
HantaNet: A New MicrobeTrace Application for Hantavirus Classification, Genomic Surveillance, Epidemiology and Outbreak Investigations
 ,
,  , and
, and 
Abstract
:1. Introduction
2. Materials and Methods
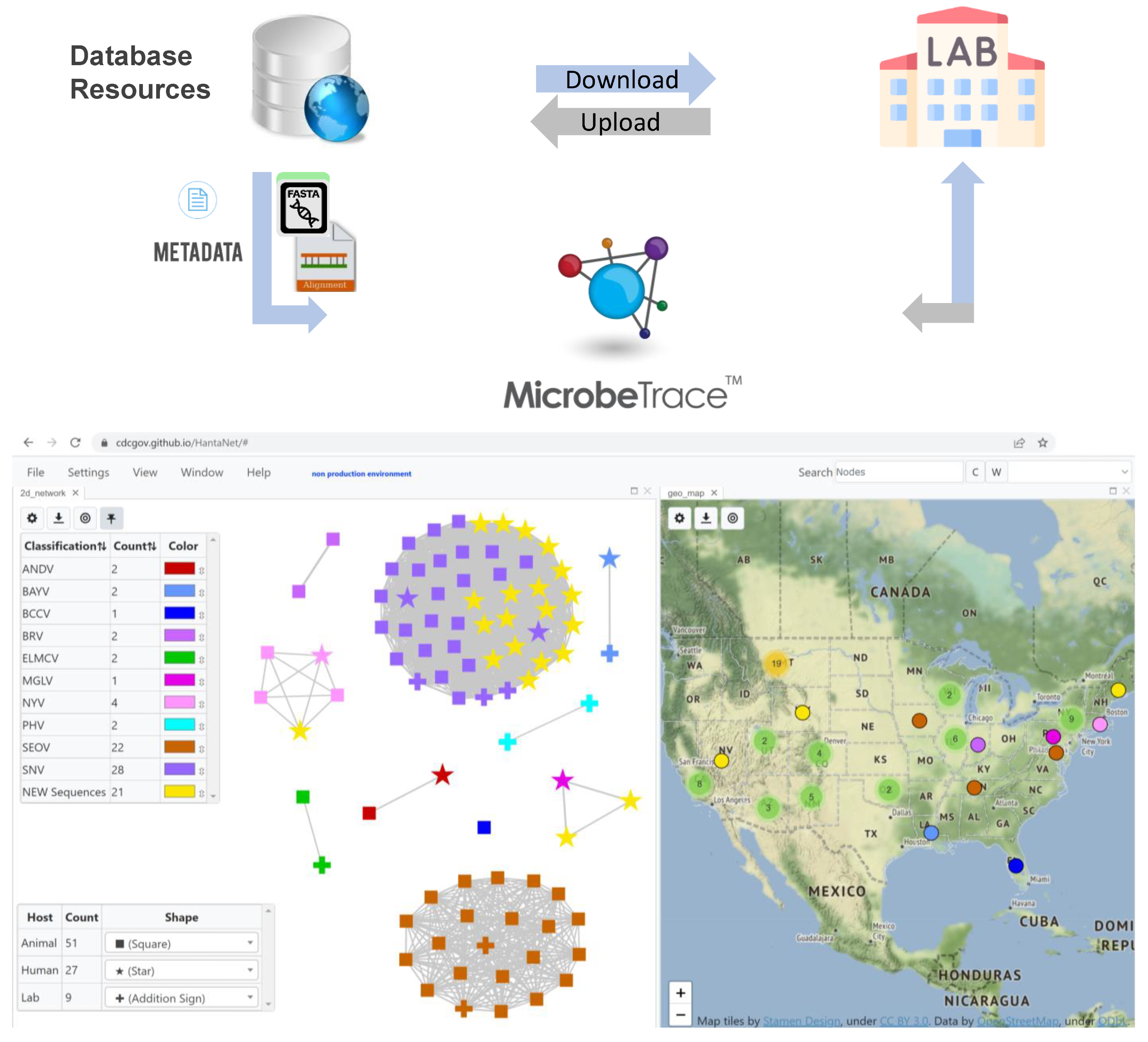
2.1. Database Search and Standardization of Hantavirus Data for Use in HantaNet
2.2. Building Hantavirus Reference Gene Modules in MicrobeTrace for HantaNet
2.3. Validation of the HantaNet Tool
2.4. HantaNet Improvements and New Features
3. Results
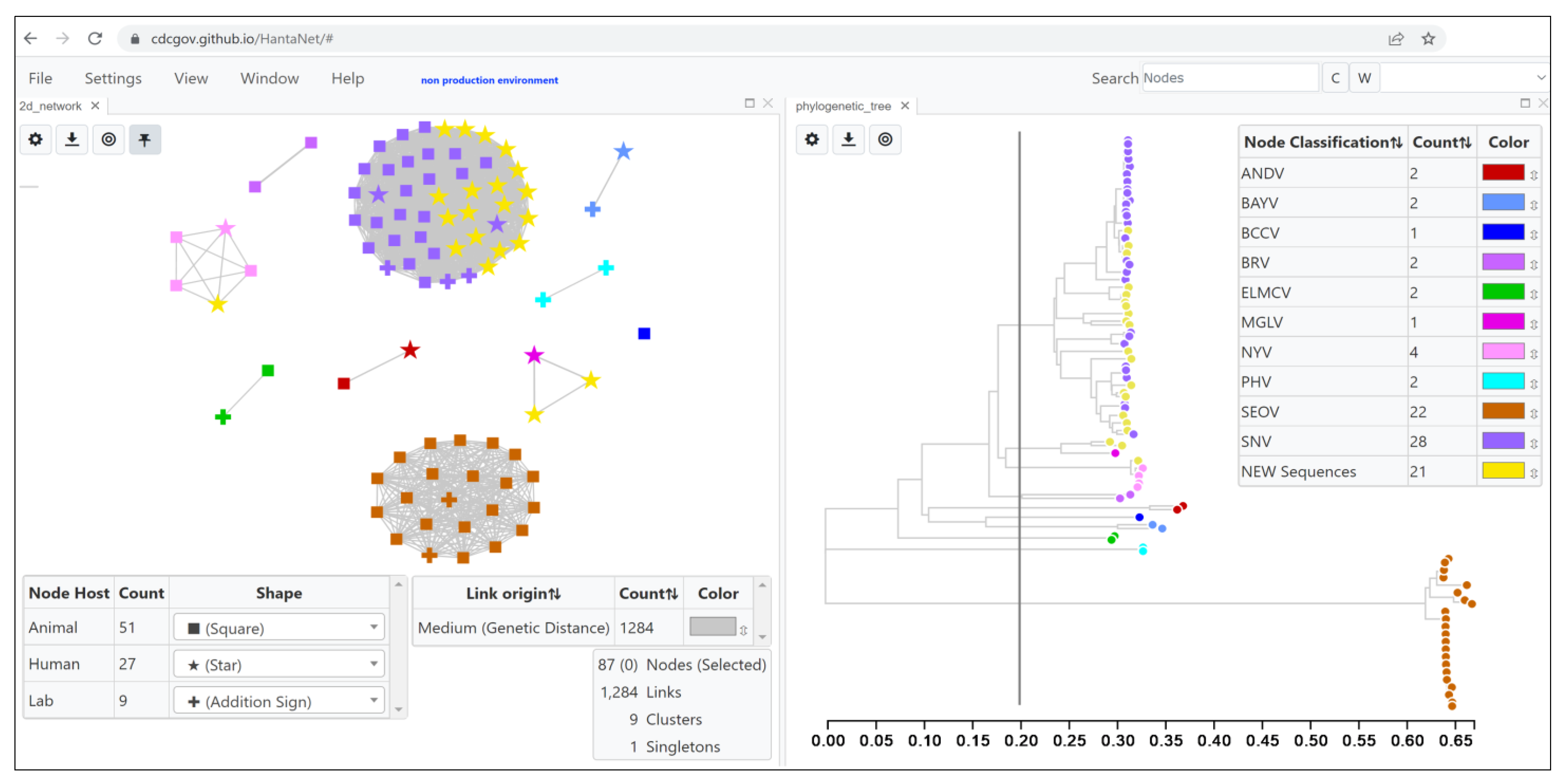
3.1. Validation of HantaNet for Classification of Hantaviruses
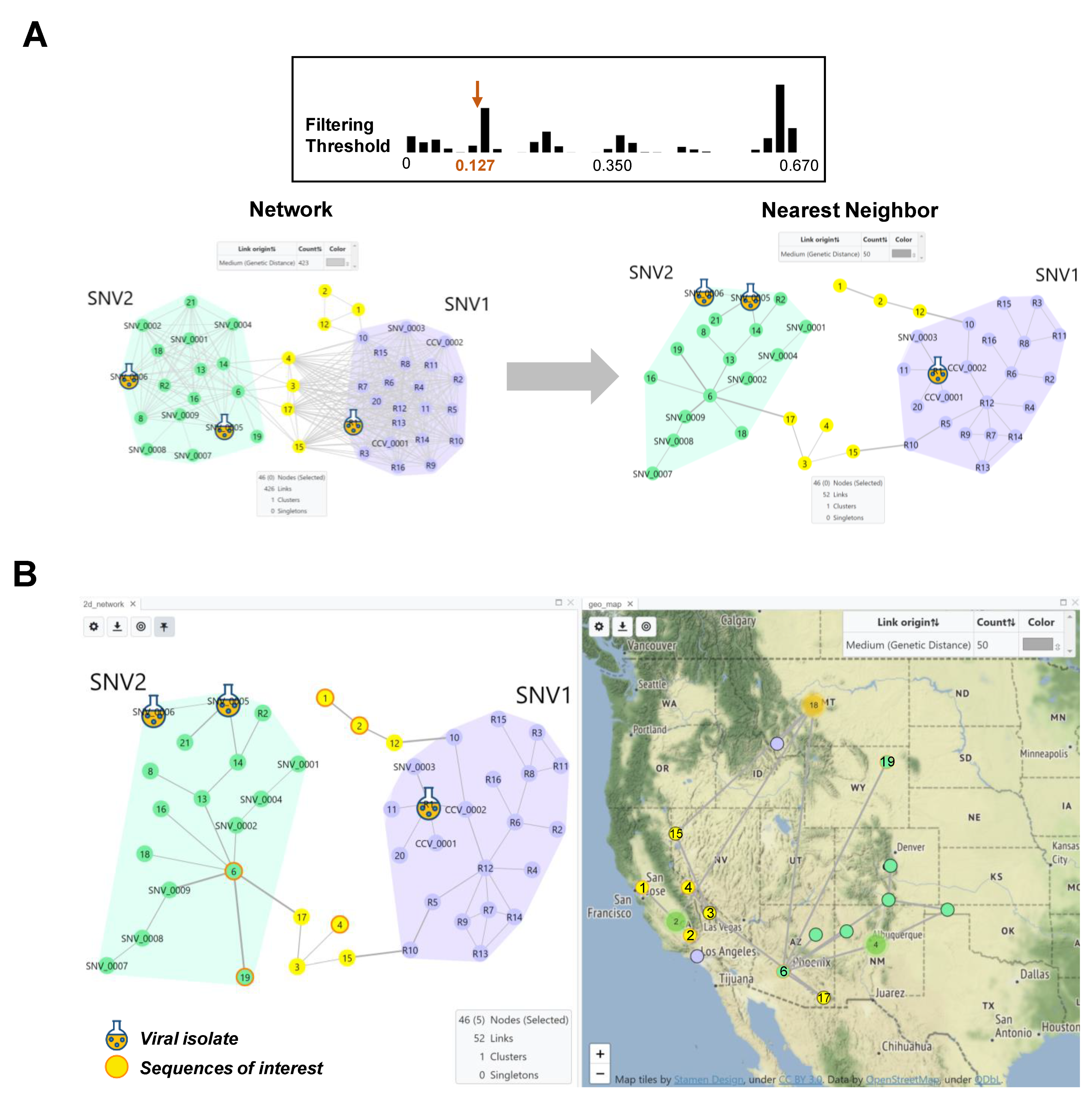
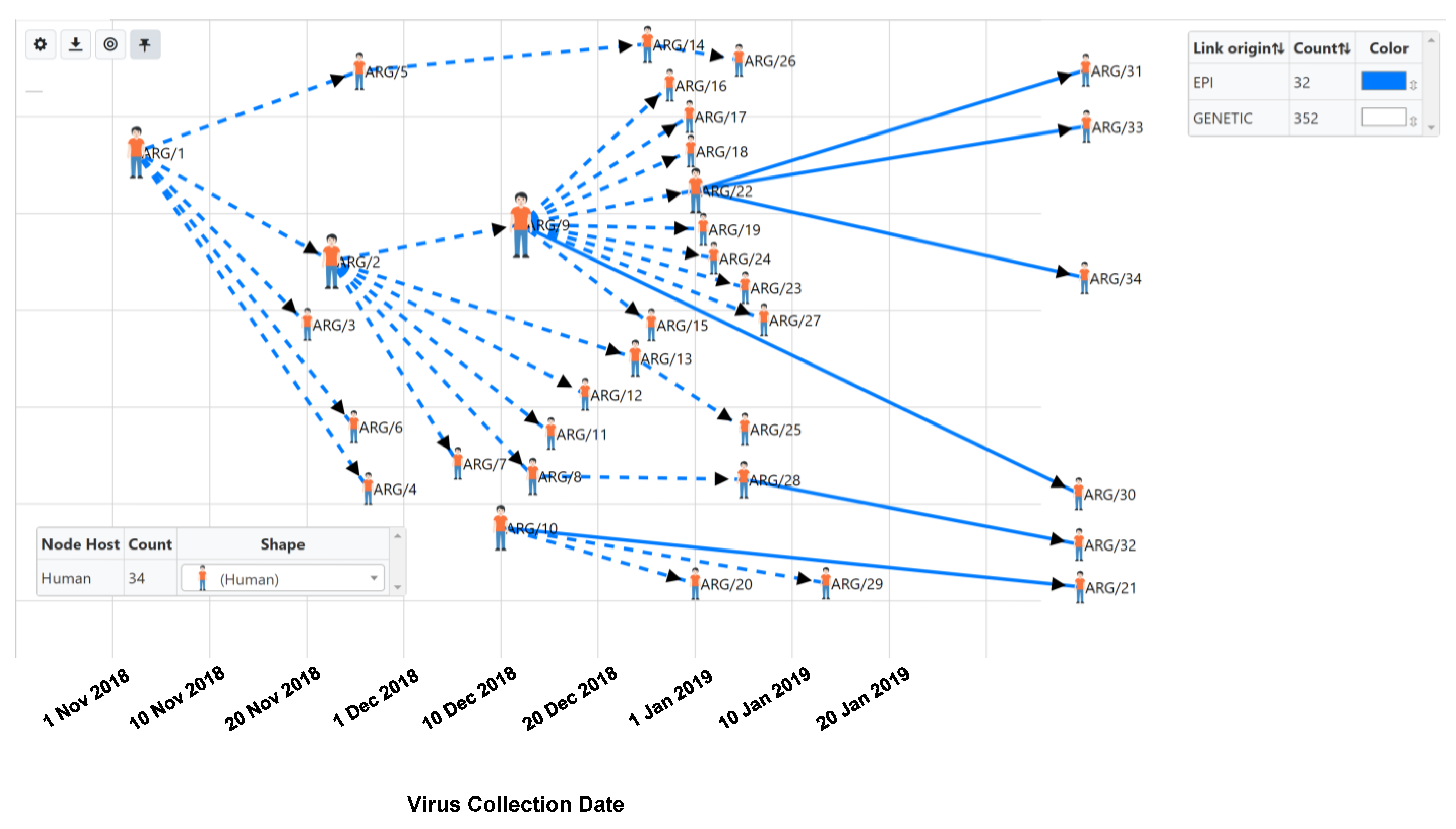
3.2. Genomic and Epidemiology Analyses of Hantaviruses Using HantaNet
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jonsson, C.B.; Figueiredo, L.T.; Vapalahti, O. A Global Perspective on Hantavirus Ecology, Epidemiology, and Disease. Clin. Microbiol. Rev. 2010, 23, 412–441. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Talwar, P.; Palaniyandi, R. Hantavirus Pulmonary Syndrome (HPS): A Concise Review Based on Current Knowledge and Emerging Concept. J. Appl. Pharm. Sci. 2014, 4, 122–130. [Google Scholar]
- Avšič-Županc, T.; Saksida, A.; Korva, M. Hantavirus Infections. Clin. Microbiol. Infect. 2019, 21s, e6–e16. [Google Scholar] [CrossRef] [PubMed]
- Kofman, A.; Eggers, P.; Kjemtrup, A.; Hall, R.; Brown, S.M.; Morales-Betoulle, M.; Graziano, J.; Zufan, S.E.; Whitmer, S.L.M.; Cannon, D.L.; et al. Notes from the Field: Contact Tracing Investigation after First Case of Andes Virus in the United States—Delaware, February 2018. MMWR Morb. Mortal. Wkly. Rep. 2018, 67, 1162–1163. [Google Scholar] [CrossRef] [PubMed]
- Alonso, D.O.; Pérez-Sautu, U.; Bellomo, C.M.; Prieto, K.; Iglesias, A.; Coelho, R.; Periolo, N.; Domenech, I.; Talmon, G.; Hansen, R.; et al. Person-to-Person Transmission of Andes Virus in Hantavirus Pulmonary Syndrome, Argentina, 2014. Emerg. Infect. Dis. 2020, 26, 756–759. [Google Scholar] [CrossRef] [PubMed]
- Martínez, V.P.; Di Paola, N.; Alonso, D.O.; Pérez-Sautu, U.; Bellomo, C.M.; Iglesias, A.A.; Coelho, R.M.; López, B.; Periolo, N.; Larson, P.A.; et al. “Super-Spreaders” and Person-to-Person Transmission of Andes Virus in Argentina. N. Engl. J. Med. 2020, 383, 2230–2241. [Google Scholar] [CrossRef] [PubMed]
- Toledo, J.; Haby, M.M.; Reveiz, L.; Sosa Leon, L.; Angerami, R.; Aldighieri, S. Evidence for Human-to-Human Transmission of Hantavirus: A Systematic Review. J. Infect. Dis. 2022, 226, 1362–1371. [Google Scholar] [CrossRef] [PubMed]
