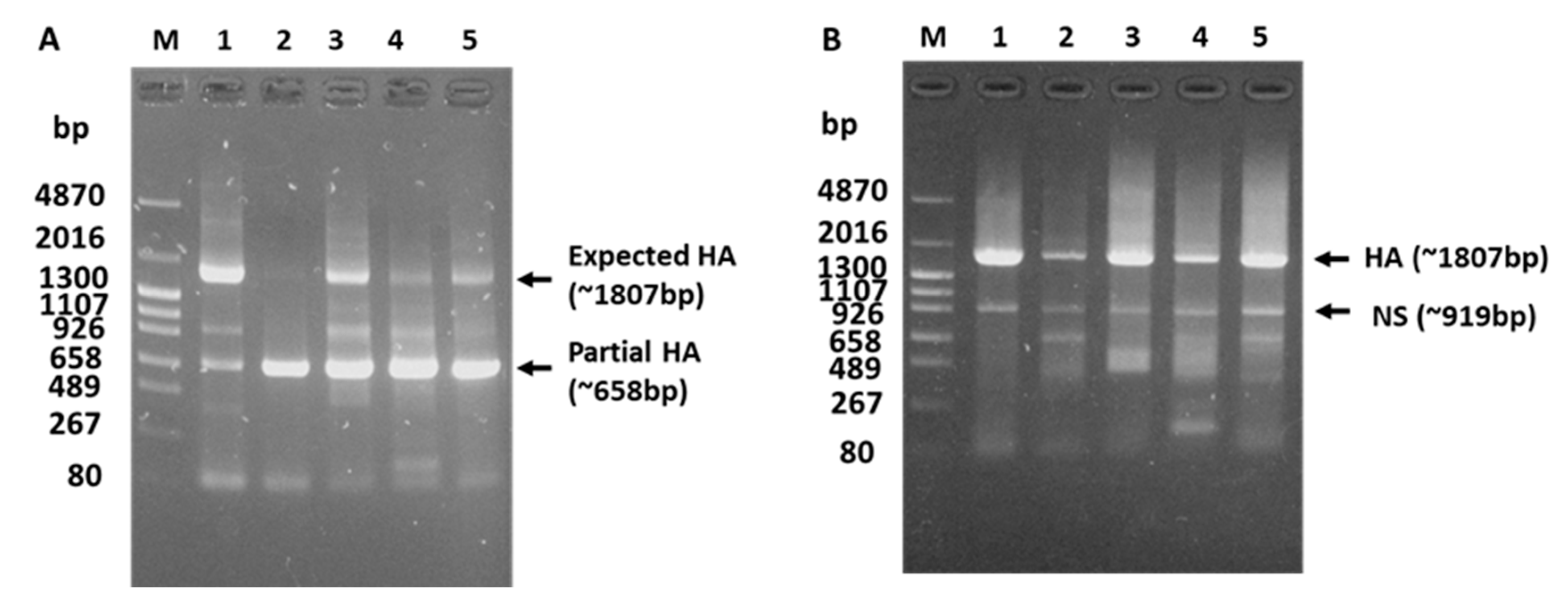
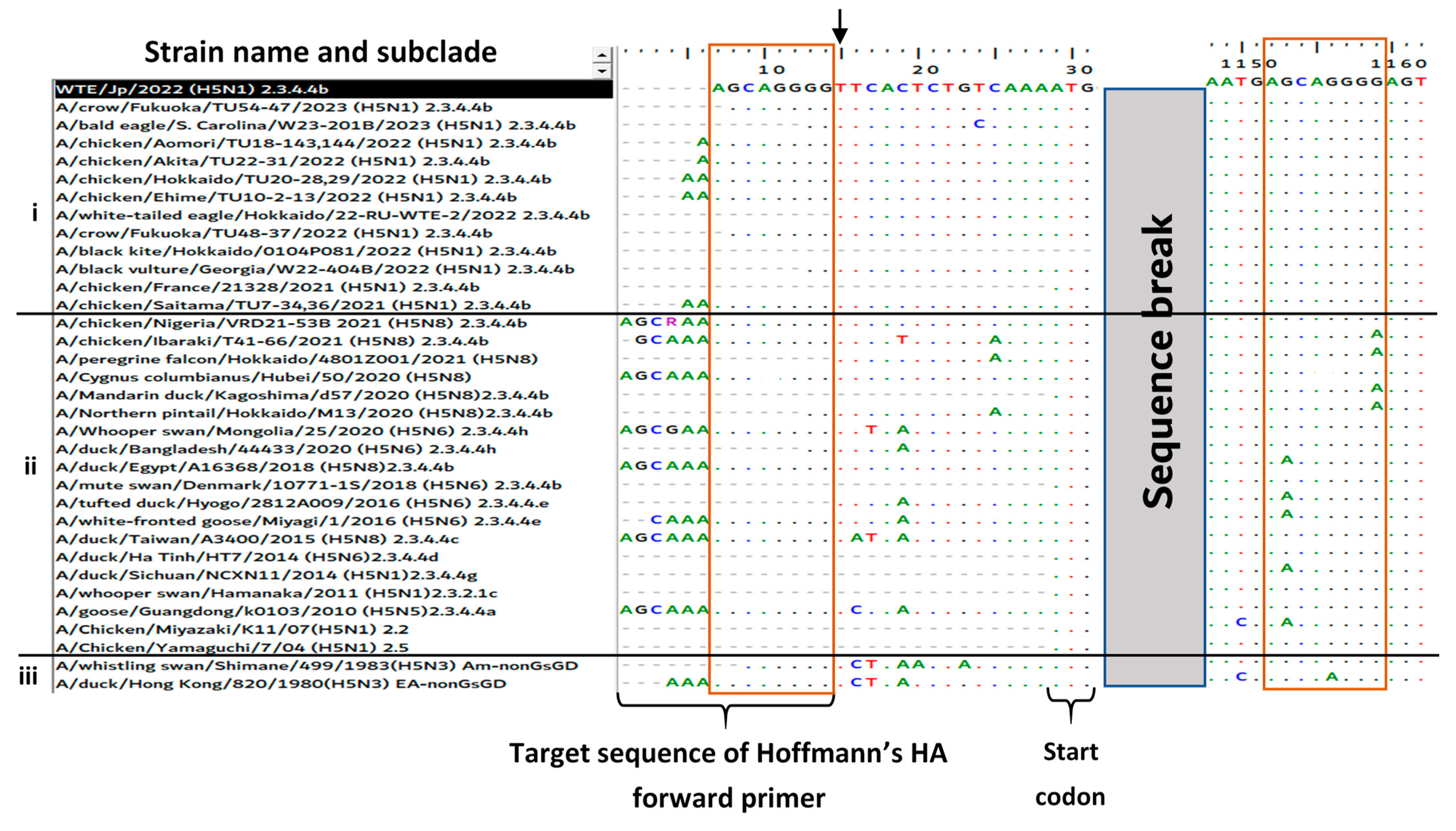
3.3. Reason for the Failed Full-Length Amplification of the HA Gene
3.4. Antigenic Characterization
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sonnberg, S.; Webby, R.J.; Webster, R.G. Natural history of highly pathogenic avian influenza H5N1. Virus Res. 2013, 178, 63–77. [Google Scholar] [CrossRef]
- Gutiérrez, R.A.; Naughtin, M.J.; Horm, S.V.; San, S.; Buchy, P. A(H5N1) virus evolution in South East Asia. Viruses 2009, 1, 335–361. [Google Scholar] [CrossRef]
- Sims, L.D.; Domenech, J.; Benigno, C.; Kahn, S.; Kamata, A.; Lubroth, J.; Martin, V.; Roeder, P. Origin and evolution of highly pathogenic H5N1 avian influenza in Asia. Vet. Rec. 2005, 157, 159–164. [Google Scholar] [CrossRef]
- WHO/OIE/FAO H5N1 Evolution Working Group. Toward a unified nomenclature system for highly pathogenic avian influenza virus (H5N1). Emerg. Infect. Dis. 2008, 14, e1. [Google Scholar] [CrossRef]
- Claes, F.; Morzaria, S.P.; Donis, R.O. Emergence and dissemination of clade 2.3.4.4 H5Nx influenza viruses—How is the Asian HPAI H5 lineage maintained. Curr. Opin. Virol. 2016, 16, 158–163. [Google Scholar] [CrossRef]
- Lee, D.H.; Bertran, K.; Kwon, J.H.; Swayne, D.E. Evolution, global spread, and pathogenicity of highly pathogenic avian influenza H5Nx clade 2.3.4.4. J. Vet. Sci. 2017, 18, 269–280. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. Highlights in the History of Avian Influenza (Bird Flu) Timeline—2020–2023. Available online: https://www.cdc.gov/flu/avianflu/timeline/avian-timeline-2020s.htm (accessed on 28 May 2023).
