
The Dynamics of Synthesis and Localization of Jumbo Phage RNA Polymerases inside Infected Cells
 ,
,  , ,
, , {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacteriophage, Bacterial Strain and Growth Conditions
2.2. Cloning
2.3. Protein Expression and Purification
2.4. Western Blotting
2.5. Co-Immunoprecipitation
2.6. Mass Spectrometry Analysis
2.7. Fluorescence Microscopy
3. Results
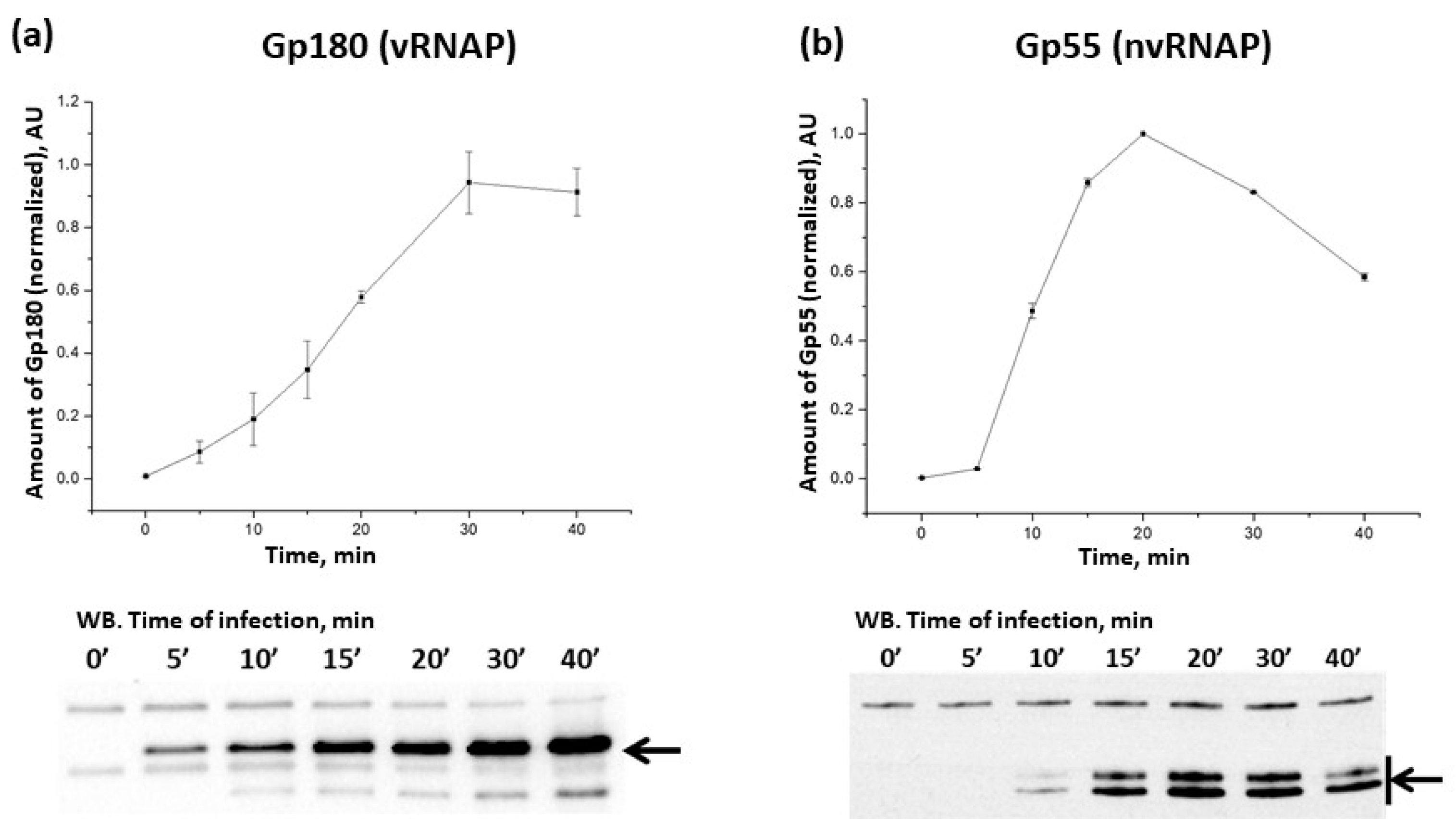
3.1. Dynamics of phiKZ RNAPs Accumulation during Infection
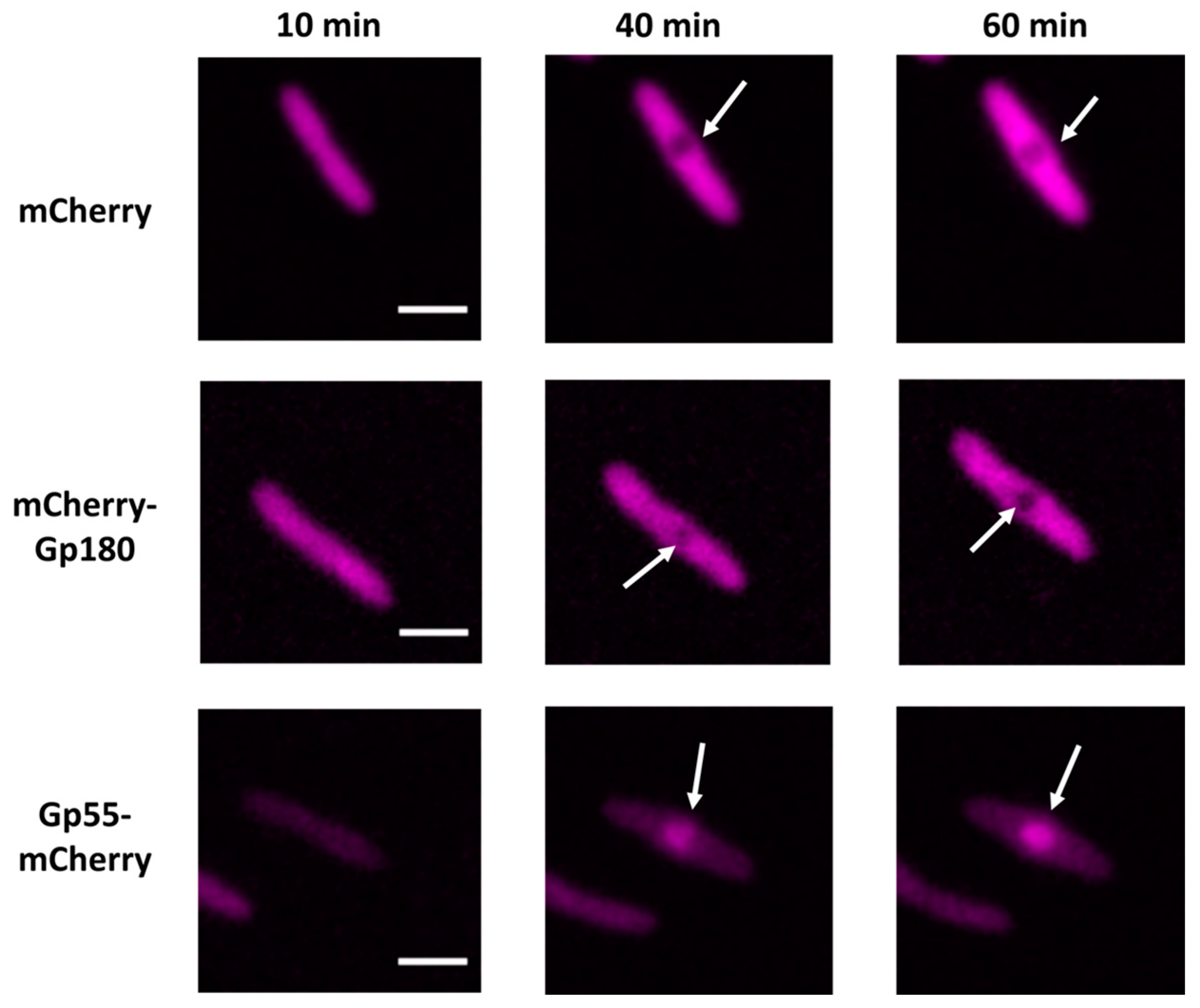
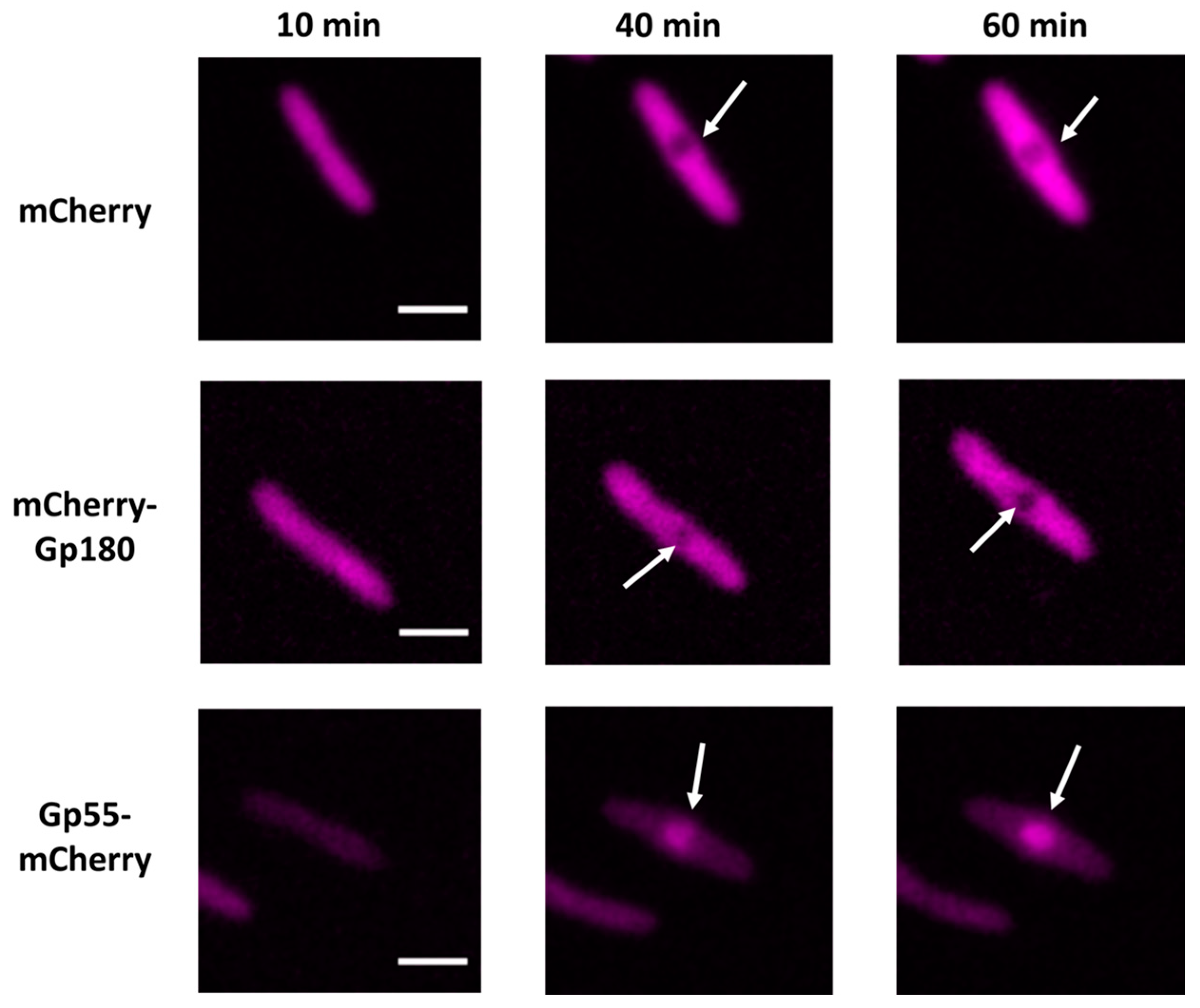
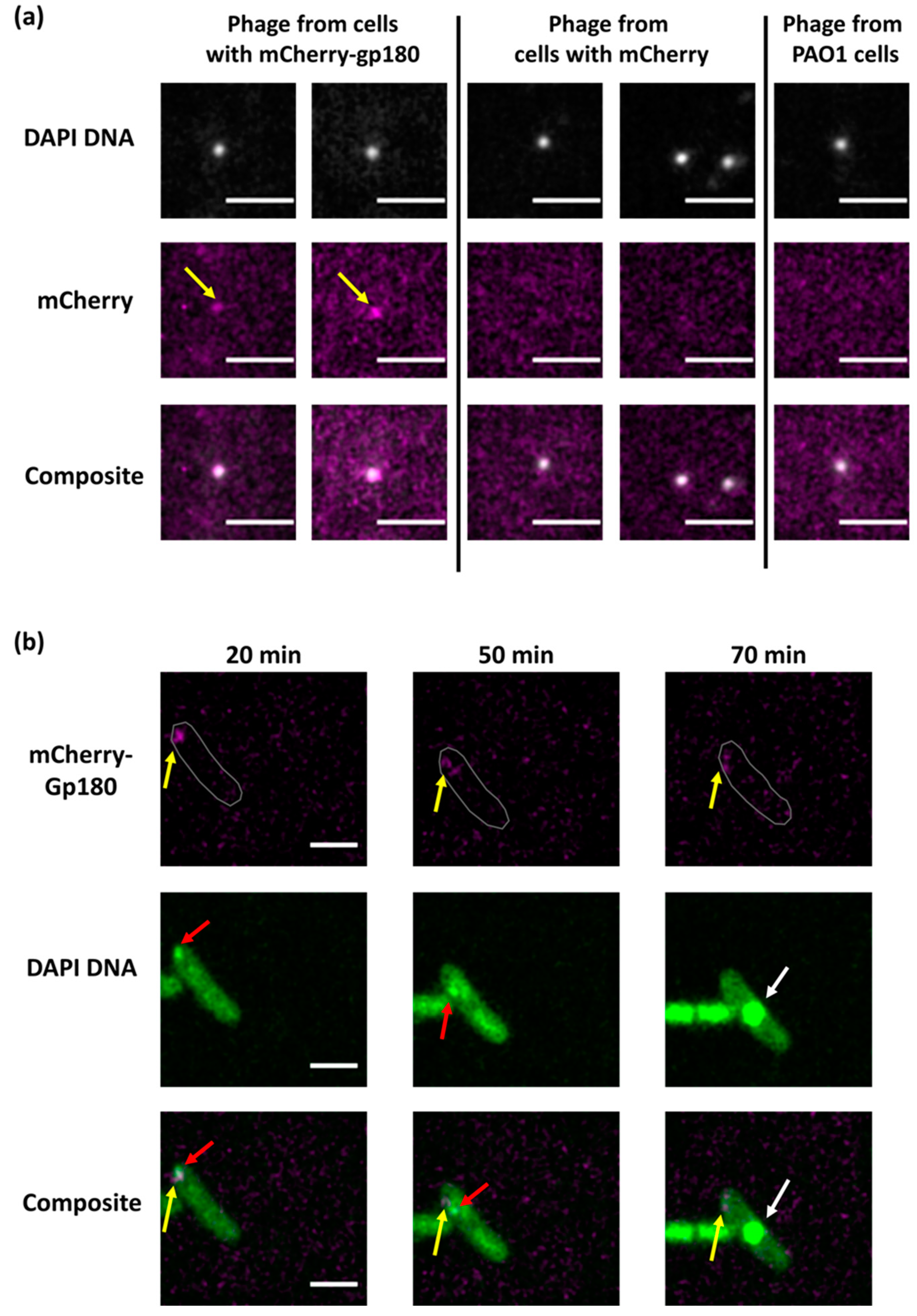
3.2. Determination of Localization of Phage RNAPs during Infection Using Fluorescence Microscopy
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Miller, E.S.; Kutter, E.; Mosig, G.; Arisaka, F.; Kunisawa, T.; Rüger, W. Bacteriophage T4 Genome. Microbiol. Mol. Biol. Rev. 2003, 67, 86–156. [Google Scholar] [CrossRef] [PubMed]
- Nechaev, S.; Severinov, K. Bacteriophage-Induced Modifications of Host RNA Polymerase. Annu. Rev. Microbiol. 2003, 57, 301–322. [Google Scholar] [CrossRef]
- Casjens, S.R.; Hendrix, R.W. Bacteriophage Lambda: Early Pioneer and Still Relevant. Virology 2015, 479–480, 310–330. [Google Scholar] [CrossRef] [PubMed]