- Bellomo, C.M.; Alonso, D.O.; Pérez-Sautu, U.; Prieto, K.; Kehl, S.; Coelho, R.M.; Periolo, N.; Di Paola, N.; Ferressini-Gerpe, N.; Kuhn, J.H.; et al. Andes Virus Genome Mutations That Are Likely Associated with Animal Model Attenuation and Human Person-to-Person Transmission. mSphere 2023, 8, e0001823. [Google Scholar] [CrossRef]
- Ermonval, M.; Baychelier, F.; Tordo, N. What Do We Know about How Hantaviruses Interact with Their Different Hosts? Viruses 2016, 8, 223. [Google Scholar] [CrossRef]
- Rissanen, I.; Stass, R.; Zeltiña, A.; Li, S.; Hepojoki, J.M.; Harlos, K.; Gilbert, R.J.C.; Huiskonen, J.T.; Bowden, T.A. Structural Transitions of the Conserved and Metastable Hantaviral Glycoprotein Envelope. J. Virol. 2017, 91, e00378-17. [Google Scholar] [CrossRef]
- Laenen, L.; Vergote, V.; Calisher, C.H.; Klempa, B.; Klingström, J.; Kuhn, J.H.; Maes, P. Hantaviridae: Current Classification and Future Perspectives. Viruses 2019, 11, 788. [Google Scholar] [CrossRef]
- Parvate, A.; Williams, E.P.; Taylor, M.K.; Chu, Y.K.; Lanman, J.; Saphire, E.O.; Jonsson, C.B. Diverse Morphology and Structural Features of Old and New World Hantaviruses. Viruses 2019, 11, 862. [Google Scholar] [CrossRef] [PubMed]
- Abudurexiti, A.; Adkins, S.; Alioto, D.; Alkhovsky, S.V.; Avšič-Županc, T.; Ballinger, M.J.; Bente, D.A.; Beer, M.; Bergeron, É.; Blair, C.D.; et al. Taxonomy of the Order Bunyavirales: Update 2019. Arch. Virol. 2019, 164, 1949–1965. [Google Scholar] [CrossRef] [PubMed]
- Vapalahti, O.; Mustonen, J.; Lundkvist, A.; Henttonen, H.; Plyusnin, A.; Vaheri, A. Hantavirus Infections in Europe. Lancet Infect. Dis. 2003, 3, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Vaheri, A.; Henttonen, H.; Voutilainen, L.; Mustonen, J.; Sironen, T.; Vapalahti, O. Hantavirus Infections in Europe and Their Impact on Public Health. Rev. Med. Virol. 2013, 23, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Tariq, M.; Kim, D.M. Hemorrhagic Fever with Renal Syndrome: Literature Review, Epidemiology, Clinical Picture and Pathogenesis. Infect. Chemother. 2022, 54, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Klena, J.D.; Chiang, C.F.; Whitmer, S.M.; Wang, Y.F.; Shieh, W.J. Hantaviruses, 12th ed.; American Society of Microbiology: Washington, DC, USA, 2023. [Google Scholar]
- Hjelle, B.; Torres-Pérez, F. Hantaviruses in the Americas and Their Role as Emerging Pathogens. Viruses 2010, 2, 2559–2586. [Google Scholar] [CrossRef] [PubMed]
- Thorp, L.; Fullerton, L.; Whitesell, A.; Dehority, W. Hantavirus Pulmonary Syndrome: 1993–2018. Pediatrics 2023, 151, e2022059352. [Google Scholar] [CrossRef] [PubMed]
