
Structural Investigations of Interactions between the Influenza a Virus NS1 and Host Cellular Proteins


Abstract
1. Introduction
2. NS1 Structure
3. NS1RBD
4. Interdomain Linker
5. NS1ED
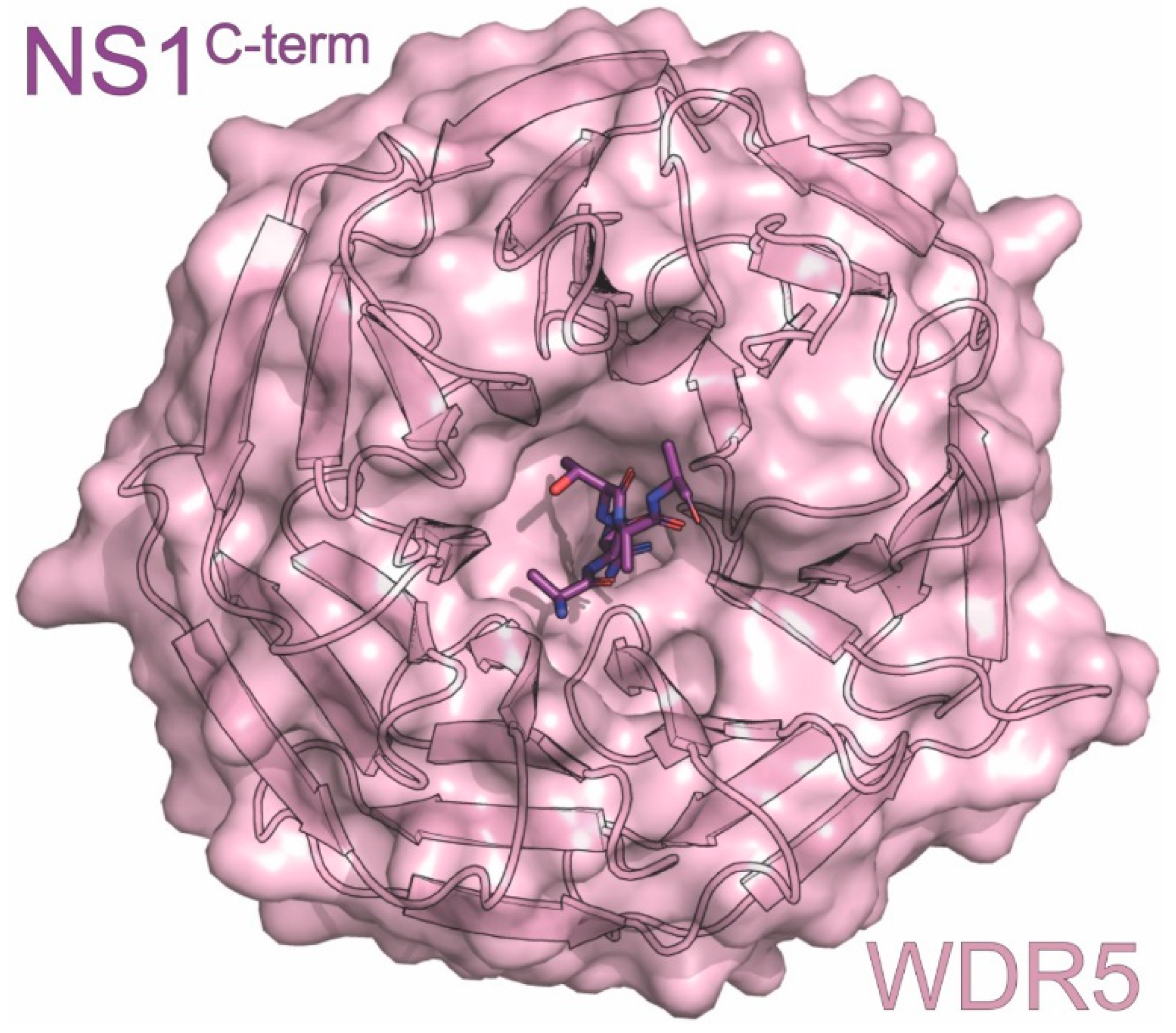
6. NS1C-term
7. CHD1
8. CrkII
9. Scribble
10. MORC3
11. CPSF30
12. NXF1-NXT1
13. PDlim2
14. PI3K
15. RIG-I
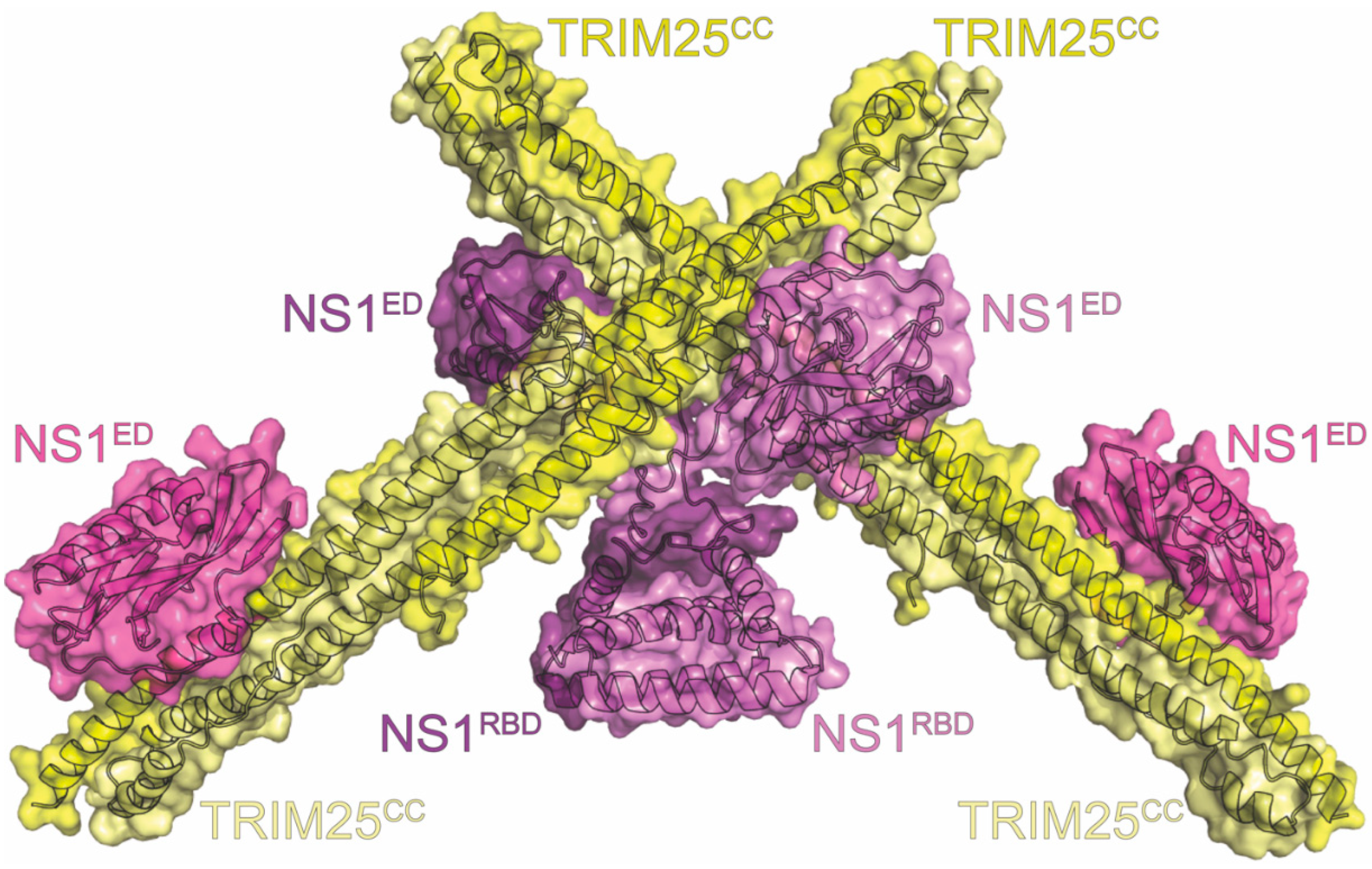
16. TRIM25
17. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Thompson, W.W.; Comanor, L.; Shay, D.K. Epidemiology of seasonal influenza: Use of surveillance data and statistical models to estimate the burden of disease. J. Infect. Dis. 2006, 194 (Suppl. S2), S82–S91. [Google Scholar] [CrossRef] [PubMed]
- Molinari, N.A.; Ortega-Sanchez, I.R.; Messonnier, M.L.; Thompson, W.W.; Wortley, P.M.; Weintraub, E.; Bridges, C.B. The annual impact of seasonal influenza in the US: Measuring disease burden and costs. Vaccine 2007, 25, 5086–5096. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Sugimoto, J.D.; Halloran, M.E.; Basta, N.E.; Chao, D.L.; Matrajt, L.; Potter, G.; Kenah, E.; Longini, I.M., Jr. The transmissibility and control of pandemic influenza A (H1N1) virus. Science 2009, 326, 729–733. [Google Scholar] [CrossRef] [PubMed]