- Okuya, K.; Mine, J.; Tokorozaki, K.; Kojima, I.; Esaki, M.; Miyazawa, K.; Tsunekuni, R.; Sakuma, S.; Kumagai, A.; Takadate, Y.; et al. Genetically diverse highly pathogenic avian influenza A(H5N1/H5N8) viruses among wild waterfowl and domestic poultry, Japan, 2021. Emerg. Infect. Dis. 2022, 28, 1451–1455. [Google Scholar] [CrossRef]
- Hiono, T.; Kobayashi, D.; Kobayashi, A.; Suzuki, T.; Satake, Y.; Harada, R.; Matsuno, K.; Sashika, M.; Ban, H.; Kobayashi, M.; et al. Virological, pathological, and glycovirological investigations of an Ezo red fox and a tanuki naturally infected with H5N1 high pathogenicity avian influenza viruses in Hokkaido, Japan. Virology 2023, 578, 35–44. [Google Scholar] [CrossRef]
- Isoda, N.; Onuma, M.; Hiono, T.; Sobolev, I.; Lim, H.Y.; Nabeshima, K.; Honjyo, H.; Yokoyama, M.; Shestopalov, A.; Sakoda, Y. Detection of new H5N1 high pathogenicity avian influenza viruses in winter 2021–2022 in the far east, which are genetically close to those in Europe. Viruses 2022, 14, 2168. [Google Scholar] [CrossRef]
- Ministry of Agriculture Forestry and Fisheries. Information on Avian Influenza in FY. 2022. Available online: https://www.maff.go.jp/j/syouan/douei/tori/220929.html#2 (accessed on 20 June 2023). (In Japanese)
- World Health Organization. 2002 WHO Manual on Animal Influenza Diagnosis and Surveillance (No. WHO/CDS/CSR/NCS/2002.5). Available online: https://apps.who.int/iris/bitstream/handle/10665/68026/who_cds?sequence=1 (accessed on 29 May 2023).
- Tseng, C.H.; Tsai, H.J.; Chang, C.M. A complete molecular diagnostic procedure for applications in surveillance and subtyping of avian influenza virus. BioMed Res. Int. 2014, 2014, 653056. [Google Scholar] [CrossRef]
- Qiu, B.F.; Liu, W.J.; Peng, D.X.; Hu, S.L.; Tang, Y.H.; Liu, X.F. A reverse transcription-PCR for subtyping of the neuraminidase of avian influenza viruses. J. Virol. Methods 2009, 155, 193–198. [Google Scholar] [CrossRef]
- Tsukamoto, K.; Ashizawa, H.; Nakanishi, K.; Kaji, N.; Suzuki, K.; Okamatsu, M.; Yamaguchi, S.; Mase, M. Subtyping of avian influenza viruses H1 to H15 on the basis of hemagglutinin genes by PCR assay and molecular determination of pathogenic potential. J. Clin. Microbiol. 2008, 46, 3048–3055. [Google Scholar] [CrossRef]
- Fereidouni, S.R.; Starick, E.; Grund, C.; Globig, A.; Mettenleiter, T.C.; Beer, M.; Harder, T. Rapid molecular subtyping by reverse transcription polymerase chain reaction of the neuraminidase gene of avian influenza A viruses. Vet. Microbiol. 2009, 135, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Chang, P.C.; Shien, J.H.; Cheng, M.C.; Shieh, H.K. Identification and subtyping of avian influenza viruses by reverse transcription-PCR. J. Virol. Methods 2001, 97, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, E.; Stech, J.; Guan, Y.; Webster, R.G.; Perez, D.R. Universal primer set for the full-length amplification of all influenza A viruses. Arch. Virol. 2001, 146, 2275–2289. [Google Scholar] [CrossRef] [PubMed]
- Ministry of Environment. Information on Avian Influenza. Available online: https://www.env.go.jp/nature/dobutsu/bird_flu/ (accessed on 25 June 2023). (In Japanese)
- Runstadler, J.A.; Happ, G.M.; Slemons, R.D.; Sheng, Z.M.; Gundlach, N.; Petrula, M.; Senne, D.; Nolting, J.; Evers, D.L.; Modrell, A.; et al. Using RRT-PCR analysis and virus isolation to determine the prevalence of avian influenza virus infections in ducks at Minto Flats State Game Refuge, Alaska, during August 2005. Arch. Virol. 2007, 152, 1901–1910. [Google Scholar] [CrossRef] [PubMed]