- Greenblatt, J.; Nodwell, J.R.; Mason, S.W. Transcriptional Antitermination. Nature 1993, 364, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Węgrzyn, G.; Licznerska, K.; Węgrzyn, A. Chapter 6—Phage λ—New Insights into Regulatory Circuits. In Advances in Virus Research; Łobocka, M., Szybalski, W.T., Eds.; Bacteriophages, Part A; Academic Press: Cambridge, MA, USA, 2012; Volume 82, pp. 155–178. [Google Scholar]
- McAllister, W.T.; Barrett, C.L. Roles of the Early Genes of Bacteriophage T7 in Shutoff of Host Macromolecular Synthesis. J. Virol. 1977, 23, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Falco, S.C.; Zehring, W.; Rothman-Denes, L.B. DNA-Dependent RNA Polymerase from Bacteriophage N4 Virions. Purification and Characterization. J. Biol. Chem. 1980, 255, 4339–4347. [Google Scholar] [CrossRef] [PubMed]
- Kazmierczak, K.M.; Davydova, E.K.; Mustaev, A.A.; Rothman-Denes, L.B. The Phage N4 Virion RNA Polymerase Catalytic Domain Is Related to Single-Subunit RNA Polymerases. EMBO J. 2002, 21, 5815–5823. [Google Scholar] [CrossRef] [PubMed]
- Molodtsov, V.; Murakami, K.S. Minimalism and Functionality: Structural Lessons from the Heterodimeric N4 Bacteriophage RNA Polymerase II. J. Biol. Chem. 2018, 293, 13616–13625. [Google Scholar] [CrossRef]
- Willis, S.H.; Kazmierczak, K.M.; Carter, R.H.; Rothman-Denes, L.B. N4 RNA Polymerase II, a Heterodimeric RNA Polymerase with Homology to the Single-Subunit Family of RNA Polymerases. J. Bacteriol. 2002, 184, 4952–4961. [Google Scholar] [CrossRef]