- Muthuri, S.G.; Venkatesan, S.; Myles, P.R.; Leonardi-Bee, J.; Al Khuwaitir, T.S.; Al Mamun, A.; Anovadiya, A.P.; Azziz-Baumgartner, E.; Baez, C.; Bassetti, M.; et al. Effectiveness of neuraminidase inhibitors in reducing mortality in patients admitted to hospital with influenza A H1N1pdm09 virus infection: A meta-analysis of individual participant data. Lancet Respir. Med. 2014, 2, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Liu, T.; Zhang, J.; Zhan, P.; Liu, X. Influenza A virus polymerase: An attractive target for next-generation anti-influenza therapeutics. Drug Discov. Today 2018, 23, 503–518. [Google Scholar] [CrossRef]
- Gamblin, S.J.; Skehel, J.J. Influenza hemagglutinin and neuraminidase membrane glycoproteins. J. Biol. Chem. 2010, 285, 28403–28409. [Google Scholar] [CrossRef]
- Wu, N.C.; Wilson, I.A. Influenza Hemagglutinin Structures and Antibody Recognition. Cold Spring Harb. Perspect. Med. 2020, 10, a038778. [Google Scholar] [CrossRef]
- Giurgea, L.T.; Morens, D.M.; Taubenberger, J.K.; Memoli, M.J. Influenza Neuraminidase: A Neglected Protein and Its Potential for a Better Influenza Vaccine. Vaccines 2020, 8, 409. [Google Scholar] [CrossRef]
- McKimm-Breschkin, J.L. Influenza neuraminidase inhibitors: Antiviral action and mechanisms of resistance. Influenza Other Respir. Viruses 2013, 7 (Suppl. 1), 25–36. [Google Scholar] [CrossRef]
- A revision of the system of nomenclature for influenza viruses: A WHO memorandum. Bull. World Health Organ. 1980, 58, 585–591.
