
Seneca Valley Virus Degrades STING via PERK and ATF6-Mediated Reticulophagy




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
2.2. Antibodies and Reagents
2.3. Pharmaceutical Treatment and Virus Infection
2.4. Construction of Plasmids
2.5. Lentivirus Packing and Infection
2.6. Western Blot
2.7. Quantitative Reverse Transcription Polymerase Chain Reaction
2.8. Statistical Analysis
3. Results
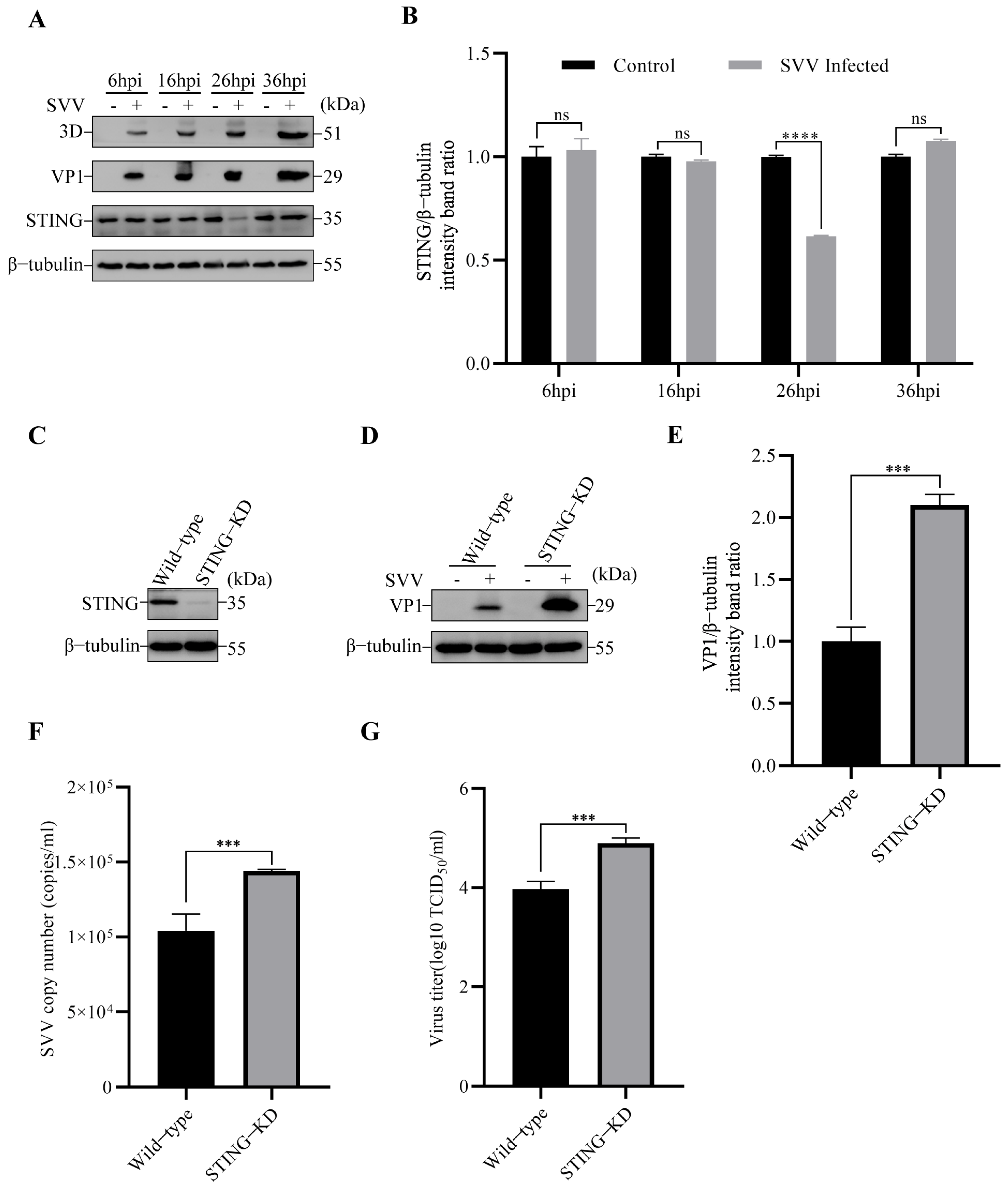
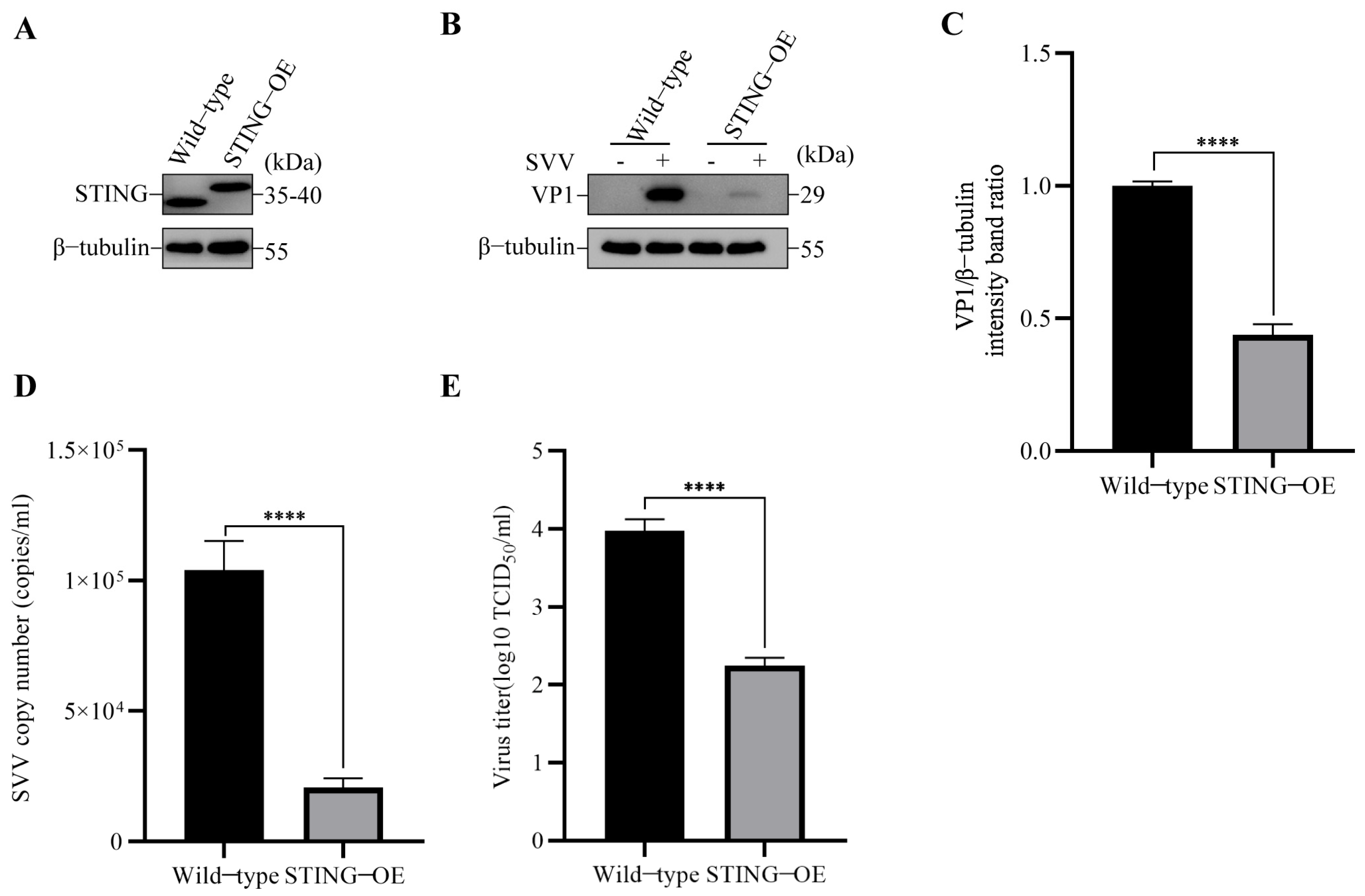
3.1. SVV Infection Reduced STING Protein Level and STING Inhibited SVV Replication
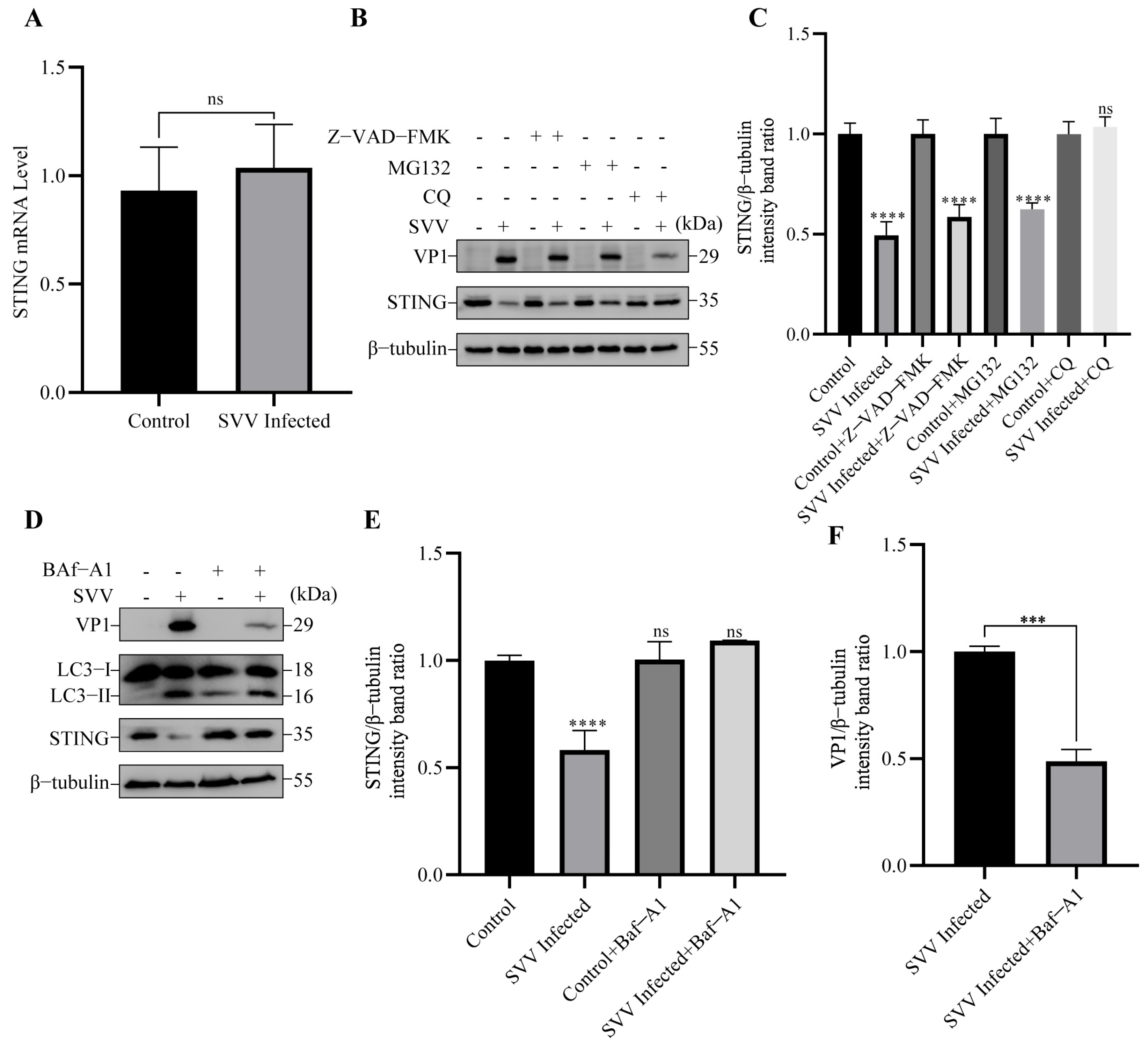
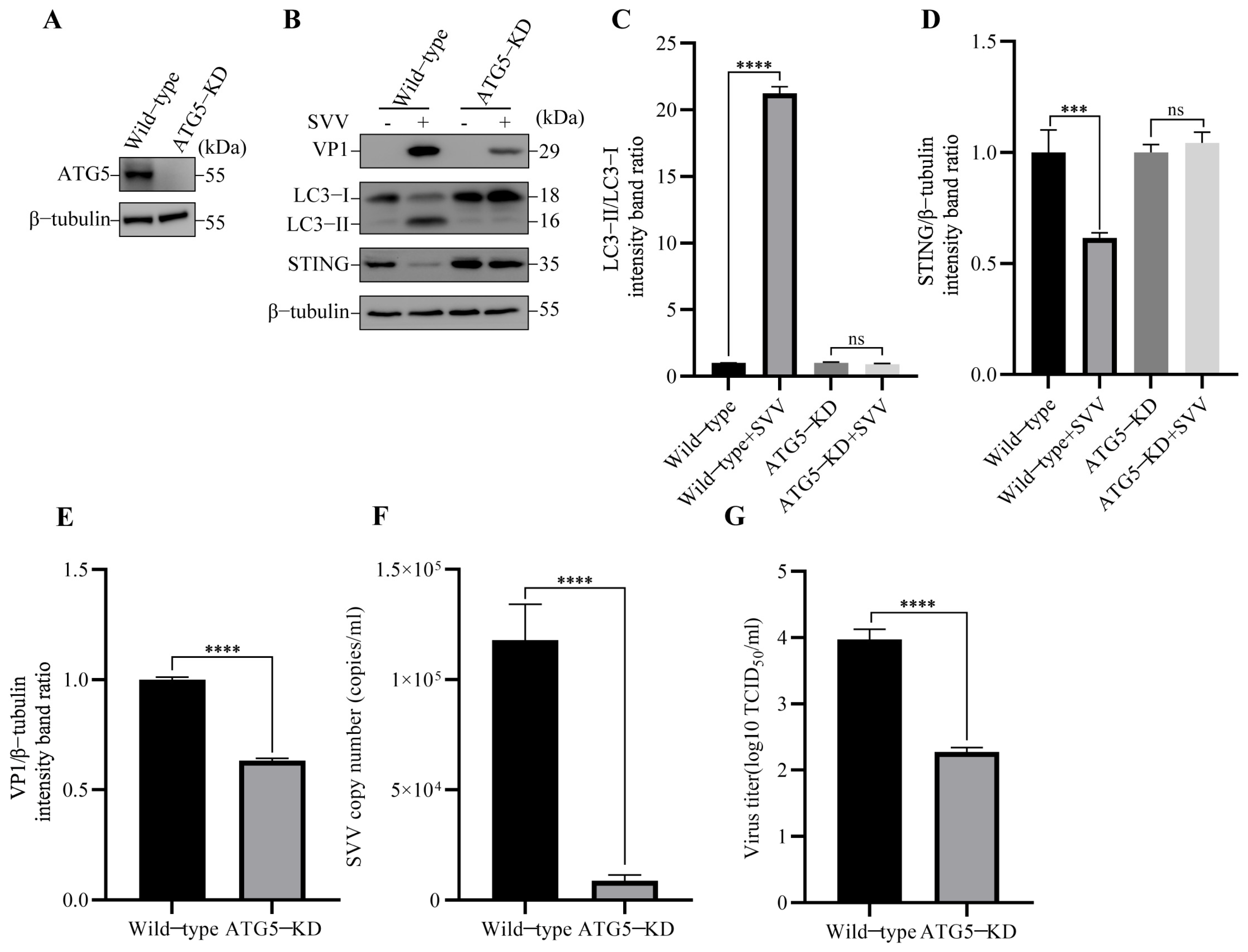
3.2. SVV Infection Leads to the Degradation of STING via Autophagy
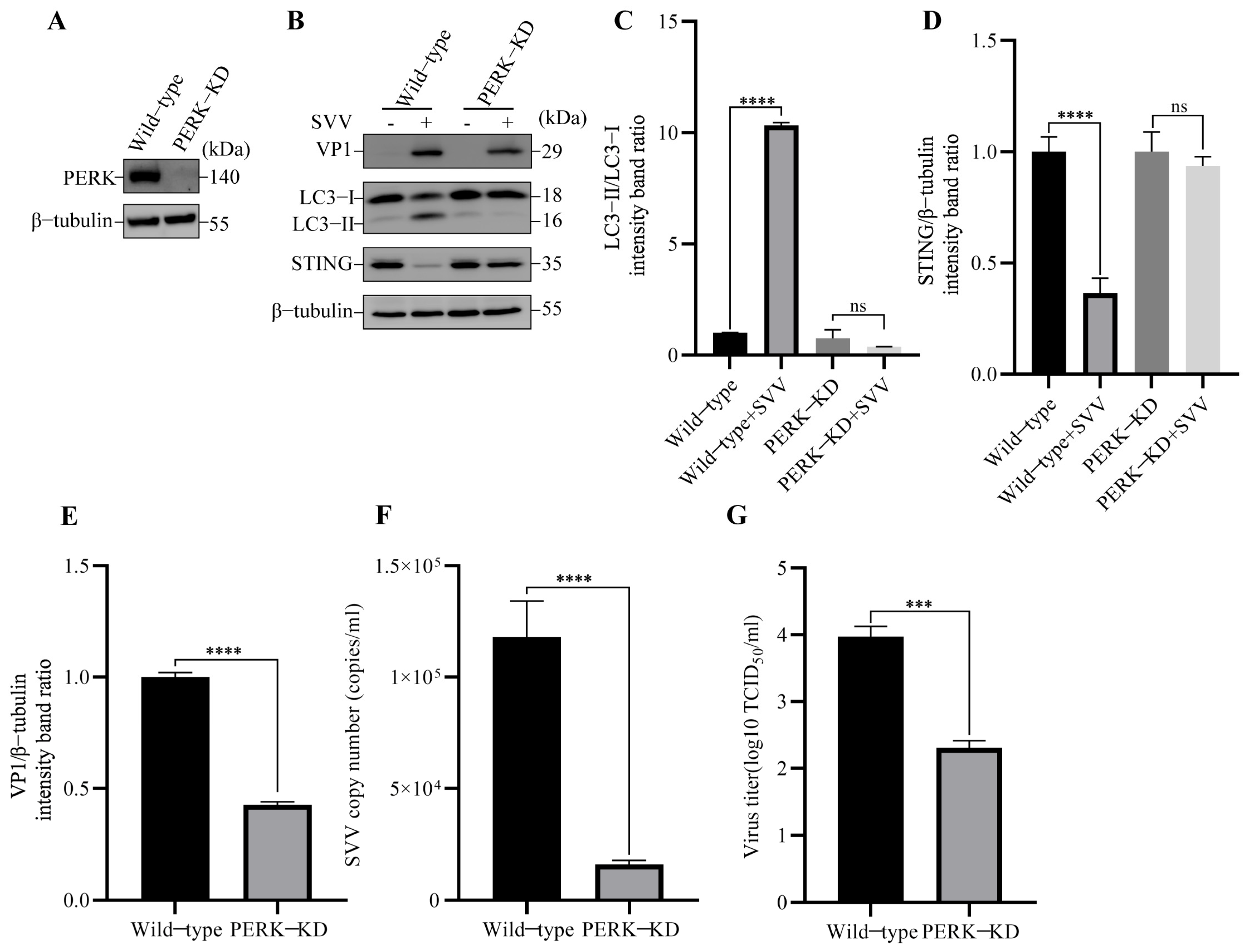
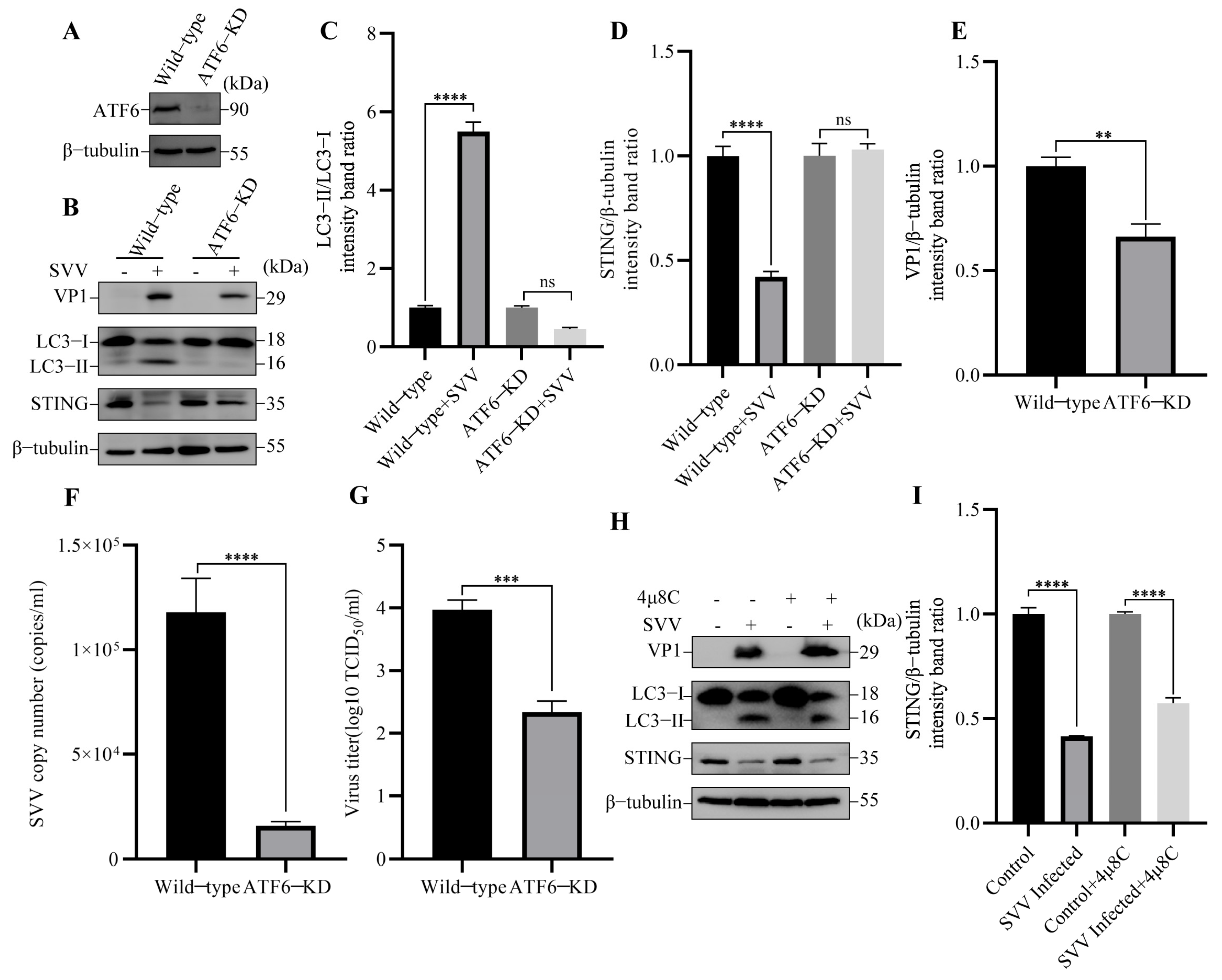
3.3. SVV-Induced Reticulophagy Degrades STING via PERK and ATF6
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hales, L.M.; Knowles, N.J.; Reddy, P.S.; Xu, L.; Hay, C.; Hallenbeck, P.L. Complete genome sequence analysis of Seneca Valley virus-001, a novel oncolytic picornavirus. J. Gen. Virol. 2008, 89, 1265–1275. [Google Scholar] [CrossRef] [PubMed]
- Buckley, A.; Kulshreshtha, V.; van Geelen, A.; Montiel, N.; Guo, B.; Yoon, K.J.; Lager, K. Experimental Seneca Valley virus infection in market-weight gilts. Vet. Microbiol. 2019, 231, 7–10. [Google Scholar] [CrossRef]
- Liu, F.; Wang, Q.; Huang, Y.; Wang, N.; Shan, H. A 5-Year Review of Senecavirus A in China since Its Emergence in 2015. Front. Vet. Sci. 2020, 7, 567792. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Mo, J.; Swanson, K.V.; Wen, H.; Petrucelli, A.; Gregory, S.M.; Zhang, Z.; Schneider, M.; Jiang, Y.; Fitzgerald, K.A.; et al. NLRC3, a member of the NLR family of proteins, is a negative regulator of innate immune signaling induced by the DNA sensor STING. Immunity 2014, 40, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Ma, Z.; Barber, G.N. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 2009, 461, 788–792. [Google Scholar] [CrossRef]
- Maringer, K.; Fernandez-Sesma, A. Message in a bottle: Lessons learned from antagonism of STING signalling during RNA virus infection. Cytokine Growth Factor Rev. 2014, 25, 669–679. [Google Scholar] [CrossRef] [PubMed]
- Barber, G.N. STING-dependent cytosolic DNA sensing pathways. Trends Immunol. 2014, 35, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Woodward, J.J.; Iavarone, A.T.; Portnoy, D.A. c-di-AMP secreted by intracellular Listeria monocytogenes activates a host type I interferon response. Science 2010, 328, 1703–1705. [Google Scholar] [CrossRef] [PubMed]