- Lenneman, B.R.; Rothman-Denes, L.B. Structural and Biochemical Investigation of Bacteriophage N4-Encoded RNA Polymerases. Biomolecules 2015, 5, 647–667. [Google Scholar] [CrossRef]
- Ceyssens, P.-J.; Minakhin, L.; Van den Bossche, A.; Yakunina, M.; Klimuk, E.; Blasdel, B.; De Smet, J.; Noben, J.-P.; Bläsi, U.; Severinov, K.; et al. Development of Giant Bacteriophage ΦKZ Is Independent of the Host Transcription Apparatus. J. Virol. 2014, 88, 10501–10510. [Google Scholar] [CrossRef] [PubMed]
- Lavysh, D.; Sokolova, M.; Minakhin, L.; Yakunina, M.; Artamonova, T.; Kozyavkin, S.; Makarova, K.S.; Koonin, E.V.; Severinov, K. The Genome of AR9, a Giant Transducing Bacillus Phage Encoding Two Multisubunit RNA Polymerases. Virology 2016, 495, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Matsui, T.; Yoshikawa, G.; Mihara, T.; Chatchawankanphanich, O.; Kawasaki, T.; Nakano, M.; Fujie, M.; Ogata, H.; Yamada, T. Replications of Two Closely Related Groups of Jumbo Phages Show Different Level of Dependence on Host-Encoded RNA Polymerase. Front. Microbiol. 2017, 8, 1010. [Google Scholar] [CrossRef] [PubMed]
- Yakunina, M.; Artamonova, T.; Borukhov, S.; Makarova, K.S.; Severinov, K.; Minakhin, L. A Non-Canonical Multisubunit RNA Polymerase Encoded by a Giant Bacteriophage. Nucleic Acids Res. 2015, 43, 10411–10420. [Google Scholar] [CrossRef] [PubMed]