- Krammer, F.; Smith, G.J.D.; Fouchier, R.A.M.; Peiris, M.; Kedzierska, K.; Doherty, P.C.; Palese, P.; Shaw, M.L.; Treanor, J.; Webster, R.G.; et al. Influenza. Nat. Rev. Dis. Primers 2018, 4, 3. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Garcia-Sastre, A.; Schwemmle, M. Expected and Unexpected Features of the Newly Discovered Bat Influenza A-like Viruses. PLoS Pathog. 2015, 11, e10048192015. [Google Scholar] [CrossRef] [PubMed]
- Tong, S.; Li, Y.; Rivailler, P.; Conrardy, C.; Castillo, D.A.; Chen, L.M.; Recuenco, S.; Ellison, J.A.; Davis, C.T.; York, I.A.; et al. A distinct lineage of influenza A virus from bats. Proc. Natl. Acad. Sci. USA 2012, 109, 4269–4274. [Google Scholar] [CrossRef] [PubMed]
- Portela, A.; Digard, P. The influenza virus nucleoprotein: A multifunctional RNA-binding protein pivotal to virus replication. J. Gen. Virol. 2002, 83, 723–734. [Google Scholar] [CrossRef]
- Esparza, M.; Bhat, P.; Fontoura, B.M. Viral-host interactions during splicing and nuclear export of influenza virus mRNAs. Curr. Opin. Virol. 2022, 55, 101254. [Google Scholar] [CrossRef]
- Peukes, J.; Xiong, X.; Briggs, J.A.G. New structural insights into the multifunctional influenza A matrix protein 1. FEBS Lett. 2021, 595, 2535–2543. [Google Scholar] [CrossRef]
- Pinto, L.H.; Lamb, R.A. The M2 proton channels of influenza A and B viruses. J. Biol. Chem. 2006, 281, 8997–9000. [Google Scholar] [CrossRef]
- Jalily, P.H.; Duncan, M.C.; Fedida, D.; Wang, J.; Tietjen, I. Put a cork in it: Plugging the M2 viral ion channel to sink influenza. Antiviral Res. 2020, 178, 104780. [Google Scholar] [CrossRef]
- Krug, R.M. Functions of the influenza A virus NS1 protein in antiviral defense. Curr. Opin. Virol. 2015, 12, 1–6. [Google Scholar] [CrossRef]
- Ayllon, J.; Garcia-Sastre, A. The NS1 protein: A multitasking virulence factor. Curr. Top. Microbiol. Immunol. 2015, 386, 73–107. [Google Scholar] [CrossRef]
- Richardson, J.C.; Akkina, R.K. NS2 protein of influenza virus is found in purified virus and phosphorylated in infected cells. Arch. Virol. 1991, 116, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Ward, A.C.; Castelli, L.A.; Lucantoni, A.C.; White, J.F.; Azad, A.A.; Macreadie, I.G. Expression and analysis of the NS2 protein of influenza A virus. Arch. Virol. 1995, 140, 2067–2073. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, J.; Nakada, S.; Kato, A.; Toyoda, T.; Ishihama, A. Molecular assembly of influenza virus: Association of the NS2 protein with virion matrix. Virology 1993, 196, 249–255. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, R.E.; Talon, J.; Palese, P. The influenza virus NEP (NS2 protein) mediates the nuclear export of viral ribonucleoproteins. EMBO J. 1998, 17, 288–296. [Google Scholar] [CrossRef] [PubMed]
- Akarsu, H.; Burmeister, W.P.; Petosa, C.; Petit, I.; Müller, C.W.; Ruigrok, R.W.H.; Baudin, F. Crystal structure of the M1 protein-binding domain of the influenza A virus nuclear export protein (NEP/NS2). EMBO J. 2003, 22, 4646–4655. [Google Scholar] [CrossRef]
- Huang, S.; Chen, J.; Chen, Q.; Wang, H.; Yao, Y.; Chen, J.; Chen, Z. A Second CRM1-Dependent Nuclear Export Signal in the Influenza A Virus NS2 Protein Contributes to the Nuclear Export of Viral Ribonucleoproteins. J. Virol. 2013, 87, 767–778. [Google Scholar] [CrossRef]
- Robb, N.C.; Smith, M.; Vreede, F.T.; Fodor, E. NS2/NEP protein regulates transcription and replication of the influenza virus RNA genome. J. General. Virol. 2009, 90, 1398–1407. [Google Scholar] [CrossRef]
- Reuther, P.; Giese, S.; Götz, V.; Kilb, N.; Mänz, B.; Brunotte, L.; Schwemmle, M. Adaptive mutations in the nuclear export protein of human-derived H5N1 strains facilitate a polymerase activity-enhancing conformation. J. Virol. 2014, 88, 263–271. [Google Scholar] [CrossRef]
- Mänz, B.; Schwemmle, M.; Brunotte, L. Adaptation of Avian Influenza A Virus Polymerase in Mammals To Overcome the Host Species Barrier. J. Virol. 2013, 87, 7200–7209. [Google Scholar] [CrossRef]
- Jagger, B.W.; Wise, H.M.; Kash, J.C.; Walters, K.A.; Wills, N.M.; Xiao, Y.L.; Dunfee, R.L.; Schwartzman, L.M.; Ozinsky, A.; Bell, G.L.; et al. An overlapping protein-coding region in influenza A virus segment 3 modulates the host response. Science 2012, 337, 199–204. [Google Scholar] [CrossRef]