- Chizhikov, V.E.; Spiropoulou, C.F.; Morzunov, S.P.; Monroe, M.C.; Peters, C.J.; Nichol, S.T. Complete Genetic Characterization and Analysis of Isolation of Sin Nombre Virus. J. Virol. 1995, 69, 8132–8136. [Google Scholar] [CrossRef]
- Huang, C.; Campbell, W.P.; Means, R.; Ackman, D.M. Hantavirus S RNA Sequence from a Fatal Case of HPS in New York. J. Med. Virol. 1996, 50, 5–8. [Google Scholar] [CrossRef]
- Bagamian, K.H.; Towner, J.S.; Mills, J.N.; Kuenzi, A.J. Increased Detection of Sin Nombre Hantavirus RNA in Antibody-Positive Deer Mice from Montana, USA: Evidence of Male Bias in RNA Viremia. Viruses 2013, 5, 2320–2328. [Google Scholar] [CrossRef] [PubMed]
- Kjemtrup, A.M.; Messenger, S.; Meza, A.M.; Feiszli, T.; Yoshimizu, M.H.; Padgett, K.; Singh, S. New Exposure Location for Hantavirus Pulmonary Syndrome Case, California, USA, 2018. Emerg. Infect. Dis. 2019, 25, 1962–1964. [Google Scholar] [CrossRef] [PubMed]
- Martinez, V.P.; Bellomo, C.M.; Cacace, M.L.; Suarez, P.; Bogni, L.; Padula, P.J. Hantavirus Pulmonary Syndrome in Argentina, 1995–2008. Emerg. Infect. Dis. 2010, 16, 1853–1860. [Google Scholar] [CrossRef] [PubMed]
- Iglesias, A.A.; Bellomo, C.M.; Martínez, V.P. Hantavirus Pulmonary Syndrome in Buenos Aires, 2009–2014. Medicina 2016, 76, 1–9. [Google Scholar] [PubMed]
- Rhodes, L.V., 3rd; Huang, C.; Sanchez, A.J.; Nichol, S.T.; Zaki, S.R.; Ksiazek, T.G.; Humphreys, J.G.; Freeman, J.J.; Knecht, K.R. Hantavirus Pulmonary Syndrome Associated with Monongahela Virus, Pennsylvania. Emerg. Infect. Dis. 2000, 6, 616–621. [Google Scholar] [CrossRef] [PubMed]
- Albariño, C.G.; Wiggleton Guerrero, L.; Chakrabarti, A.K.; Rollin, P.E.; Nichol, S.T. Complete Genome Sequences of Monongahela Hantavirus from Pennsylvania, USA. Microbiol. Resour. Announc. 2018, 7, e00928-18. [Google Scholar] [CrossRef] [PubMed]
- Fernando, R.; Capone, D.; Elrich, S.; Mantovani, R.; Quarles, L., 3rd; D’Amato, A.; Lowe, N.; Malhotra, A.; Khoo, T.; Zufan, S.; et al. Infection with New York Orthohantavirus and Associated Respiratory Failure and Multiple Cerebral Complications. Emerg. Infect. Dis. 2019, 25, 1241–1243. [Google Scholar] [CrossRef] [PubMed]
- Doyle, T.J.; Bryan, R.T.; Peters, C.J. Viral Hemorrhagic Fevers and Hantavirus Infections in the Americas. Infect. Dis. Clin. North Am. 1998, 12, 95–110. [Google Scholar] [CrossRef]
- Witkowski, P.T.; Klempa, B.; Ithete, N.L.; Auste, B.; Mfune, J.K.; Hoveka, J.; Matthee, S.; Preiser, W.; Kruger, D.H. Hantaviruses in Africa. Virus Res. 2014, 187, 34–42. [Google Scholar] [CrossRef]
- Reported Cases of Hantavirus Disease. Available online: https://www.cdc.gov/hantavirus/surveillance/index.html (accessed on 25 April 2023).