- Sokolova, M.; Borukhov, S.; Lavysh, D.; Artamonova, T.; Khodorkovskii, M.; Severinov, K. A Non-Canonical Multisubunit RNA Polymerase Encoded by the AR9 Phage Recognizes the Template Strand of Its Uracil-Containing Promoters. Nucleic Acids Res. 2017, 45, 5958–5967. [Google Scholar] [CrossRef] [PubMed]
- Danilova, Y.A.; Belousova, V.V.; Moiseenko, A.V.; Vishnyakov, I.E.; Yakunina, M.V.; Sokolova, O.S. Maturation of Pseudo-Nucleus Compartment in P. aeruginosa, Infected with Giant PhiKZ Phage. Viruses 2020, 12, 1197. [Google Scholar] [CrossRef] [PubMed]
- Chaikeeratisak, V.; Nguyen, K.; Egan, M.E.; Erb, M.L.; Vavilina, A.; Pogliano, J. The Phage Nucleus and Tubulin Spindle Are Conserved among Large Pseudomonas Phages. Cell Rep. 2017, 20, 1563–1571. [Google Scholar] [CrossRef]
- Nieweglowska, E.S.; Brilot, A.F.; Méndez-Moran, M.; Kokontis, C.; Baek, M.; Li, J.; Cheng, Y.; Baker, D.; Bondy-Denomy, J.; Agard, D.A. The ΦPA3 Phage Nucleus Is Enclosed by a Self-Assembling 2D Crystalline Lattice. Nat. Commun. 2023, 14, 927. [Google Scholar] [CrossRef]
- Laughlin, T.G.; Deep, A.; Prichard, A.M.; Seitz, C.; Gu, Y.; Enustun, E.; Suslov, S.; Khanna, K.; Birkholz, E.A.; Armbruster, E.; et al. Architecture and Self-Assembly of the Jumbo Bacteriophage Nuclear Shell. Nature 2022, 608, 429–435. [Google Scholar] [CrossRef]
- Liu, Z.; Xiang, Y. Structural Studies of the Nucleus-like Assembly of Jumbo Bacteriophage 201φ2-1. Front. Microbiol. 2023, 14, 1170112. [Google Scholar] [CrossRef]