- Muramoto, Y.; Noda, T.; Kawakami, E.; Akkina, R.; Kawaoka, Y. Identification of novel influenza A virus proteins translated from PA mRNA. J. Virol. 2013, 87, 2455–2462. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Calvo, P.A.; Malide, D.; Gibbs, J.; Schubert, U.; Bacik, I.; Basta, S.; O’Neill, R.; Schickli, J.; Palese, P.; et al. A novel influenza A virus mitochondrial protein that induces cell death. Nat. Med. 2001, 7, 1306–1312. [Google Scholar] [CrossRef] [PubMed]
- Wise, H.M.; Foeglein, A.; Sun, J.; Dalton, R.M.; Patel, S.; Howard, W.; Anderson, E.C.; Barclay, W.S.; Digard, P. A complicated message: Identification of a novel PB1-related protein translated from influenza A virus segment 2 mRNA. J. Virol. 2009, 83, 8021–8031. [Google Scholar] [CrossRef]
- Yamayoshi, S.; Watanabe, M.; Goto, H.; Kawaoka, Y. Identification of a Novel Viral Protein Expressed from the PB2 Segment of Influenza A Virus. J. Virol. 2016, 90, 444–456. [Google Scholar] [CrossRef] [PubMed]
- Wise, H.M.; Hutchinson, E.C.; Jagger, B.W.; Stuart, A.D.; Kang, Z.H.; Robb, N.; Schwartzman, L.M.; Kash, J.C.; Fodor, E.; Firth, A.E.; et al. Identification of a novel splice variant form of the influenza A virus M2 ion channel with an antigenically distinct ectodomain. PLoS Pathog. 2012, 8, e1002998. [Google Scholar] [CrossRef] [PubMed]
- Selman, M.; Dankar, S.K.; Forbes, N.E.; Jia, J.J.; Brown, E.G. Adaptive mutation in influenza A virus non-structural gene is linked to host switching and induces a novel protein by alternative splicing. Emerg. Microbes Infect. 2012, 1, e42. [Google Scholar] [CrossRef]
- Robb, N.C.; Jackson, D.; Vreede, F.T.; Fodor, E. Splicing of influenza A virus NS1 mRNA is independent of the viral NS1 protein. J. Gen. Virol. 2010, 91, 2331–2340. [Google Scholar] [CrossRef]
- Chua, M.A.; Schmid, S.; Perez, J.T.; Langlois, R.A.; Tenoever, B.R. Influenza A virus utilizes suboptimal splicing to coordinate the timing of infection. Cell Rep. 2013, 3, 23–29. [Google Scholar] [CrossRef]
- Garcia-Sastre, A.; Egorov, A.; Matassov, D.; Brandt, S.; Levy, D.E.; Durbin, J.E.; Palese, P.; Muster, T. Influenza A virus lacking the NS1 gene replicates in interferon-deficient systems. Virology 1998, 252, 324–330. [Google Scholar] [CrossRef]
- Das, K.; Ma, L.C.; Xiao, R.; Radvansky, B.; Aramini, J.; Zhao, L.; Marklund, J.; Kuo, R.L.; Twu, K.Y.; Arnold, E.; et al. Structural basis for suppression of a host antiviral response by influenza A virus. Proc. Natl. Acad. Sci. USA 2008, 105, 13093–13098. [Google Scholar] [CrossRef]
- Nemeroff, M.E.; Barabino, S.M.; Li, Y.; Keller, W.; Krug, R.M. Influenza virus NS1 protein interacts with the cellular 30 kDa subunit of CPSF and inhibits 3′end formation of cellular pre-mRNAs. Mol. Cell 1998, 1, 991–1000. [Google Scholar] [CrossRef] [PubMed]
- Noah, D.L.; Twu, K.Y.; Krug, R.M. Cellular antiviral responses against influenza A virus are countered at the posttranscriptional level by the viral NS1A protein via its binding to a cellular protein required for the 3′ end processing of cellular pre-mRNAS. Virology 2003, 307, 386–395. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Chen, L.M.; Zeng, H.; Gomez, J.A.; Plowden, J.; Fujita, T.; Katz, J.M.; Donis, R.O.; Sambhara, S. NS1 protein of influenza A virus inhibits the function of intracytoplasmic pathogen sensor, RIG-I. Am. J. Respir. Cell Mol. Biol. 2007, 36, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Mibayashi, M.; Martínez-Sobrido, L.; Loo, Y.M.; Cárdenas, W.B.; Gale, M., Jr.; García-Sastre, A. Inhibition of retinoic acid-inducible gene I-mediated induction of beta interferon by the NS1 protein of influenza A virus. J. Virol. 2007, 81, 514–524. [Google Scholar] [CrossRef]
- Opitz, B.; Rejaibi, A.; Dauber, B.; Eckhard, J.; Vinzing, M.; Schmeck, B.; Hippenstiel, S.; Suttorp, N.; Wolff, T. IFNbeta induction by influenza A virus is mediated by RIG-I which is regulated by the viral NS1 protein. Cell. Microbiol. 2007, 9, 930–938. [Google Scholar] [CrossRef]
- Pichlmair, A.; Schulz, O.; Tan, C.P.; Näslund, T.I.; Liljeström, P.; Weber, F.; Reis e Sousa, C. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science 2006, 314, 997–1001. [Google Scholar] [CrossRef]
- Li, S.; Min, J.Y.; Krug, R.M.; Sen, G.C. Binding of the influenza A virus NS1 protein to PKR mediates the inhibition of its activation by either PACT or double-stranded RNA. Virology 2006, 349, 13–21. [Google Scholar] [CrossRef]