- Wan, D.; Jiang, W.; Hao, J. Research Advances in How the cGAS-STING Pathway Controls the Cellular Inflammatory Response. Front. Immunol. 2020, 11, 615. [Google Scholar] [CrossRef]
- Li, Y.; He, M.; Wang, Z.; Duan, Z.; Guo, Z.; Wang, Z.; Gong, R.; Chu, T.; Cai, J.; Gao, B. STING signaling activation inhibits HBV replication and attenuates the severity of liver injury and HBV-induced fibrosis. Cell. Mol. Immunol. 2022, 19, 92–107. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zhao, M.; Chang, B.; Zhou, Y.; Wu, X.; Ma, M.; Liu, S.; Cao, Y.; Zheng, M.; Dang, Y.; et al. Cytoplasmic PARP1 links the genome instability to the inhibition of antiviral immunity through PARylating cGAS. Mol. Cell 2022, 82, 2032–2049.e2037. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Liu, C.; Yue, R.; El-Ashram, S.; Wang, J.; He, X.; Zhao, D.; Zhou, X.; Xu, L. cGAS/STING/TBK1/IRF3 Signaling Pathway Activates BMDCs Maturation Following Mycobacterium bovis Infection. Int. J. Mol. Sci. 2019, 20, 895. [Google Scholar] [CrossRef]
- Alonso Paiva, I.M.; Santos, A.R.; Brito, C.B.; Ferrero, M.C.; Ortiz Wilczyñski, J.M.; Carrera Silva, E.A.; Oliveira, S.C.; Baldi, P.C. Role of the cGAS/STING pathway in the control of Brucella abortus infection acquired through the respiratory route. Front. Immunol. 2023, 14, 1116811. [Google Scholar] [CrossRef] [PubMed]
- Nazmi, A.; Mukhopadhyay, R.; Dutta, K.; Basu, A. STING mediates neuronal innate immune response following Japanese encephalitis virus infection. Sci. Rep. 2012, 2, 347. [Google Scholar] [CrossRef] [PubMed]
- Zevini, A.; Olagnier, D.; Hiscott, J. Crosstalk between Cytoplasmic RIG-I and STING Sensing Pathways. Trends Immunol. 2017, 38, 194–205. [Google Scholar] [CrossRef]
- Neufeldt, C.J.; Cerikan, B.; Cortese, M.; Frankish, J.; Lee, J.Y.; Plociennikowska, A.; Heigwer, F.; Prasad, V.; Joecks, S.; Burkart, S.S.; et al. SARS-CoV-2 infection induces a pro-inflammatory cytokine response through cGAS-STING and NF-κB. Commun. Biol. 2022, 5, 45. [Google Scholar] [CrossRef] [PubMed]
- Tian, M.; Liu, W.; Zhang, Q.; Huang, Y.; Li, W.; Wang, W.; Zhao, P.; Huang, S.; Song, Y.; Shereen, M.A.; et al. MYSM1 Represses Innate Immunity and Autoimmunity through Suppressing the cGAS-STING Pathway. Cell Rep. 2020, 33, 108297. [Google Scholar] [CrossRef]