- Whitmer, S.L.M.; Whitesell, A.; Mobley, M.; Talundzic, E.; Shedroff, E.; Cossaboom, C.M.; Messenger, S.; Deldari, M.; Bhatnagar, J.; Estetter, L.; et al. Human Orthohantavirus Disease Prevalence and Genotype Distribution in the U.S., 2008–2020. Lancet ID, 2023; in submission. [Google Scholar]
- Goodfellow, S.M.; Nofchissey, R.A.; Schwalm, K.C.; Cook, J.A.; Dunnum, J.L.; Guo, Y.; Ye, C.; Mertz, G.J.; Chandran, K.; Harkins, M.; et al. Tracing Transmission of Sin Nombre Virus and Discovery of Infection in Multiple Rodent Species. J. Virol. 2021, 95, e0153421. [Google Scholar] [CrossRef] [PubMed]
- Knust, B.; Brown, S.; de St. Maurice, A.; Whitmer, S.; Koske, S.E.; Ervin, E.; Patel, K.; Graziano, J.; Morales-Betoulle, M.E.; House, J.; et al. Seoul Virus Infection and Spread in United States Home-Based Ratteries: Rat and Human Testing Results from a Multistate Outbreak Investigation. J. Infect. Dis. 2020, 222, 1311–1319. [Google Scholar] [PubMed]
- Hecht, G.; Dale, A.P.; Ruberto, I.; Adame, G.; Close, R.; Snyder, S.J.; Pink, K.; Lemmon, N.; Rudolfo, J.; Madsen, M.; et al. Detection of Hantavirus During the COVID-19 Pandemic, Arizona, USA, 2020. Emerg. Infect. Dis. 2023, 29, 1663–1667. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.K.; No, J.S.; Lee, D.; Jung, J.; Park, H.; Yi, Y.; Kim, J.A.; Lee, S.H.; Kim, Y.; Park, S.; et al. Active Targeted Surveillance to Identify Sites of Emergence of Hantavirus. Clin. Infect. Dis. 2020, 70, 464–473. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.K.; Cho, S.; Lee, S.H.; No, J.S.; Lee, G.Y.; Park, K.; Lee, D.; Jeong, S.T.; Song, J.W. Genomic Epidemiology and Active Surveillance to Investigate Outbreaks of Hantaviruses. Front. Cell. Infect. Microbiol. 2020, 10, 532388. [Google Scholar] [CrossRef] [PubMed]
- Campbell, E.M.; Boyles, A.; Shankar, A.; Kim, J.; Knyazev, S.; Cintron, R.; Switzer, W.M. Microbetrace: Retooling Molecular Epidemiology for Rapid Public Health Response. PLoS Comput. Biol. 2021, 17, e1009300. [Google Scholar] [CrossRef] [PubMed]
- Bradley, H.; Hogan, V.; Agnew-Brune, C.; Armstrong, J.; Broussard, D.; Buchacz, K.; Burton, K.; Cope, S.; Dawson, E.; De La Garza, G.; et al. Increased Hiv Diagnoses in West Virginia Counties Highly Vulnerable to Rapid Hiv Dissemination through Injection Drug Use: A cautionary Tale. Ann. Epidemiol. 2019, 34, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Campbell, E.M.; Patala, A.; Shankar, A.; Li, J.F.; Johnson, J.A.; Westheimer, E.; Gay, C.L.; Cohen, S.E.; Switzer, W.M.; Peters, P.J. Phylodynamic Analysis Complements Partner Services by Identifying Acute and Unreported Hiv Transmission. Viruses 2020, 12, 145. [Google Scholar] [CrossRef]
- Brenner, B.G.; Ibanescu, R.I.; Osman, N.; Cuadra-Foy, E.; Oliveira, M.; Chaillon, A.; Stephens, D.; Hardy, I.; Routy, J.P.; Thomas, R.; et al. The Role of Phylogenetics in Unravelling Patterns of Hiv Transmission Towards Epidemic Control: The Quebec Experience (2002–2020). Viruses 2021, 13, 1643. [Google Scholar] [CrossRef]
- Rich, S.N.; Prosperi, M.C.F.; Dellicour, S.; Vrancken, B.; Cook, R.L.; Spencer, E.C.; Salemi, M.; Mavian, C. Molecular Epidemiology of Hiv-1 Subtype B Infection across Florida Reveals Few Large Superclusters with Metropolitan Origin. Microbiol. Spectr. 2022, 10, e0188922. [Google Scholar] [CrossRef]
- Alexiev, I.; Shankar, A.; Pan, Y.; Grigorova, L.; Partsuneva, A.; Dimitrova, R.; Gancheva, A.; Kostadinova, A.; Elenkov, I.; Yancheva, N.; et al. Transmitted Hiv Drug Resistance in Bulgaria Occurs in Clusters of Individuals from Different Transmission Groups and Various Subtypes (2012–2020). Viruses 2023, 15, 941. [Google Scholar] [CrossRef] [PubMed]
- Longmire, A.G.; Sims, S.; Rytsareva, I.; Campo, D.S.; Skums, P.; Dimitrova, Z.; Ramachandran, S.; Medrzycki, M.; Thai, H.; Ganova-Raeva, L.; et al. Ghost: Global Hepatitis Outbreak and Surveillance Technology. BMC Genom. 2017, 18 (Suppl. 10), 916. [Google Scholar] [CrossRef] [PubMed]
- Rao, J.R.; Rafique, S.; Ali, A.; Rehman, G.; Ilyas, M.; Idrees, M. Next-Generation Sequencing Studies on the E1-Hvr1 Region of Hepatitis C Virus (Hcv) from Non-High-Risk Hcv Patients Living in Punjab and Khyber Pakhtunkhwa, Pakistan. Arch. Virol. 2021, 166, 3049–3059. [Google Scholar] [CrossRef] [PubMed]
- Labuda, S.M.; McDaniel, C.J.; Talwar, A.; Braumuller, A.; Parker, S.; McGaha, S.; Blissett, C.; Wortham, J.M.; Mukasa, L.; Stewart, R.J. Tuberculosis Outbreak Associated with Delayed Diagnosis and Long Infectious Periods in Rural Arkansas, 2010–2018. Public Health Rep. 2022, 137, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Houwaart, T.; Belhaj, S.; Tawalbeh, E.; Nagels, D.; Fröhlich, Y.; Finzer, P.; Ciruela, P.; Sabrià, A.; Herrero, M.; Andrés, C.; et al. Integrated Genomic Surveillance Enables Tracing of Person-to-Person SARS-CoV-2 Transmission Chains During Community Transmission and Reveals Extensive Onward Transmission of Travel-Imported Infections, Germany, June to July 2021. Eurosurveillance 2022, 27, 2101089. [Google Scholar] [CrossRef] [PubMed]
- Yaglom, H.D.; Gebhardt, M.; Pfeiffer, A.; Ormsby, M.E.; Jasso-Selles, D.E.; Lemmer, D.; Folkerts, M.L.; French, C.; Maurer, M.; Bowers, J.R.; et al. Applying Genomic Epidemiology to Characterize a COVID-19 Outbreak in a Developmentally Disabled Adult Group Home Setting, Arizona. Front. Public Health 2021, 9, 668214. [Google Scholar] [CrossRef] [PubMed]
- Jayroe, M.; Aguilar, D.R.; Porter, A.; Cima, M.; Chai, S.; Hayman, K. Transmission Analysis of COVID-19 Outbreaks Associated with Places of Worship, Arkansas, May 2020–December 2020. J. Relig. Health 2023, 62, 650–661. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. Mafft: A Novel Method for Rapid Multiple Sequence Alignment Based on Fast Fourier Transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. Mafft Online Service: Multiple Sequence Alignment, Interactive Sequence Choice and Visualization. Brief. Bioinform. 2017, 20, 1160–1166. [Google Scholar] [CrossRef]
- Hall, T.A. Bioedit: A User-Friendly Biological Sequence Alignment Editor and Analysis Program for Windows 95/98/Nt. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Tamura, K.; Nei, M. Estimation of the Number of Nucleotide Substitutions in the Control Region of Mitochondrial DNA in Humans and Chimpanzees. Mol. Biol. Evol. 1993, 10, 512–526. [Google Scholar] [PubMed]
- Seqruler: Gui for Calculating TN93 between Set of Sequences. Available online: https://github.com/Sergey-Knyazev/SeqRuler (accessed on 3 February 2023).