- Min, J.Y.; Li, S.; Sen, G.C.; Krug, R.M. A site on the influenza A virus NS1 protein mediates both inhibition of PKR activation and temporal regulation of viral RNA synthesis. Virology 2007, 363, 236–243. [Google Scholar] [CrossRef]
- Hale, B.G.; Kerry, P.S.; Jackson, D.; Precious, B.L.; Gray, A.; Killip, M.J.; Randall, R.E.; Russell, R.J. Structural insights into phosphoinositide 3-kinase activation by the influenza A virus NS1 protein. Proc. Natl. Acad. Sci. USA 2010, 107, 1954–1959. [Google Scholar] [CrossRef]
- Ehrhardt, C.; Wolff, T.; Pleschka, S.; Planz, O.; Beermann, W.; Bode, J.G.; Schmolke, M.; Ludwig, S. Influenza A virus NS1 protein activates the PI3K/Akt pathway to mediate antiapoptotic signaling responses. J. Virol. 2007, 81, 3058–3067. [Google Scholar] [CrossRef]
- Jackson, D.; Killip, M.J.; Galloway, C.S.; Russell, R.J.; Randall, R.E. Loss of function of the influenza A virus NS1 protein promotes apoptosis but this is not due to a failure to activate phosphatidylinositol 3-kinase (PI3K). Virology 2010, 396, 94–105. [Google Scholar] [CrossRef]
- Li, W.; Wang, G.; Zhang, H.; Shen, Y.; Dai, J.; Wu, L.; Zhou, J.; Jiang, Z.; Li, K. Inability of NS1 protein from an H5N1 influenza virus to activate PI3K/Akt signaling pathway correlates to the enhanced virus replication upon PI3K inhibition. Vet. Res. 2012, 43, 36. [Google Scholar] [CrossRef]
- Lu, X.; Masic, A.; Li, Y.; Shin, Y.; Liu, Q.; Zhou, Y. The PI3K/Akt pathway inhibits influenza A virus-induced Bax-mediated apoptosis by negatively regulating the JNK pathway via ASK1. J. Gen. Virol. 2010, 91, 1439–1449. [Google Scholar] [CrossRef]
- Hale, B.G.; Randall, R.E.; Ortin, J.; Jackson, D. The multifunctional NS1 protein of influenza A viruses. J. Gen. Virol. 2008, 89, 2359–2376. [Google Scholar] [CrossRef]
- Marc, D. Influenza virus non-structural protein NS1: Interferon antagonism and beyond. J. Gen. Virol. 2014, 95, 2594–2611. [Google Scholar] [CrossRef]
- Khaperskyy, D.A.; McCormick, C. Timing Is Everything: Coordinated Control of Host Shutoff by Influenza A Virus NS1 and PA-X Proteins. J. Virol. 2015, 89, 6528–6531. [Google Scholar] [CrossRef]
- Egorov, A.; Brandt, S.; Sereinig, S.; Romanova, J.; Ferko, B.; Katinger, D.; Grassauer, A.; Alexandrova, G.; Katinger, H.; Muster, T. Transfectant influenza A viruses with long deletions in the NS1 protein grow efficiently in Vero cells. J. Virol. 1998, 72, 6437–6441. [Google Scholar] [CrossRef]
- Dankar, S.K.; Wang, S.; Ping, J.; Forbes, N.E.; Keleta, L.; Li, Y.; Brown, E.G. Influenza A virus NS1 gene mutations F103L and M106I increase replication and virulence. Virol. J. 2011, 8, 13. [Google Scholar] [CrossRef]
- Cauthen, A.N.; Swayne, D.E.; Sekellick, M.J.; Marcus, P.I.; Suarez, D.L. Amelioration of influenza virus pathogenesis in chickens attributed to the enhanced interferon-inducing capacity of a virus with a truncated NS1 gene. J. Virol. 2007, 81, 1838–1847. [Google Scholar] [CrossRef]
- Basler, C.F.; Reid, A.H.; Dybing, J.K.; Janczewski, T.A.; Fanning, T.G.; Zheng, H.; Salvatore, M.; Perdue, M.L.; Swayne, D.E.; Garcia-Sastre, A.; et al. Sequence of the 1918 pandemic influenza virus nonstructural gene (NS) segment and characterization of recombinant viruses bearing the 1918 NS genes. Proc. Natl. Acad. Sci. USA 2001, 98, 2746–2751. [Google Scholar] [CrossRef]
- Kim, I.H.; Kwon, H.J.; Lee, S.H.; Kim, D.Y.; Kim, J.H. Effects of different NS genes of avian influenza viruses and amino acid changes on pathogenicity of recombinant A/Puerto Rico/8/34 viruses. Vet. Microbiol. 2015, 175, 17–25. [Google Scholar] [CrossRef]
- Dankar, S.K.; Miranda, E.; Forbes, N.E.; Pelchat, M.; Tavassoli, A.; Selman, M.; Ping, J.; Jia, J.; Brown, E.G. Influenza A/Hong Kong/156/1997(H5N1) virus NS1 gene mutations F103L and M106I both increase IFN antagonism, virulence and cytoplasmic localization but differ in binding to RIG-I and CPSF30. Virol. J. 2013, 10, 243. [Google Scholar] [CrossRef] [PubMed]
- Chauche, C.; Nogales, A.; Zhu, H.; Goldfarb, D.; Ahmad Shanizza, A.I.; Gu, Q.; Parrish, C.R.; Martinez-Sobrido, L.; Marshall, J.F.; Murcia, P.R. Mammalian Adaptation of an Avian Influenza A Virus Involves Stepwise Changes in NS1. J. Virol. 2018, 92, e01875-17. [Google Scholar] [CrossRef] [PubMed]