- Lou, M.; Huang, D.; Zhou, Z.; Shi, X.; Wu, M.; Rui, Y.; Su, J.; Zheng, W.; Yu, X.F. DNA virus oncoprotein HPV18 E7 selectively antagonizes cGAS-STING-triggered innate immune activation. J. Med. Virol. 2023, 95, e28310. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Lee, M.S. Autophagy--a key player in cellular and body metabolism. Nat. Rev. Endocrinol. 2014, 10, 322–337. [Google Scholar] [CrossRef]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 2011, 27, 107–132. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B. Autophagy in mammalian development and differentiation. Nat. Cell Biol. 2010, 12, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Yao, R.Q.; Ren, C.; Xia, Z.F.; Yao, Y.M. Organelle-specific autophagy in inflammatory diseases: A potential therapeutic target underlying the quality control of multiple organelles. Autophagy 2021, 17, 385–401. [Google Scholar] [CrossRef] [PubMed]
- Uno, N.; Ross, T.M. Dengue virus and the host innate immune response. Emerg. Microbes Infect. 2018, 7, 167. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Wang, J.; Wang, X.; Wang, B.; Zhao, Z.; Fu, J.; Wang, Y.; Zhang, X.; Zhu, P.; Jiang, M.; et al. Degradation of HDAC10 by autophagy promotes IRF3-mediated antiviral innate immune responses. Sci. Signal. 2022, 15, eabo4356. [Google Scholar] [CrossRef] [PubMed]
- Zhai, X.; Kong, N.; Zhang, Y.; Song, Y.; Qin, W.; Yang, X.; Ye, C.; Ye, M.; Tong, W.; Liu, C.; et al. N protein of PEDV plays chess game with host proteins by selective autophagy. Autophagy 2023, 19, 2338–2352. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Zhong, G.; Ding, S.; Xu, K.; Peng, X.; Dong, W.; Zhou, J. African swine fever virus protein p17 promotes mitophagy by facilitating the interaction of SQSTM1 with TOMM70. Virulence 2023, 14, 2232707. [Google Scholar] [CrossRef]
- Zhang, R.; Qin, X.; Yang, Y.; Zhu, X.; Zhao, S.; Zhang, Z.; Su, Q.; Zhao, Z.; Yin, X.; Meng, X.; et al. STING1 is essential for an RNA-virus triggered autophagy. Autophagy 2022, 18, 816–828. [Google Scholar] [CrossRef]
- Wen, W.; Li, X.; Yin, M.; Wang, H.; Qin, L.; Li, H.; Liu, W.; Zhao, Z.; Zhao, Q.; Chen, H.; et al. Selective autophagy receptor SQSTM1/p62 inhibits Seneca Valley virus replication by targeting viral VP1 and VP3. Autophagy 2021, 17, 3763–3775. [Google Scholar] [CrossRef] [PubMed]