- Seqruler: Gui for Calculating TN93 or SNP Distances for a Set of Sequences. Available online: https://github.com/CDCGov/SeqRuler (accessed on 25 October 2023).
- Monroe, M.C.; Morzunov, S.P.; Johnson, A.M.; Bowen, M.D.; Artsob, H.; Yates, T.; Peters, C.J.; Rollin, P.E.; Ksiazek, T.G.; Nichol, S.T. Genetic Diversity and Distribution of Peromyscus-Borne Hantaviruses in North America. Emerg. Infect. Dis. 1999, 5, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Ramsden, C.; Holmes, E.C.; Charleston, M.A. Hantavirus Evolution in Relation to Its Rodent and Insectivore Hosts: No Evidence for Codivergence. Mol. Biol. Evol. 2009, 26, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Tidytree: Uncompromisingly Flexible Phylogenetic Trees in Javascript. Available online: https://github.com/CDCgov/TidyTree (accessed on 25 October 2023).
- Kumar, S.; Tamura, K.; Nei, M. Mega: Molecular Evolutionary Genetics Analysis Software for Microcomputers. Comput. Appl. Biosci. 1994, 10, 189–191. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. Mega X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Fourment, M.; Gibbs, M.J. Patristic: A Program for Calculating Patristic Distances and Graphically Comparing the Components of Genetic Change. BMC Evol. Biol. 2006, 6, 1. [Google Scholar] [CrossRef] [PubMed]
- Patristic: A Phylogenetics Toolkit for Javascript. Available online: https://github.com/CDCgov/patristic (accessed on 25 October 2023).
- Kruskal, J.B. On the Shortest Spanning Subtree of a Graph and the Traveling Salesman Problem. Proc. Am. Math. Soc. 1956, 7, 48–50. [Google Scholar] [CrossRef]
- Emst: Epsilon Minimal Spanning Trees. Available online: https://github.com/Sergey-Knyazev/eMST (accessed on 3 February 2023).
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-Time Tracking of Pathogen Evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef]
- Cintron, R.; Gigante, C.M.; Li, Y.; Kelly, R.; Caravas, J.; Switzer, W.M. Implementation of Nextstrain and Microbetrace Pipeline for Mpox Genomic Surveillance and Rapid Public Health Action. In Proceedings of the 2023 APHL/CDC Advanced Molecular Detection (AMD) Days, Atlanta, GA, USA, 12–14 September 2023. [Google Scholar]
- Van Iersel, L.; Kelk, S.; Rupp, R.; Huson, D. Phylogenetic Networks Do Not Need to Be Complex: Using Fewer Reticulations to Represent Conflicting Clusters. Bioinformatics 2010, 26, i124–i131. [Google Scholar] [CrossRef]
- Mardulyn, P. Trees and/or Networks to Display Intraspecific DNA Sequence Variation? Mol. Ecol. 2012, 21, 3385–3390. [Google Scholar] [CrossRef]
- Schliep, K.P.; Potts, A.J.; Morrison, D.A.; Grimm, G.W. Intertwining Phylogenetic Trees and Networks. Methods Ecol. Evol. 2017, 8, 1212–1220. [Google Scholar] [CrossRef]
- Blair, C.; Ané, C. Phylogenetic Trees and Networks Can Serve as Powerful and Complementary Approaches for Analysis of Genomic Data. Syst. Biol. 2020, 69, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Vijaykrishna, D.; Mukerji, R.; Smith, G.J. RNA Virus Reassortment: An Evolutionary Mechanism for Host Jumps and Immune Evasion. PLoS Pathog. 2015, 11, e1004902. [Google Scholar] [CrossRef] [PubMed]
- McDonald, S.M.; Nelson, M.I.; Turner, P.E.; Patton, J.T. Reassortment in Segmented RNA Viruses: Mechanisms and Outcomes. Nat. Rev. Microbiol. 2016, 14, 448–460. [Google Scholar] [CrossRef] [PubMed]
- Klempa, B. Reassortment Events in the Evolution of Hantaviruses. Virus Genes. 2018, 54, 638–646. [Google Scholar] [CrossRef] [PubMed]
- Varsani, A.; Lefeuvre, P.; Roumagnac, P.; Martin, D. Notes on Recombination and Reassortment in Multipartite/Segmented Viruses. Curr. Opin. Virol. 2018, 33, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Barrat-Charlaix, P.; Vaughan, T.G.; Neher, R.A. Treeknit: Inferring Ancestral Reassortment Graphs of Influenza Viruses. PLoS Comput. Biol. 2022, 18, e1010394. [Google Scholar] [CrossRef]
- Seqcombo. Available online: https://github.com/YuLab-SMU/seqcombo (accessed on 21 March 2023).
- Rasmussen, D.A.; Grünwald, N.J. Phylogeographic Approaches to Characterize the Emergence of Plant Pathogens. Phytopathology 2021, 111, 68–77. [Google Scholar] [CrossRef]
- Drummond, A.J.; Rambaut, A. Beast: Bayesian Evolutionary Analysis by Sampling Trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef]
- Bouckaert, R.A.-O.; Vaughan, T.G.; Barido-Sottani, J.A.-O.; Duchêne, S.A.-O.; Fourment, M.A.-O.; Gavryushkina, A.; Heled, J.A.-O.; Jones, G.A.-O.; Kühnert, D.A.-O.X.; De Maio, N.A.-O.; et al. Beast 2.5: An Advanced Software Platform for Bayesian Evolutionary Analysis. PLoS Comput. Biol. 2019, 15, e1006650. [Google Scholar] [CrossRef]
- Fisher, A.A.-O.; Hassler, G.W.; Ji, X.; Baele, G.; Suchard, M.A.; Lemey, P. Scalable Bayesian Phylogenetics. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2022, 377, 20210242. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. Iq-Tree: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Sagulenko, P.; Puller, V.; Neher, R.A. Treetime: Maximum-Likelihood Phylodynamic Analysis. Virus Evol. 2018, 4, vex042. [Google Scholar] [CrossRef]
- Treetime. Available online: https://github.com/neherlab/treetime (accessed on 21 March 2023).