- Nogales, A.; Martinez-Sobrido, L.; Chiem, K.; Topham, D.J.; DeDiego, M.L. Functional Evolution of the 2009 Pandemic H1N1 Influenza Virus NS1 and PA in Humans. J. Virol. 2018, 92, 10-1128. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Brenner, D.; Wang, Z.; Dauber, B.; Ehrhardt, C.; Hogner, K.; Herold, S.; Ludwig, S.; Wolff, T.; Yu, K.; et al. The NS segment of an H5N1 highly pathogenic avian influenza virus (HPAIV) is sufficient to alter replication efficiency, cell tropism, and host range of an H7N1 HPAIV. J. Virol. 2010, 84, 2122–2133. [Google Scholar] [CrossRef]
- Engel, D.A. The influenza virus NS1 protein as a therapeutic target. Antiviral Res. 2013, 99, 409–416. [Google Scholar] [CrossRef]
- Darapaneni, V.; Prabhaker, V.K.; Kukol, A. Large-scale analysis of influenza A virus sequences reveals potential drug target sites of non-structural proteins. J. Gen. Virol. 2009, 90, 2124–2133. [Google Scholar] [CrossRef]
- Twu, K.Y.; Noah, D.L.; Rao, P.; Kuo, R.L.; Krug, R.M. The CPSF30 binding site on the NS1A protein of influenza A virus is a potential antiviral target. J. Virol. 2006, 80, 3957–3965. [Google Scholar] [CrossRef]
- Rosario-Ferreira, N.; Preto, A.J.; Melo, R.; Moreira, I.S.; Brito, R.M.M. The Central Role of Non-Structural Protein 1 (NS1) in Influenza Biology and Infection. Int. J. Mol. Sci. 2020, 21, 1511. [Google Scholar] [CrossRef]
- Kim, H.J.; Jeong, M.S.; Jang, S.B. Structure and Activities of the NS1 Influenza Protein and Progress in the Development of Small-Molecule Drugs. Int. J. Mol. Sci. 2021, 22, 4242. [Google Scholar] [CrossRef]
- Trigueiro-Louro, J.; Santos, L.A.; Almeida, F.; Correia, V.; Brito, R.M.M.; Rebelo-de-Andrade, H. NS1 protein as a novel anti-influenza target: Map-and-mutate antiviral rationale reveals new putative druggable hot spots with an important role on viral replication. Virology 2022, 565, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Naceri, S.; Marc, D.; Blot, R.; Flatters, D.; Camproux, A.C. Druggable Pockets at the RNA Interface Region of Influenza A Virus NS1 Protein Are Conserved across Sequence Variants from Distinct Subtypes. Biomolecules 2022, 13, 64. [Google Scholar] [CrossRef] [PubMed]
- Abi Hussein, H.; Geneix, C.; Cauvin, C.; Marc, D.; Flatters, D.; Camproux, A.C. Molecular Dynamics Simulations of Influenza A Virus NS1 Reveal a Remarkably Stable RNA-Binding Domain Harboring Promising Druggable Pockets. Viruses 2020, 12, 837. [Google Scholar] [CrossRef] [PubMed]
- Wathen, M.W.; Barro, M.; Bright, R.A. Antivirals in seasonal and pandemic influenza--future perspectives. Influenza Other Respir. Viruses 2013, 7 (Suppl. S1), 76–80. [Google Scholar] [CrossRef] [PubMed]
- Koszalka, P.; Tilmanis, D.; Hurt, A.C. Influenza antivirals currently in late-phase clinical trial. Influenza Other Respir. Viruses 2017, 11, 240–246. [Google Scholar] [CrossRef] [PubMed]
- de Chassey, B.; Aublin-Gex, A.; Ruggieri, A.; Meyniel-Schicklin, L.; Pradezynski, F.; Davoust, N.; Chantier, T.; Tafforeau, L.; Mangeot, P.E.; Ciancia, C.; et al. The interactomes of influenza virus NS1 and NS2 proteins identify new host factors and provide insights for ADAR1 playing a supportive role in virus replication. PLoS Pathog. 2013, 9, e1003440. [Google Scholar] [CrossRef] [PubMed]
- Kuo, R.L.; Chen, C.J.; Tam, E.H.; Huang, C.G.; Li, L.H.; Li, Z.H.; Su, P.C.; Liu, H.P.; Wu, C.C. Interactome Analysis of NS1 Protein Encoded by Influenza A H7N9 Virus Reveals an Inhibitory Role of NS1 in Host mRNA Maturation. J. Proteome Res. 2018, 17, 1474–1484. [Google Scholar] [CrossRef]
- Kuo, R.L.; Li, Z.H.; Li, L.H.; Lee, K.M.; Tam, E.H.; Liu, H.M.; Liu, H.P.; Shih, S.R.; Wu, C.C. Interactome Analysis of the NS1 Protein Encoded by Influenza A H1N1 Virus Reveals a Positive Regulatory Role of Host Protein PRP19 in Viral Replication. J. Proteome Res. 2016, 15, 1639–1648. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, Q.; Zheng, M.; Wen, J.; Li, Q.; Zhao, G. Viral-Host Interactome Analysis Reveals Chicken STAU2 Interacts With Non-structural Protein 1 and Promotes the Replication of H5N1 Avian Influenza Virus. Front. Immunol. 2021, 12, 590679. [Google Scholar] [CrossRef]
- Krug, R.M.; García-Sastre, A. The NS1 Protein: A Master Regulator of Host and Viral Functions. In Textbook of Influenza; 2013; pp. 114–132. Available online: https://onlinelibrary.wiley.com/doi/abs/10.1002/9781118636817.ch7 (accessed on 1 August 2023).