- Hou, L.; Dong, J.; Zhu, S.; Yuan, F.; Wei, L.; Wang, J.; Quan, R.; Chu, J.; Wang, D.; Jiang, H.; et al. Seneca valley virus activates autophagy through the PERK and ATF6 UPR pathways. Virology 2019, 537, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Hou, L.; Quan, R.; Wang, D.; Jiang, H.; Liu, J. Synergetic Contributions of Viral VP1, VP3, and 3C to Activation of the AKT-AMPK-MAPK-MTOR Signaling Pathway for Seneca Valley Virus-Induced Autophagy. J. Virol. 2022, 96, e0155021. [Google Scholar] [CrossRef]
- Lee, Y.R.; Kuo, S.H.; Lin, C.Y.; Fu, P.J.; Lin, Y.S.; Yeh, T.M.; Liu, H.S. Dengue virus-induced ER stress is required for autophagy activation, viral replication, and pathogenesis both in vitro and in vivo. Sci. Rep. 2018, 8, 489. [Google Scholar] [CrossRef]
- Chen, Q.; Men, Y.; Wang, D.; Xu, D.; Liu, S.; Xiao, S.; Fang, L. Porcine reproductive and respiratory syndrome virus infection induces endoplasmic reticulum stress, facilitates virus replication, and contributes to autophagy and apoptosis. Sci. Rep. 2020, 10, 13131. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.Y.; Huang, H.I. Autophagy is induced and supports virus replication in Enterovirus A71-infected human primary neuronal cells. Sci. Rep. 2020, 10, 15234. [Google Scholar] [CrossRef] [PubMed]
- Diao, F.; Jiang, C.; Sun, Y.; Gao, Y.; Bai, J.; Nauwynck, H.; Wang, X.; Yang, Y.; Jiang, P.; Liu, X. Porcine reproductive and respiratory syndrome virus infection triggers autophagy via ER stress-induced calcium signaling to facilitate virus replication. PLoS Pathog. 2023, 19, e1011295. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Yang, F.; Chen, P.; Liu, H.; Cao, W.; Zhang, K.; Liu, X.; Zheng, H. Emergence of novel Seneca Valley virus strains in China, 2017. Transbound. Emerg. Dis. 2017, 64, 1024–1029. [Google Scholar] [CrossRef]
- Tang, J.; Abdullah, S.W.; Li, P.; Wu, J.; Pei, C.; Mu, S.; Wang, Y.; Sun, S.; Guo, H. Heat Shock Protein 60 Is Involved in Viral Replication Complex Formation and Facilitates Foot and Mouth Virus Replication by Stabilizing Viral Nonstructural Proteins 3A and 2C. mBio 2022, 13, e0143422. [Google Scholar] [CrossRef]
- Ishikawa, H.; Barber, G.N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 2008, 455, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Waterman, P.M.; Jonscher, K.R.; Short, C.M.; Reisdorph, N.A.; Cambier, J.C. MPYS, a novel membrane tetraspanner, is associated with major histocompatibility complex class II and mediates transduction of apoptotic signals. Mol. Cell Biol. 2008, 28, 5014–5026. [Google Scholar] [CrossRef]