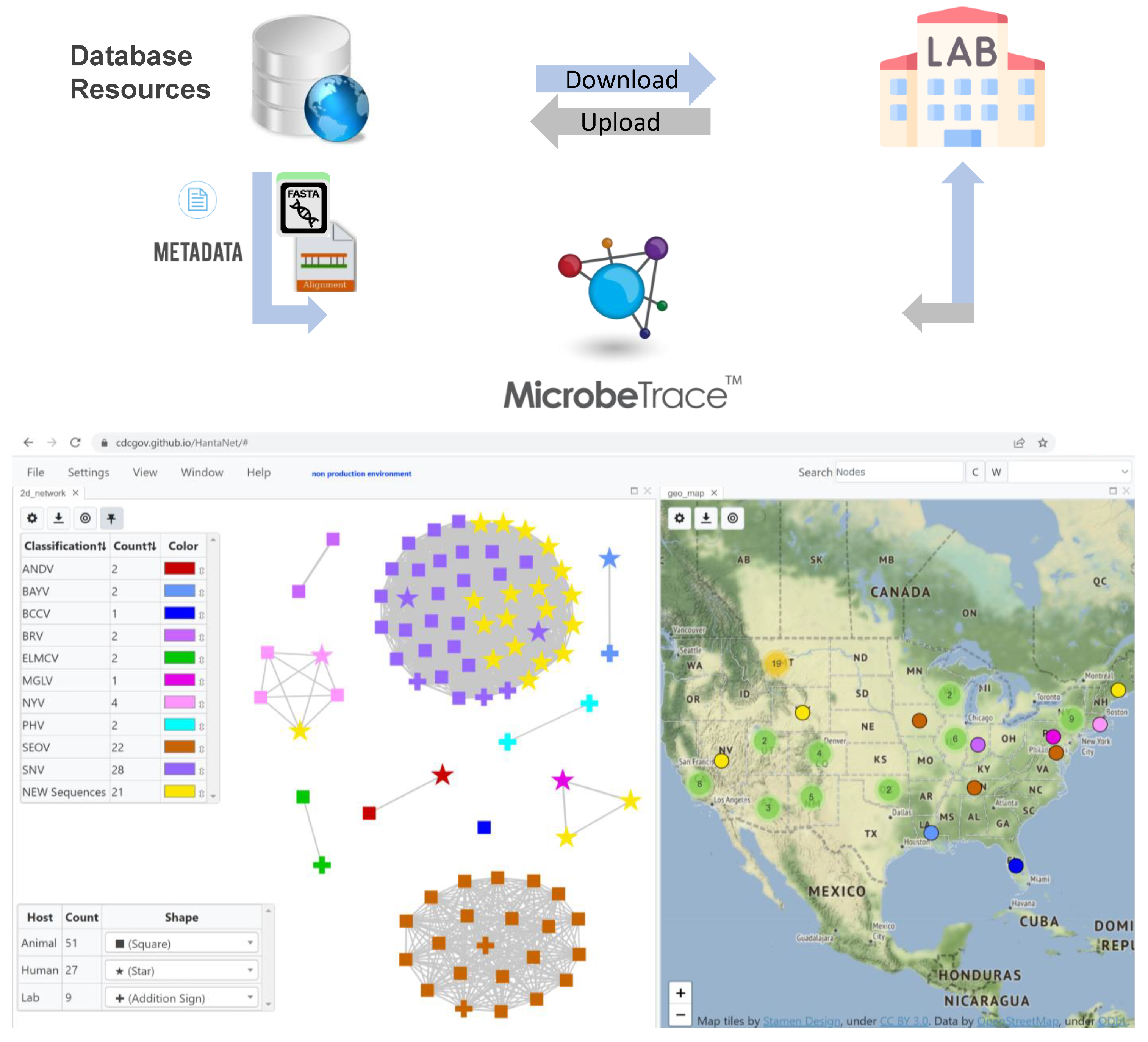

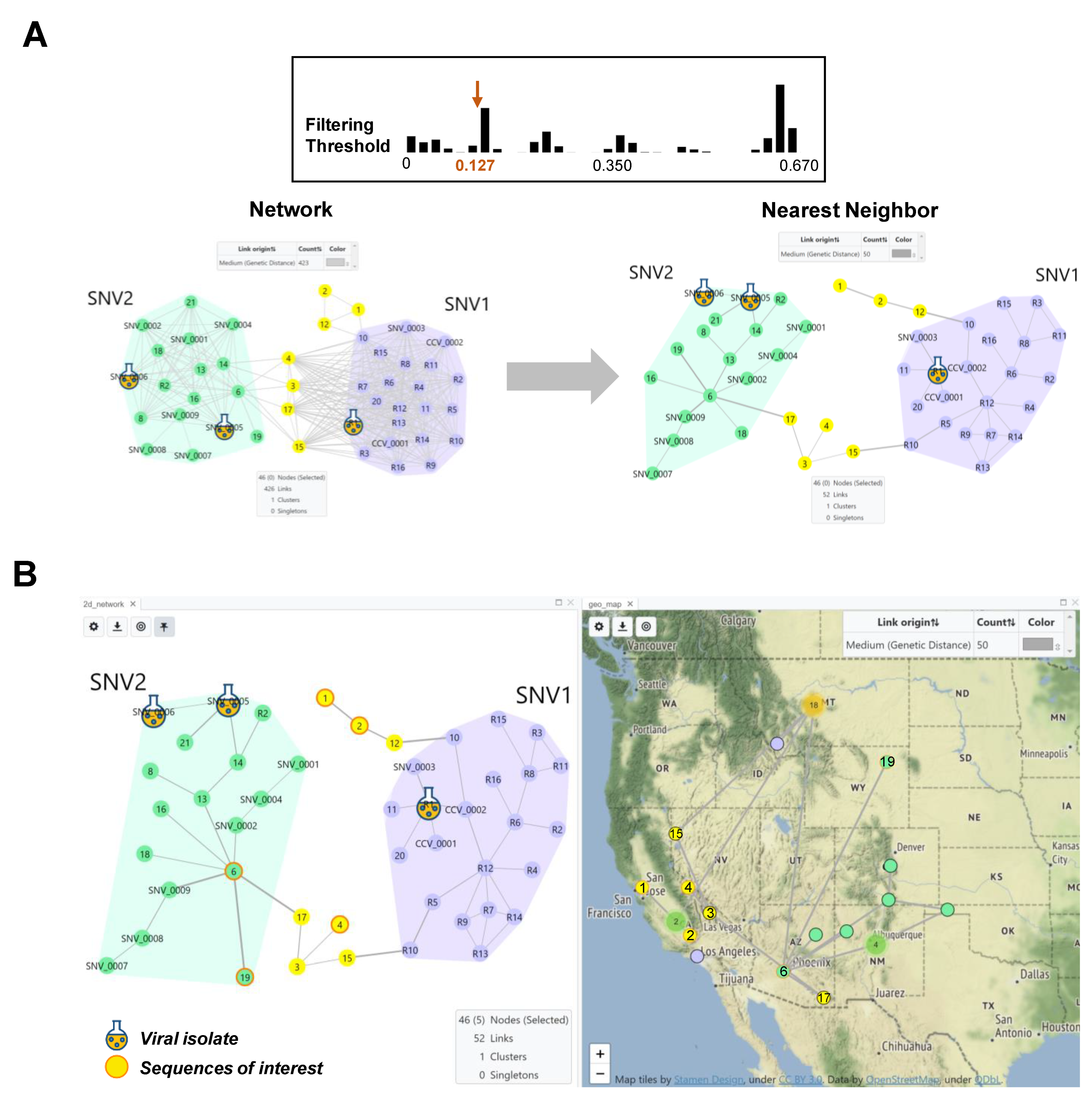
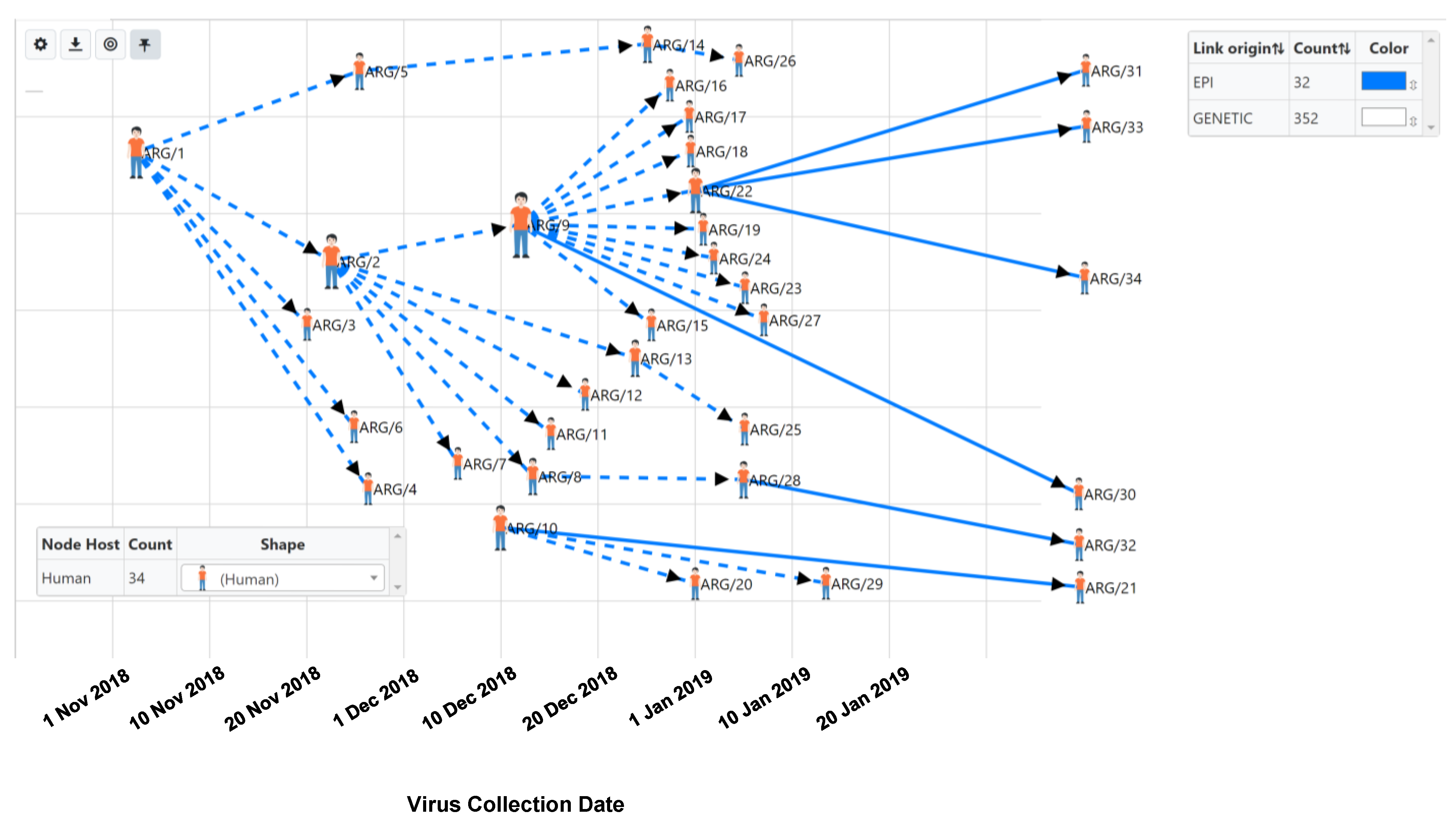
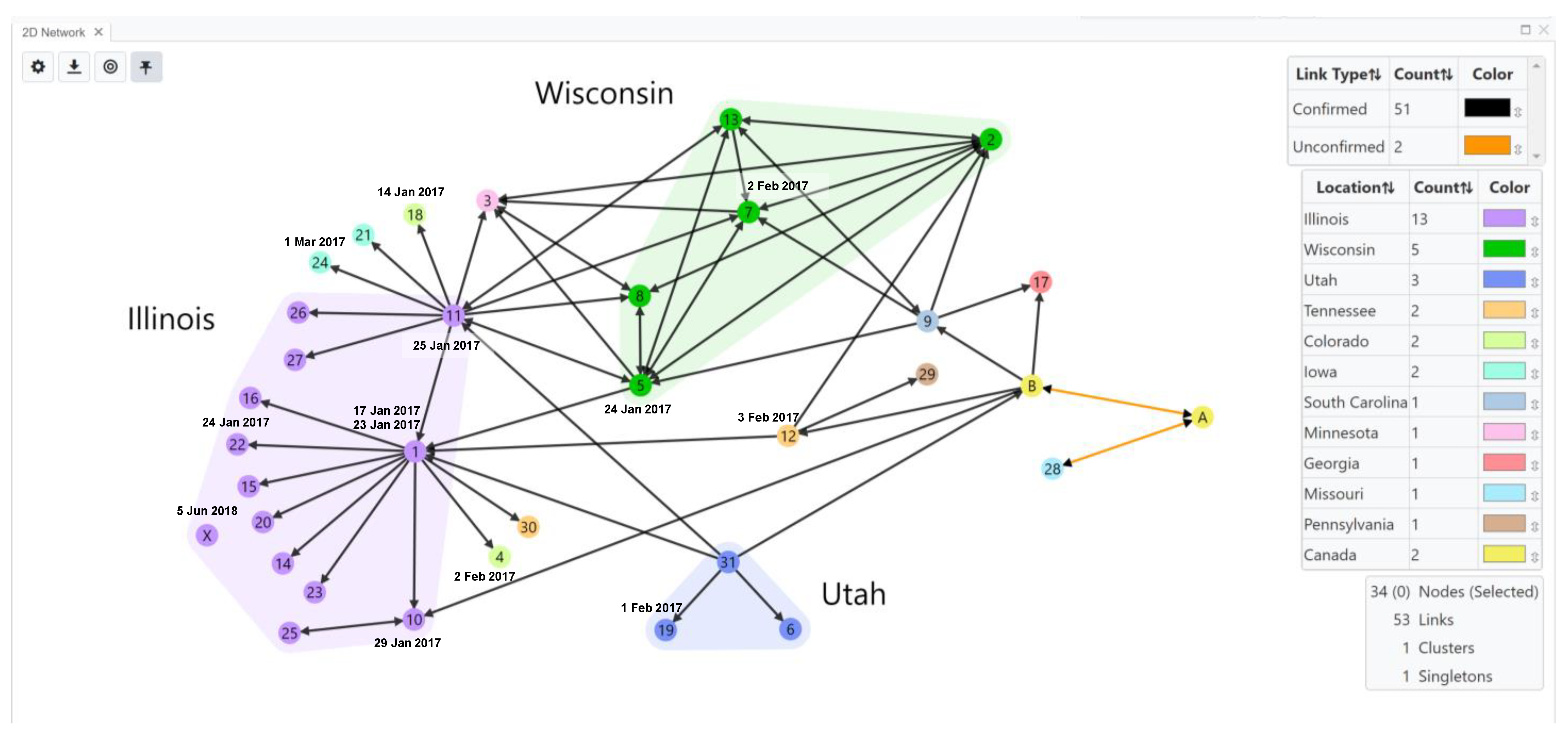

{kind=link}
{kind=link}
{kind=link}
{kind=link}
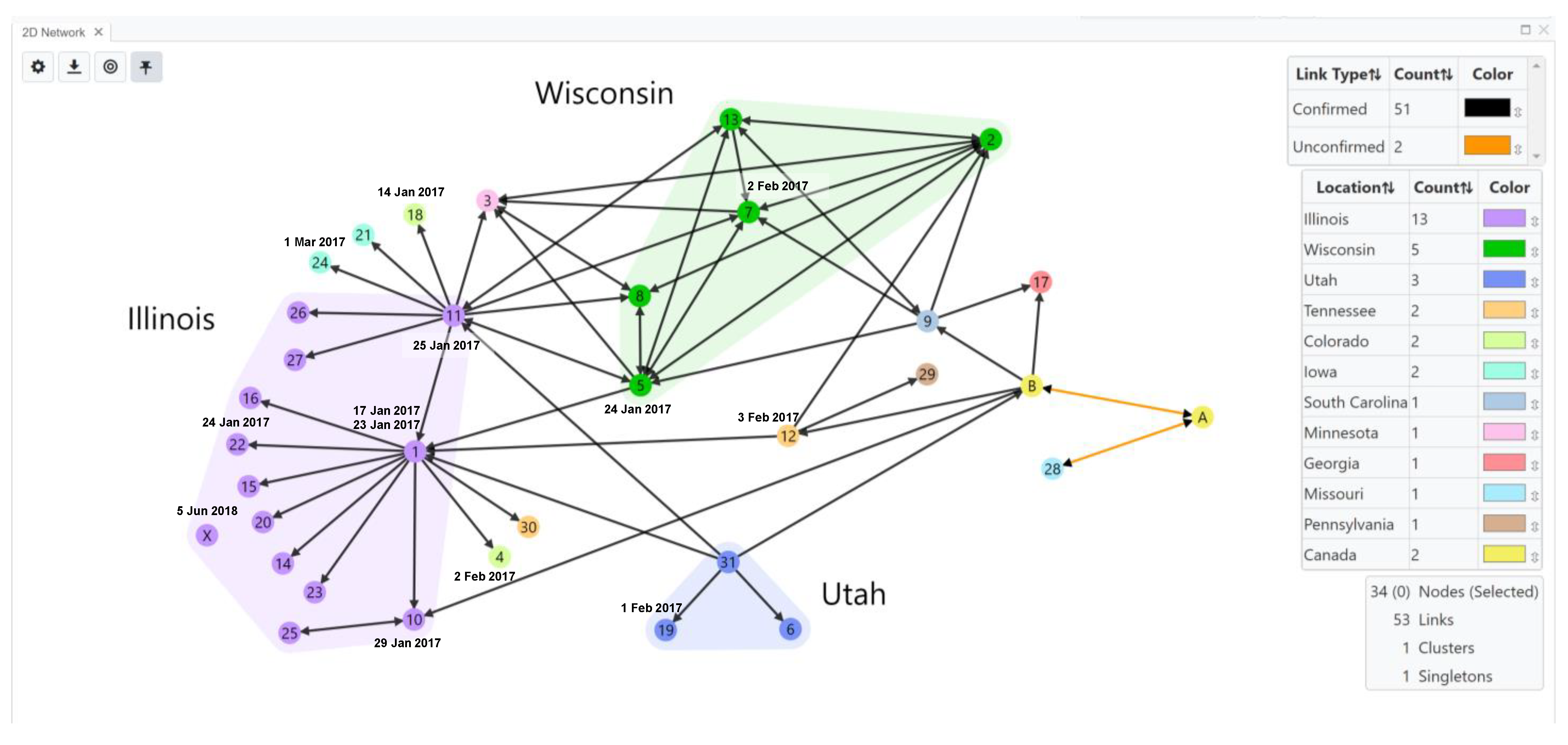{kind=link}
| Classification a | Virus Name b | Virus ID c | Collection Date d | Host e | Isolation Country f | State g | Segment h | Accession ID i |
|---|---|---|---|---|---|---|---|---|
| NDV | Andes virus | ANDV_0001 | 1997 | Rice rat | Chile | Unknown | S M L | AF291702 AF291703 AF291704 |
| ANDV_0002 | 2016 | Human | Switzerland | Unknown | S M L | KY659432 KY604962 KY659431 | ||
| BAYV | Bayou virus | BAYV_0001 | 1993 | Human | United States of America | Louisiana | S M | L36929 L36930 |
| BAYV_0002 | Viral isolate | S M L | GQ200820 GQ244521 GQ244526 | |||||
| BAYV_0003 | Viral isolate | S L | NC_038298 NC_038299 | |||||
| BCCV | Black Creek Canal virus | BCCV_0001 | 1994 | Cotton rat | United States of America | Florida | S M | L39949 L39950 |
| BCCV_0002 | Viral isolate | S | NC_043075 | |||||
| BRV | Blue River virus | BRV_0001 | 2005 | Deer mice | United States of America | Missouri | S | DQ090888 |
| BRV_0002 | 2005 | Deer mice | United States of America | Missouri | S | DQ090890 | ||
| BRV_0003 | 1997 | Deer mice | United States of America | Indiana | M | AF030551 | ||
| BRV_0004 | 1997 | Deer mice | United States of America | Oklahoma | M | AF030552 | ||
| ELMCV | El Moro Canyon virus | ELMCV_0001 | 1994 | Harvest mice | United States of America | California | S M | U11427 U26828 |
| ELMCV_0002 | Viral isolate | S M | NC_038423 NC_038424 | |||||
| ILV | Isla Vista virus | ILV_0001 | 1994 | Vole | United States of America | California | S | U19302 |
| ILV_0002 | 1995 | Vole | United States of America | California | S | U31534 | ||
| ILV_0003 | 1995 | Deer mice | United States of America | California | S | U31535 | ||
| MGLV | Monongahela virus | MGLV_0001 | 1985 | Deer mice | United States of America | West Virginia | S | U32591 |