- Palese, P.; Shaw, M.L. Orthomyxoviridae: The Viruses and Their Replication. In Fields Viology, 5th ed.; Knipe, D.M., Katze, M.G., Eds.; Lippincott, Williams, & Wilkins: Philadelphia, PA, USA, 2006; pp. 1647–1689. [Google Scholar]
- Chien, C.Y.; Tejero, R.; Huang, Y.; Zimmerman, D.E.; Rios, C.B.; Krug, R.M.; Montelione, G.T. A novel RNA-binding motif in influenza A virus non-structural protein 1. Nat. Struct. Biol. 1997, 4, 891–895. [Google Scholar] [CrossRef]
- Wang, W.; Riedel, K.; Lynch, P.; Chien, C.Y.; Montelione, G.T.; Krug, R.M. RNA binding by the novel helical domain of the influenza virus NS1 protein requires its dimer structure and a small number of specific basic amino acids. RNA 1999, 5, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Hatada, E.; Saito, S.; Okishio, N.; Fukuda, R. Binding of the influenza virus NS1 protein to model genome RNAs. J. Gen. Virol. 1997, 78 Pt 5, 1059–1063. [Google Scholar] [CrossRef]
- Hatada, E.; Takizawa, T.; Fukuda, R. Specific binding of influenza A virus NS1 protein to the virus minus-sense RNA in vitro. J. Gen. Virol. 1992, 73 Pt 1, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Trapp, S.; Soubieux, D.; Marty, H.; Esnault, E.; Hoffmann, T.W.; Chandenier, M.; Lion, A.; Kut, E.; Quere, P.; Larcher, T.; et al. Shortening the unstructured, interdomain region of the non-structural protein NS1 of an avian H1N1 influenza virus increases its replication and pathogenicity in chickens. J. Gen. Virol. 2014, 95, 1233–1243. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Marazzi, I.; Ho, J.S.; Kim, J.; Manicassamy, B.; Dewell, S.; Albrecht, R.A.; Seibert, C.W.; Schaefer, U.; Jeffrey, K.L.; Prinjha, R.K.; et al. Suppression of the antiviral response by an influenza histone mimic. Nature 2012, 483, 428–433. [Google Scholar] [CrossRef]
- Golebiewski, L.; Liu, H.; Javier, R.T.; Rice, A.P. The avian influenza virus NS1 ESEV PDZ binding motif associates with Dlg1 and Scribble to disrupt cellular tight junctions. J. Virol. 2011, 85, 10639–10648. [Google Scholar] [CrossRef]
- Thomas, M.; Kranjec, C.; Nagasaka, K.; Matlashewski, G.; Banks, L. Analysis of the PDZ binding specificities of Influenza A Virus NS1 proteins. Virol. J. 2011, 8, 25. [Google Scholar] [CrossRef]
- Jackson, D.; Hossain, M.J.; Hickman, D.; Perez, D.R.; Lamb, R.A. A new influenza virus virulence determinant: The NS1 protein four C-terminal residues modulate pathogenicity. Proc. Natl. Acad. Sci. USA 2008, 105, 4381. [Google Scholar] [CrossRef]
- Soubies, S.M.; Volmer, C.; Croville, G.; Loupias, J.; Peralta, B.; Costes, P.; Lacroux, C.; Guérin, J.-L.; Volmer, R. Species-Specific Contribution of the Four C-Terminal Amino Acids of Influenza A Virus NS1 Protein to Virulence. J. Virol. 2010, 84, 6733. [Google Scholar] [CrossRef]
- Bornholdt, Z.A.; Prasad, B.V. X-ray structure of influenza virus NS1 effector domain. Nat. Struct. Mol. Biol. 2006, 13, 559–560. [Google Scholar] [CrossRef]
- Bullough, P.A.; Hughson, F.M.; Skehel, J.J.; Wiley, D.C. Structure of influenza haemagglutinin at the pH of membrane fusion. Nature 1994, 371, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Hale, B.G.; Barclay, W.S.; Randall, R.E.; Russell, R.J. Structure of an avian influenza A virus NS1 protein effector domain. Virology 2008, 378, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Jureka, A.S.; Kleinpeter, A.B.; Cornilescu, G.; Cornilescu, C.C.; Petit, C.M. Structural Basis for a Novel Interaction between the NS1 Protein Derived from the 1918 Influenza Virus and RIG-I. Structure 2015, 23, 2001–2010. [Google Scholar] [CrossRef] [PubMed]
- Kerry, P.S.; Ayllon, J.; Taylor, M.A.; Hass, C.; Lewis, A.; García-Sastre, A.; Randall, R.E.; Hale, B.G.; Russell, R.J. A Transient Homotypic Interaction Model for the Influenza A Virus NS1 Protein Effector Domain. PLoS ONE 2011, 6, e17946. [Google Scholar] [CrossRef]
- Kerry, P.S.; Long, E.; Taylor, M.A.; Russell, R.J. Conservation of a crystallographic interface suggests a role for beta-sheet augmentation in influenza virus NS1 multifunctionality. Acta Crystallo. Sect. F Struct. Biol. Cryst. Commun. 2011, 67, 858–861. [Google Scholar] [CrossRef]
- Liu, J.; Lynch, P.A.; Chien, C.Y.; Montelione, G.T.; Krug, R.M.; Berman, H.M. Crystal structure of the unique RNA-binding domain of the influenza virus NS1 protein. Nat. Struct. Biol. 1997, 4, 896–899. [Google Scholar] [CrossRef]
- Turkington, H.L.; Juozapaitis, M.; Kerry, P.S.; Aydillo, T.; Ayllon, J.; García-Sastre, A.; Schwemmle, M.; Hale, B.G. Novel Bat Influenza Virus NS1 Proteins Bind Double-Stranded RNA and Antagonize Host Innate Immunity. J. Virol. 2015, 89, 10696. [Google Scholar] [CrossRef]
- Xia, S.; Monzingo, A.F.; Robertus, J.D. Structure of NS1A effector domain from the influenza A/Udorn/72 virus. Acta Crystallo. Sect. D Biol. Crystallogr. 2009, 65, 11–17. [Google Scholar] [CrossRef]
- Xia, S.; Robertus, J.D. X-ray structures of NS1 effector domain mutants. Arch. Biochem. Biophys. 2010, 494, 198–204. [Google Scholar] [CrossRef]
- Kleinpeter, A.B.; Jureka, A.S.; Falahat, S.M.; Green, T.J.; Petit, C.M. Structural analyses reveal the mechanism of inhibition of influenza virus NS1 by two antiviral compounds. J. Biol. Chem. 2018, 293, 14659–14668. [Google Scholar] [CrossRef]