- Sun, W.; Li, Y.; Chen, L.; Chen, H.; You, F.; Zhou, X.; Zhou, Y.; Zhai, Z.; Chen, D.; Jiang, Z. ERIS, an endoplasmic reticulum IFN stimulator, activates innate immune signaling through dimerization. Proc. Natl. Acad. Sci. USA 2009, 106, 8653–8658. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Xu, P. Cellular functions of cGAS-STING signaling. Trends Cell Biol. 2023, 33, 630–648. [Google Scholar] [CrossRef]
- Aguirre, S.; Luthra, P.; Sanchez-Aparicio, M.T.; Maestre, A.M.; Patel, J.; Lamothe, F.; Fredericks, A.C.; Tripathi, S.; Zhu, T.; Pintado-Silva, J.; et al. Dengue virus NS2B protein targets cGAS for degradation and prevents mitochondrial DNA sensing during infection. Nat. Microbiol. 2017, 2, 17037. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yang, X.; Zheng, Y.; Yang, Y.; Xing, Y.; Chen, Z. SARS coronavirus papain-like protease inhibits the type I interferon signaling pathway through interaction with the STING-TRAF3-TBK1 complex. Protein Cell 2014, 5, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Zhong, B.; Yang, Y.; Li, S.; Wang, Y.Y.; Li, Y.; Diao, F.; Lei, C.; He, X.; Zhang, L.; Tien, P.; et al. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity 2008, 29, 538–550. [Google Scholar] [CrossRef] [PubMed]
- Fu, F.; Shao, Q.; Zhang, J.; Wang, J.; Wang, Z.; Ma, J.; Yan, Y.; Sun, J.; Cheng, Y. Bat STING drives IFN-beta production in anti-RNA virus innate immune response. Front. Microbiol. 2023, 14, 1232314. [Google Scholar] [CrossRef]
- Franz, K.M.; Neidermyer, W.J.; Tan, Y.J.; Whelan, S.P.J.; Kagan, J.C. STING-dependent translation inhibition restricts RNA virus replication. Proc. Natl. Acad. Sci. USA 2018, 115, E2058–E2067. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Xia, N.; Luo, J.; Zhang, Y.; Cao, Q.; Zhang, J.; Wang, Y.; Zhao, Y.; Zheng, W.; Chen, N.; et al. The Porcine Cyclic GMP-AMP Synthase-STING Pathway Exerts an Unusual Antiviral Function Independent of Interferon and Autophagy. J. Virol. 2022, 96, e0147622. [Google Scholar] [CrossRef]
- Ulasli, M.; Verheije, M.H.; de Haan, C.A.; Reggiori, F. Qualitative and quantitative ultrastructural analysis of the membrane rearrangements induced by coronavirus. Cell. Microbiol. 2010, 12, 844–861. [Google Scholar] [CrossRef]
- Snijder, E.J.; Limpens, R.; de Wilde, A.H.; de Jong, A.W.M.; Zevenhoven-Dobbe, J.C.; Maier, H.J.; Faas, F.; Koster, A.J.; Bárcena, M. A unifying structural and functional model of the coronavirus replication organelle: Tracking down RNA synthesis. PLoS Biol. 2020, 18, e3000715. [Google Scholar] [CrossRef]