- Gronsang, D.; Bui, A.N.; Trinh, D.Q.; Bui, V.N.; Nguyen, K.V.; Can, M.X.; Omatsu, T.; Mizutani, T.; Nagai, M.; Katayama, Y.; et al. Characterization of cross-clade monoclonal antibodies against H5N1 highly pathogenic avian influenza virus and their application to the antigenic analysis of diverse H5 subtype viruses. Arch. Virol. 2017, 162, 2257–2269. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993, 10, 512–526. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Ministry of Environment. Red List 2020, Appendix 3. Available online: https://www.env.go.jp/press/107905.html (accessed on 23 September 2023). (In Japanese)
- Liang, Y.; Krog, J.S.; Ryt-Hansen, P.; Pedersen, A.G.; Kvisgaard, L.K.; Holm, E.; Nielsen, P.D.; Hammer, A.S.; Madsen, J.J.; Thorup, K.; et al. Molecular characterization of highly pathogenic avian influenza viruses H5N6 detected in Denmark in 2018–2019. Viruses 2021, 13, 1052. [Google Scholar] [CrossRef]
- Fujimoto, Y.; Ogasawara, K.; Isoda, N.; Hatai, H.; Okuya, K.; Watanabe, Y.; Takada, A.; Sakoda, Y.; Saito, K.; Ozawa, M. Experimental and natural infections of white-tailed sea eagles (Haliaeetus albicilla) with high pathogenicity avian influenza virus of H5 subtype. Front. Microbiol. 2022, 13, 1007350. [Google Scholar] [CrossRef] [PubMed]
- Krone, O.; Globig, A.; Ulrich, R.; Harder, T.; Schinköthe, J.; Herrmann, C.; Gerst, S.; Conraths, F.; Beer, M. White-tailed sea eagle (Haliaeetus albicilla) die-off due to infection with highly pathogenic avian influenza virus, subtype H5N8, in Germany. Viruses 2018, 10, 478. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Report for the 11th Meeting of the WHO Working Group for the Molecular Detection and Subtyping of Influenza Viruses and the Use of NGS in GISRS. 2021. Available online: https://www.who.int/publications/m/item/report-for-the-11th-meeting-of-the-who-working-group-for-the-molecular-detection-and-subtyping-of-influenza-viruses-and-the-use-of-ngs-in-gisrs (accessed on 28 July 2023).
- Swayne David, E. (Ed.) Avian Influenza, 1st ed.; John Wiley & Sons: Ames, IA, USA, 2009. [Google Scholar]
- Bui, V.N.; Ogawa, H.; Ngo, L.H.; Baatartsogt, T.; Abao, L.N.B.; Tamaki, S.; Saito, K.; Watanabe, Y.; Runstadler, J.; Imai, K. H5N1 highly pathogenic avian influenza virus isolated from conjunctiva of a whooper swan with neurological signs. Arch. Virol. 2013, 158, 451–455. [Google Scholar] [CrossRef]
- Bui, V.N.; Dao, T.D.; Nguyen, T.T.H.; Nguyen, L.T.; Bui, A.N.; Trinh, D.Q.; Pham, N.T.; Inui, K.; Runstadler, J.; Ogawa, H.; et al. Pathogenicity of an H5N1 avian influenza virus isolated in Vietnam in 2012 and reliability of conjunctival samples for diagnosis of infection. Virus Res. 2014, 179, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Soda, K.; Tomioka, Y.; Hidaka, C.; Matsushita, M.; Usui, T.; Yamaguchi, T. Susceptibility of common family Anatidae bird species to clade 2.3.4.4e H5N6 high pathogenicity avian influenza virus: An experimental infection study. BMC Vet. Res. 2022, 18, 127. [Google Scholar] [CrossRef]
- Usui, T.; Soda, K.; Sumi, K.; Ozaki, H.; Tomioka, Y.; Ito, H.; Murase, T.; Kawamoto, T.; Miura, M.; Komatsu, M.; et al. Outbreaks of highly pathogenic avian influenza in zoo birds caused by HA clade 2.3.4.4 H5N6 subtype viruses in Japan in winter 2016. Transbound. Emerg. Dis. 2020, 67, 686–697. [Google Scholar] [CrossRef]