- Chaikeeratisak, V.; Nguyen, K.; Khanna, K.; Brilot, A.F.; Erb, M.L.; Coker, J.K.C.; Vavilina, A.; Newton, G.L.; Buschauer, R.; Pogliano, K.; et al. Assembly of a Nucleus-like Structure during Viral Replication in Bacteria. Science 2017, 355, 194–197. [Google Scholar] [CrossRef] [PubMed]
- Carroll-Portillo, A.; Coffman, C.N.; Varga, M.G.; Alcock, J.; Singh, S.B.; Lin, H.C. Standard Bacteriophage Purification Procedures Cause Loss in Numbers and Activity. Viruses 2021, 13, 328. [Google Scholar] [CrossRef] [PubMed]
- Chuanchuen, R.; Narasaki, C.T.; Schweizer, H.P. Benchtop and Microcentrifuge Preparation of Pseudomonas Aeruginosa Competent Cells. BioTechniques 2002, 33, 760–763. [Google Scholar] [CrossRef] [PubMed]
- Orekhova, M.; Koreshova, A.; Artamonova, T.; Khodorkovskii, M.; Yakunina, M. The Study of the PhiKZ Phage Non-Canonical Non-Virion RNA Polymerase. Biochem. Biophys. Res. Commun. 2019, 511, 759–764. [Google Scholar] [CrossRef] [PubMed]
- Morozova, N.; Sabantsev, A.; Bogdanova, E.; Fedorova, Y.; Maikova, A.; Vedyaykin, A.; Rodic, A.; Djordjevic, M.; Khodorkovskii, M.; Severinov, K. Temporal Dynamics of Methyltransferase and Restriction Endonuclease Accumulation in Individual Cells after Introducing a Restriction-Modification System. Nucleic Acids Res. 2016, 44, 790–800. [Google Scholar] [CrossRef] [PubMed]