- Cortese, M.; Lee, J.Y.; Cerikan, B.; Neufeldt, C.J.; Oorschot, V.M.J.; Köhrer, S.; Hennies, J.; Schieber, N.L.; Ronchi, P.; Mizzon, G.; et al. Integrative Imaging Reveals SARS-CoV-2-Induced Reshaping of Subcellular Morphologies. Cell Host Microbe 2020, 28, 853–866.e855. [Google Scholar] [CrossRef]
- Klein, S.; Cortese, M.; Winter, S.L.; Wachsmuth-Melm, M.; Neufeldt, C.J.; Cerikan, B.; Stanifer, M.L.; Boulant, S.; Bartenschlager, R.; Chlanda, P. SARS-CoV-2 structure and replication characterized by in situ cryo-electron tomography. Nat. Commun. 2020, 11, 5885. [Google Scholar] [CrossRef]
- Webb, L.G.; Veloz, J.; Pintado-Silva, J.; Zhu, T.; Rangel, M.V.; Mutetwa, T.; Zhang, L.; Bernal-Rubio, D.; Figueroa, D.; Carrau, L.; et al. Chikungunya virus antagonizes cGAS-STING mediated type-I interferon responses by degrading cGAS. PLoS Pathog. 2020, 16, e1008999. [Google Scholar] [CrossRef]
- Sun, M.; Yu, S.; Ge, H.; Wang, T.; Li, Y.; Zhou, P.; Pan, L.; Han, Y.; Yang, Y.; Sun, Y.; et al. The A137R Protein of African Swine Fever Virus Inhibits Type I Interferon Production via the Autophagy-Mediated Lysosomal Degradation of TBK1. J. Virol. 2022, 96, e0195721. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wu, X.; Wang, H.; Wei, J.; Wu, Q.; Wang, X.; Yan, Y.; Cui, J.; Min, J.; Wang, F.; et al. HFE inhibits type I IFNs signaling by targeting the SQSTM1-mediated MAVS autophagic degradation. Autophagy 2021, 17, 1962–1977. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.; Kanyema, M.M.; Sun, Y.; Zhao, W.; Lu, Y.; Wang, J.; Li, X.; Shi, C.; Wang, J.; Wang, N.; et al. African Swine Fever Virus L83L Negatively Regulates the cGAS-STING-Mediated IFN-I Pathway by Recruiting Tollip to Promote STING Autophagic Degradation. J. Virol. 2023, 97, e0192322. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Yang, W.; Li, L.; Li, P.; Ma, Z.; Zhang, J.; Qi, X.; Ren, J.; Ru, Y.; Niu, Q.; et al. African Swine Fever Virus MGF-505-7R Negatively Regulates cGAS-STING-Mediated Signaling Pathway. J. Immunol. 2021, 206, 1844–1857. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.L.; Li, Z.C.; Zhang, C.; Jiang, J.Y.; Han, K.J.; Tong, J.F.; Yang, X.L.; Chen, D.D.; Lu, L.F.; Li, S. Spring Viremia of Carp Virus N Protein Negatively Regulates IFN Induction through Autophagy-Lysosome-Dependent Degradation of STING. J. Immunol. 2023, 210, 72–81. [Google Scholar] [CrossRef]