| MGLV_0002 | 1997 | Human | United States of America | Pennsylvania | S M L | MH539867 MH539866 MH539865 | ||
| MULV | Muleshoe virus | MULV_0001 | 1995 | Cotton rat | United States of America | Texas | S | U54575 |
| MULV_0002 | 1999 | Cotton rat | United States of America | Texas | S | KX066124 | ||
| NYV | New York virus | NYV_0001 | 1994 | Deer mice | United States of America | New York | S M L | MG717391 MG717392 MG717393 |
| NYV_0002 | 1994 | Human | United States of America | Rhode Island | S M | U09488 U36801 | ||
| PHV | Prospect Hill virus | PHV_0001 | Viral isolate | S M L | NC_038938 NC_038940 NC_038939 | |||
| SEOV | Seoul virus | SEOV_0001 | 1993 | Rat | South Korea | Unknown | S M L | NC_005236 S47716 X56492 |
| SEOV_0002 | 2013 | Rat | United States of America | New York | S M | KJ950866 KJ950862 | ||
| SEOV_0003 | 2002 | Rat | United States of America | Maryland | S M L | KT897726 KT897725 KT897724 | ||
| SEOV_0004 | Viral isolate | S M L | KU204960 KU204959 KU204958 | |||||
| SEOV_0005 | 2013 | Rat | United States of America | New York | S M | KJ950867 KJ950863 | ||
| SEOV_0006 | 2013 | Rat | United States of America | New York | S M | KJ950868 KJ950864 | ||
| SEOV_0007 | 2013 | Rat | United States of America | New York | S M | KJ950869 KJ950865 | ||
| SNV | Sin Nombre virus | SNV_0001 | 1993 | Human | United States of America | New Mexico | S M L | NC_005216 NC_005215 L37901 |
| SNV_0002 | 1993 | Deer mice | United States of America | New Mexico | S M L | L37904 L37903 L37902 | ||
| SNV_0003 | 2009 | Deer mice | United States of America | Montana | S M | JQ690281 JQ690283 | ||
| SNV_0004 | 1993 | Human | United States of America | New Mexico | S M | L25784 L25783 | ||
| SNV_0005 | Viral isolate | S M L | KF537003 KF537002 KF537001 | |||||
| SNV_0006 | Viral isolate | S M L | KF537006 KF537005 KF537004 | |||||
| SNV_0007 | 2008 | Deer mice | United States of America | Montana | S M | JQ690276 JQ690279 | ||
| SNV_0008 | 2008 | Deer mice | United States of America | Montana | S M | JQ690277 JQ690280 | ||