- Ohkawara, A.; Okamatsu, M.; Ozawa, M.; Chu, D.H.; Nguyen, L.T.; Hiono, T.; Matsuno, K.; Kida, H.; Sakoda, Y. Antigenic diversity of H5 highly pathogenic avian influenza viruses of clade 2.3.4.4 isolated in Asia. Microbiol. Immunol. 2017, 61, 149–158. [Google Scholar] [CrossRef]
- Arafa, A.S.; Selim, A.A.; Hassan, M.K.; Aly, M.M. Genetic characterization of variant strains of highly pathogenic avian influenza H5N1 that escaped detection by real-time reverse transcriptase–PCR diagnostic tests. Avian Dis. 2010, 54 (Suppl. S1), 673–676. [Google Scholar] [CrossRef]
- Klungthong, C.; Chinnawirotpisan, P.; Hussem, K.; Phonpakobsin, T.; Manasatienkij, W.; Ajariyakhajorn, C.; Rungrojcharoenkit, K.; Gibbons, R.V.; Jarman, R.G. The impact of primer and probe-template mismatches on the sensitivity of pandemic influenza A/H1N1/2009 virus detection by real-time RT-PCR. J. Clin. Virol. 2010, 48, 91–95. [Google Scholar] [CrossRef]
- Slomka, M.J.; To, T.L.; Tong, H.H.; Coward, V.J.; Hanna, A.; Shell, W.; Pavlidis, T.; Densham, A.L.E.; Kargiolakis, G.; Arnold, M.E.; et al. Challenges for accurate and prompt molecular diagnosis of clades of highly pathogenic avian influenza H5N1 viruses emerging in Vietnam. Avian Pathol. 2012, 41, 177–193. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Swab Sample | Ct Value | Hemagglutination Titer | |
|---|---|---|---|
| Original | AF | AF | |
| Oropharyngeal | 36.35 | 21.81 | 256 |
| Conjunctival | 33.51 | 20.93 | 512 |
| Cloacal | 34.86 | * NT | <2 |
| Virus | Subtype | Year Isolated | Clade | Antiserum | mAbs | ||||
|---|---|---|---|---|---|---|---|---|---|
| * Dk/Hk | Ck/Mz | WTE/Hk | 3B5.1 | 3B5.2 | 1G5 | ||||
| Dk/Hk | H5N3 | 1980 | nonGs/GD | 1280 | 1280 | 320 | 1280 | 160 | 80 |
| Ck/Ym | H5N1 | 2004 | 2.5 | 640 | >5120 | 80 | 2560 | 640 | 80 |
| Ck/Mz | H5N1 | 2007 | 2.2 | 1280 | 2560 | 80 | 640 | 640 | 320 |
| Ws/Hm | H5N1 | 2011 | 2.3.2.1 | 80 | 160 | <40 | 320 | 20 | <10 |
| WTE/Jp | H5N1 | 2022 | 2.3.4.4b | <10 | 80 | 1280 | <10 | <10 | <10 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Komu, J.G.; Nguyen, H.D.; Takeda, Y.; Fukumoto, S.; Imai, K.; Takemae, H.; Mizutani, T.; Ogawa, H. Challenges for Precise Subtyping and Sequencing of a H5N1 Clade 2.3.4.4b Highly Pathogenic Avian Influenza Virus Isolated in Japan in the 2022–2023 Season Using Classical Serological and Molecular Methods. Viruses 2023, 15, 2274. https://doi.org/10.3390/v15112274
Komu JG, Nguyen HD, Takeda Y, Fukumoto S, Imai K, Takemae H, Mizutani T, Ogawa H. Challenges for Precise Subtyping and Sequencing of a H5N1 Clade 2.3.4.4b Highly Pathogenic Avian Influenza Virus Isolated in Japan in the 2022–2023 Season Using Classical Serological and Molecular Methods. Viruses. 2023; 15(11):2274. https://doi.org/10.3390/v15112274
Chicago/Turabian StyleKomu, James G., Hiep Dinh Nguyen, Yohei Takeda, Shinya Fukumoto, Kunitoshi Imai, Hitoshi Takemae, Tetsuya Mizutani, and Haruko Ogawa. 2023. "Challenges for Precise Subtyping and Sequencing of a H5N1 Clade 2.3.4.4b Highly Pathogenic Avian Influenza Virus Isolated in Japan in the 2022–2023 Season Using Classical Serological and Molecular Methods" Viruses 15, no. 11: 2274. https://doi.org/10.3390/v15112274
APA StyleKomu, J. G., Nguyen, H. D., Takeda, Y., Fukumoto, S., Imai, K., Takemae, H., Mizutani, T., & Ogawa, H. (2023). Challenges for Precise Subtyping and Sequencing of a H5N1 Clade 2.3.4.4b Highly Pathogenic Avian Influenza Virus Isolated in Japan in the 2022–2023 Season Using Classical Serological and Molecular Methods. Viruses, 15(11), 2274. https://doi.org/10.3390/v15112274