- Edelstein, A.D.; Tsuchida, M.A.; Amodaj, N.; Pinkard, H.; Vale, R.D.; Stuurman, N. Advanced Methods of Microscope Control Using ΜManager Software. J. Biol. Methods 2014, 1, e10. [Google Scholar] [CrossRef] [PubMed]
- de Martín Garrido, N.; Orekhova, M.; Lai Wan Loong, Y.T.E.; Litvinova, A.; Ramlaul, K.; Artamonova, T.; Melnikov, A.S.; Serdobintsev, P.; Aylett, C.H.S.; Yakunina, M. Structure of the Bacteriophage PhiKZ Non-Virion RNA Polymerase. Nucleic Acids Res. 2021, 49, 7732–7739. [Google Scholar] [CrossRef] [PubMed]
- Sokolova, M.L.; Misovetc, I.; V Severinov, K. Multisubunit RNA Polymerases of Jumbo Bacteriophages. Viruses 2020, 12, 1064. [Google Scholar] [CrossRef]
- Krylov, V.N.; Zhazykov, I.Z. Pseudomonas bacteriophage phiKZ—possible model for studying the genetic control of morphogenesis. Genetika 1978, 14, 678–685. [Google Scholar]
- Van den Bossche, A.; Hardwick, S.W.; Ceyssens, P.-J.; Hendrix, H.; Voet, M.; Dendooven, T.; Bandyra, K.J.; De Maeyer, M.; Aertsen, A.; Noben, J.-P.; et al. Structural Elucidation of a Novel Mechanism for the Bacteriophage-Based Inhibition of the RNA Degradosome. eLife 2016, 5, e16413. [Google Scholar] [CrossRef]
- Chaikeeratisak, V.; Khanna, K.; Nguyen, K.T.; Sugie, J.; Egan, M.E.; Erb, M.L.; Vavilina, A.; Nonejuie, P.; Nieweglowska, E.; Pogliano, K.; et al. Viral Capsid Trafficking along Treadmilling Tubulin Filaments in Bacteria. Cell 2019, 177, 1771–1780.e12. [Google Scholar] [CrossRef]