- Liu, H.; Zhu, Z.; Xue, Q.; Yang, F.; Li, Z.; Xue, Z.; Cao, W.; He, J.; Guo, J.; Liu, X.; et al. Innate sensing of picornavirus infection involves cGAS-STING-mediated antiviral responses triggered by mitochondrial DNA release. PLoS Pathog. 2023, 19, e1011132. [Google Scholar] [CrossRef]
- Huang, S.C.; Chang, C.L.; Wang, P.S.; Tsai, Y.; Liu, H.S. Enterovirus 71-induced autophagy detected in vitro and in vivo promotes viral replication. J. Med. Virol. 2009, 81, 1241–1252. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Huang, L.; Sun, W.; Chen, Y.; He, M.L.; Yue, J.; Ballard, H. Saikosaponin D suppresses enterovirus A71 infection by inhibiting autophagy. Signal Transduct. Target. Ther. 2019, 4, 4. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.; Zhang, J.; Si, X.; Gao, G.; Mao, I.; McManus, B.M.; Luo, H. Autophagosome supports coxsackievirus B3 replication in host cells. J. Virol. 2008, 82, 9143–9153. [Google Scholar] [CrossRef]
- Tabor-Godwin, J.M.; Tsueng, G.; Sayen, M.R.; Gottlieb, R.A.; Feuer, R. The role of autophagy during coxsackievirus infection of neural progenitor and stem cells. Autophagy 2012, 8, 938–953. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, V.; Pacheco, J.M.; LaRocco, M.; Burrage, T.; Jackson, W.; Rodriguez, L.L.; Borca, M.V.; Baxt, B. Foot-and-mouth disease virus utilizes an autophagic pathway during viral replication. Virology 2011, 410, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Corona, A.K.; Saulsbery, H.M.; Corona Velazquez, A.F.; Jackson, W.T. Enteroviruses Remodel Autophagic Trafficking through Regulation of Host SNARE Proteins to Promote Virus Replication and Cell Exit. Cell Rep. 2018, 22, 3304–3314. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, G.; Sgadari, M.; Longobardi, C.; Iovane, G.; Pagnini, U.; Montagnaro, S. Autophagy up-regulation upon FeHV-1 infection on permissive cells. Front. Vet. Sci. 2023, 10, 1174681. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Wang, M.; Song, Z.; Wang, Z.; Liu, Q.; Jiang, P.; Bai, J.; Li, Y.; Wang, X. Pseudorabies virus induces autophagy to enhance viral replication in mouse neuro-2a cells in vitro. Virus Res. 2018, 248, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, G.; Longobardi, C.; Damiano, S.; Ciarcia, R.; Pagnini, U.; Montagnaro, S. Modifications of the PI3K/Akt/mTOR axis during FeHV-1 infection in permissive cells. Front. Vet. Sci. 2023, 10, 1157350. [Google Scholar] [CrossRef]