| SNV_0009 | 2009 | Deer mice | United States of America | Montana | S M | JQ690282 JQ690284 | ||
| Convict Creek virus | CCV_0001 | 1993 | Deer mice | United States of America | California | S M L | L33683 L33474 AF425256 | |
| CCV_0002 | 1995 | Deer mice | United States of America | California | S M | L33816 L33684 |
| SNV Group | Pairwise Genetic Distance |
|---|---|
| SNV1 | 0–0.05714 |
| SNV1-like | 0.07099–0.11967 |
| SNV2 | 0.00029–0.06914 |
| SNV2-like | 0.12026–0.12295 |
| Other | 0.00693–0.03564 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cintron, R.; Whitmer, S.L.M.; Moscoso, E.; Campbell, E.M.; Kelly, R.; Talundzic, E.; Mobley, M.; Chiu, K.W.; Shedroff, E.; Shankar, A.; et al. HantaNet: A New MicrobeTrace Application for Hantavirus Classification, Genomic Surveillance, Epidemiology and Outbreak Investigations. Viruses 2023, 15, 2208. https://doi.org/10.3390/v15112208
Cintron R, Whitmer SLM, Moscoso E, Campbell EM, Kelly R, Talundzic E, Mobley M, Chiu KW, Shedroff E, Shankar A, et al. HantaNet: A New MicrobeTrace Application for Hantavirus Classification, Genomic Surveillance, Epidemiology and Outbreak Investigations. Viruses. 2023; 15(11):2208. https://doi.org/10.3390/v15112208
Chicago/Turabian StyleCintron, Roxana, Shannon L. M. Whitmer, Evan Moscoso, Ellsworth M. Campbell, Reagan Kelly, Emir Talundzic, Melissa Mobley, Kuo Wei Chiu, Elizabeth Shedroff, Anupama Shankar, and et al. 2023. "HantaNet: A New MicrobeTrace Application for Hantavirus Classification, Genomic Surveillance, Epidemiology and Outbreak Investigations" Viruses 15, no. 11: 2208. https://doi.org/10.3390/v15112208
APA StyleCintron, R., Whitmer, S. L. M., Moscoso, E., Campbell, E. M., Kelly, R., Talundzic, E., Mobley, M., Chiu, K. W., Shedroff, E., Shankar, A., Montgomery, J. M., Klena, J. D., & Switzer, W. M. (2023). HantaNet: A New MicrobeTrace Application for Hantavirus Classification, Genomic Surveillance, Epidemiology and Outbreak Investigations. Viruses, 15(11), 2208. https://doi.org/10.3390/v15112208