- Lecoutere, E.; Ceyssens, P.-J.; Miroshnikov, K.A.; Mesyanzhinov, V.V.; Krylov, V.N.; Noben, J.-P.; Robben, J.; Hertveldt, K.; Volckaert, G.; Lavigne, R. Identification and Comparative Analysis of the Structural Proteomes of ΦKZ and EL, Two Giant Pseudomonas Aeruginosa Bacteriophages. Proteomics 2009, 9, 3215–3219. [Google Scholar] [CrossRef]
- Thomas, J.A.; Weintraub, S.T.; Wu, W.; Winkler, D.C.; Cheng, N.; Steven, A.C.; Black, L.W. Extensive Proteolysis of Head and Inner Body Proteins by a Morphogenetic Protease in the Giant Pseudomonas Aeruginosa Phage ΦKZ. Mol. Microbiol. 2012, 84, 324–339. [Google Scholar] [CrossRef]
- Mendoza, S.D.; Nieweglowska, E.S.; Govindarajan, S.; Leon, L.M.; Berry, J.D.; Tiwari, A.; Chaikeeratisak, V.; Pogliano, J.; Agard, D.A.; Bondy-Denomy, J. A Bacteriophage Nucleus-like Compartment Shields DNA from CRISPR Nucleases. Nature 2020, 577, 244–248. [Google Scholar] [CrossRef]
- Armbruster, E.G.; Lee, J.; Hutchings, J.; VanderWal, A.R.; Enustun, E.; Adler, B.A.; Aindow, A.; Deep, A.; Rodriguez, Z.K.; Morgan, C.J.; et al. Sequential Membrane- and Protein-Bound Organelles Compartmentalize Genomes during Phage Infection. bioRxiv 2023. [Google Scholar] [CrossRef]
- Kiljunen, S.; Hakala, K.; Pinta, E.; Huttunen, S.; Pluta, P.; Gador, A.; Lönnberg, H.; Skurnik, M. Yersiniophage PhiR1-37 Is a Tailed Bacteriophage Having a 270 Kb DNA Genome with Thymidine Replaced by Deoxyuridine. Microbiology 2005, 151, 4093–4102. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Antonova, D.; Belousova, V.V.; Zhivkoplias, E.; Sobinina, M.; Artamonova, T.; Vishnyakov, I.E.; Kurdyumova, I.; Arseniev, A.; Morozova, N.; Severinov, K.; et al. The Dynamics of Synthesis and Localization of Jumbo Phage RNA Polymerases inside Infected Cells. Viruses 2023, 15, 2096. https://doi.org/10.3390/v15102096
Antonova D, Belousova VV, Zhivkoplias E, Sobinina M, Artamonova T, Vishnyakov IE, Kurdyumova I, Arseniev A, Morozova N, Severinov K, et al. The Dynamics of Synthesis and Localization of Jumbo Phage RNA Polymerases inside Infected Cells. Viruses. 2023; 15(10):2096. https://doi.org/10.3390/v15102096
Chicago/Turabian StyleAntonova, Daria, Viktoriia V. Belousova, Erik Zhivkoplias, Mariia Sobinina, Tatyana Artamonova, Innokentii E. Vishnyakov, Inna Kurdyumova, Anatoly Arseniev, Natalia Morozova, Konstantin Severinov, and et al. 2023. "The Dynamics of Synthesis and Localization of Jumbo Phage RNA Polymerases inside Infected Cells" Viruses 15, no. 10: 2096. https://doi.org/10.3390/v15102096
APA StyleAntonova, D., Belousova, V. V., Zhivkoplias, E., Sobinina, M., Artamonova, T., Vishnyakov, I. E., Kurdyumova, I., Arseniev, A., Morozova, N., Severinov, K., Khodorkovskii, M., & Yakunina, M. V. (2023). The Dynamics of Synthesis and Localization of Jumbo Phage RNA Polymerases inside Infected Cells. Viruses, 15(10), 2096. https://doi.org/10.3390/v15102096