- Wang, C.; Hu, R.; Duan, L.; Hou, Q.; Yang, M.; Wang, T.; Liu, H.; Xiao, S.; Dang, R.; Wang, J.; et al. The canonical Wnt/β-catenin signaling pathway facilitates pseudorabies virus proliferation and enhances virus-induced autophagy. Vet. Microbiol. 2022, 272, 109502. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Luo, S.; Wang, X.; Li, C.; Yang, J.; Zhu, X.; Xiao, L.; Sun, L. ER-Phagy: A New Regulator of ER Homeostasis. Front. Cell Dev. Biol. 2021, 9, 684526. [Google Scholar] [CrossRef]
- Smith, M.; Wilkinson, S. ER homeostasis and autophagy. Essays Biochem. 2017, 61, 625–635. [Google Scholar] [CrossRef]
- Wang, J.; Kang, R.; Huang, H.; Xi, X.; Wang, B.; Wang, J.; Zhao, Z. Hepatitis C virus core protein activates autophagy through EIF2AK3 and ATF6 UPR pathway-mediated MAP1LC3B and ATG12 expression. Autophagy 2014, 10, 766–784. [Google Scholar] [CrossRef] [PubMed]
- Shi, D.; Zhou, L.; Shi, H.; Zhang, J.; Zhang, J.; Zhang, L.; Liu, D.; Feng, T.; Zeng, M.; Chen, J.; et al. Autophagy is induced by swine acute diarrhea syndrome coronavirus through the cellular IRE1-JNK-Beclin 1 signaling pathway after an interaction of viral membrane-associated papain-like protease and GRP78. PLoS Pathog. 2023, 19, e1011201. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Jin, J.; Wang, L.; Wang, J.; Zhou, H.; Zhang, Q.; Xu, X. Porcine epidemic diarrhea virus infections induce autophagy in Vero cells via ROS-dependent endoplasmic reticulum stress through PERK and IRE1 pathways. Vet. Microbiol. 2021, 253, 108959. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bai, L.; Zhang, R.; Zheng, H.; Zhang, Z.; Zhang, Z.; Li, Y. Seneca Valley Virus Degrades STING via PERK and ATF6-Mediated Reticulophagy. Viruses 2023, 15, 2209. https://doi.org/10.3390/v15112209
Bai L, Zhang R, Zheng H, Zhang Z, Zhang Z, Li Y. Seneca Valley Virus Degrades STING via PERK and ATF6-Mediated Reticulophagy. Viruses. 2023; 15(11):2209. https://doi.org/10.3390/v15112209
Chicago/Turabian StyleBai, Ling, Rui Zhang, Haixue Zheng, Zhixiong Zhang, Zhidong Zhang, and Yanmin Li. 2023. "Seneca Valley Virus Degrades STING via PERK and ATF6-Mediated Reticulophagy" Viruses 15, no. 11: 2209. https://doi.org/10.3390/v15112209
APA StyleBai, L., Zhang, R., Zheng, H., Zhang, Z., Zhang, Z., & Li, Y. (2023). Seneca Valley Virus Degrades STING via PERK and ATF6-Mediated Reticulophagy. Viruses, 15(11), 2209. https://doi.org/10.3390/v15112209