- Aramini, J.M.; Ma, L.C.; Zhou, L.; Schauder, C.M.; Hamilton, K.; Amer, B.R.; Mack, T.R.; Lee, H.W.; Ciccosanti, C.T.; Zhao, L.; et al. Dimer interface of the effector domain of non-structural protein 1 from influenza A virus: An interface with multiple functions. J. Biol. Chem. 2011, 286, 26050–26060. [Google Scholar] [CrossRef] [PubMed]
- Carrillo, B.; Choi, J.-M.; Bornholdt, Z.A.; Sankaran, B.; Rice, A.P.; Prasad, B.V.V. The influenza A virus protein NS1 displays structural polymorphism. J. Virol. 2014, 88, 4113–4122. [Google Scholar] [CrossRef]
- Bornholdt, Z.A.; Prasad, B.V. X-ray structure of NS1 from a highly pathogenic H5N1 influenza virus. Nature 2008, 456, 985–988. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.; Wong, S.M.; Yuan, Y.A. Structural basis for dsRNA recognition by NS1 protein of influenza A virus. Cell Res. 2009, 19, 187–195. [Google Scholar] [CrossRef]
- Chien, C.Y.; Xu, Y.; Xiao, R.; Aramini, J.M.; Sahasrabudhe, P.V.; Krug, R.M.; Montelione, G.T. Biophysical characterization of the complex between double-stranded RNA and the N-terminal domain of the NS1 protein from influenza A virus: Evidence for a novel RNA-binding mode. Biochemistry 2004, 43, 1950–1962. [Google Scholar] [CrossRef] [PubMed]
- Wacquiez, A.; Coste, F.; Kut, E.; Gaudon, V.; Trapp, S.; Castaing, B.; Marc, D. Structure and Sequence Determinants Governing the Interactions of RNAs with Influenza A Virus Non-Structural Protein NS1. Viruses 2020, 12, 947. [Google Scholar] [CrossRef] [PubMed]
- Jureka, A.S.; Kleinpeter, A.B.; Tipper, J.L.; Harrod, K.S.; Petit, C.M. The influenza NS1 protein modulates RIG-I activation via a strain-specific direct interaction with the second CARD of RIG-I. J. Biol. Chem. 2020, 295, 1153–1164. [Google Scholar] [CrossRef]
- Li, W.; Noah, J.W.; Noah, D.L. Alanine substitutions within a linker region of the influenza A virus non-structural protein 1 alter its subcellular localization and attenuate virus replication. J. Gen. Virol. 2011, 92, 1832–1842. [Google Scholar] [CrossRef]
- Neumann, G.; Macken, C.A.; Kawaoka, Y. Identification of amino acid changes that may have been critical for the genesis of A(H7N9) influenza viruses. J. Virol. 2014, 88, 4877–4896. [Google Scholar] [CrossRef]
- Kanrai, P.; Mostafa, A.; Madhugiri, R.; Lechner, M.; Wilk, E.; Schughart, K.; Ylosmaki, L.; Saksela, K.; Ziebuhr, J.; Pleschka, S. Identification of specific residues in avian influenza A virus NS1 that enhance viral replication and pathogenicity in mammalian systems. J. Gen. Virol. 2016, 97, 2135–2148. [Google Scholar] [CrossRef]
- Aramini, J.M.; Hamilton, K.; Ma, L.C.; Swapna, G.V.T.; Leonard, P.G.; Ladbury, J.E.; Krug, R.M.; Montelione, G.T. (19)F NMR reveals multiple conformations at the dimer interface of the nonstructural protein 1 effector domain from influenza A virus. Structure 2014, 22, 515–525. [Google Scholar] [CrossRef] [PubMed]
- Koliopoulos, M.G.; Lethier, M.; van der Veen, A.G.; Haubrich, K.; Hennig, J.; Kowalinski, E.; Stevens, R.V.; Martin, S.R.; Reis e Sousa, C.; Cusack, S.; et al. Molecular mechanism of influenza A NS1-mediated TRIM25 recognition and inhibition. Nat. Commun. 2018, 9, 1820. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Liu, Y.; Tempel, W.; Eram, M.S.; Bian, C.; Liu, K.; Senisterra, G.; Crombet, L.; Vedadi, M.; Min, J. Structural basis for histone mimicry and hijacking of host proteins by influenza virus protein NS1. Nat. Commun. 2014, 5, 3952. [Google Scholar] [CrossRef]
- Shen, Q.; Zeng, D.; Zhao, B.; Bhatt, V.S.; Li, P.; Cho, J.H. The Molecular Mechanisms Underlying the Hijack of Host Proteins by the 1918 Spanish Influenza Virus. ACS Chem. Biol. 2017, 12, 1199–1203. [Google Scholar] [CrossRef] [PubMed]
- Lusser, A.; Urwin, D.L.; Kadonaga, J.T. Distinct activities of CHD1 and ACF in ATP-dependent chromatin assembly. Nat. Struct. Mol. Biol. 2005, 12, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Konev, A.Y.; Tribus, M.; Park, S.Y.; Podhraski, V.; Lim, C.Y.; Emelyanov, A.V.; Vershilova, E.; Pirrotta, V.; Kadonaga, J.T.; Lusser, A.; et al. CHD1 motor protein is required for deposition of histone variant H3.3 into chromatin in vivo. Science 2007, 317, 1087–1090. [Google Scholar] [CrossRef] [PubMed]
- Tran, H.G.; Steger, D.J.; Iyer, V.R.; Johnson, A.D. The chromo domain protein Chd1p from budding yeast is an ATP-dependent chromatin-modifying factor. EMBO J. 2000, 19, 2323–2331. [Google Scholar] [CrossRef]
- Sundaramoorthy, R.; Hughes, A.L.; El-Mkami, H.; Norman, D.G.; Ferreira, H.; Owen-Hughes, T. Structure of the chromatin remodelling enzyme Chd1 bound to a ubiquitinylated nucleosome. eLife 2018, 7, e35720. [Google Scholar] [CrossRef]
- Sims, R.J.; Millhouse, S.; Chen, C.-F.; Lewis, B.A.; Erdjument-Bromage, H.; Tempst, P.; Manley, J.L.; Reinberg, D. Recognition of Trimethylated Histone H3 Lysine 4 Facilitates the Recruitment of Transcription Postinitiation Factors and Pre-mRNA Splicing. Mol. Cell 2007, 28, 665–676. [Google Scholar] [CrossRef]
- Tu, J.; Guo, J.; Zhang, A.; Zhang, W.; Zhao, Z.; Zhou, H.; Liu, C.; Chen, H.; Jin, M. Effects of the C-terminal truncation in NS1 protein of the 2009 pandemic H1N1 influenza virus on host gene expression. PLoS ONE 2011, 6, e26175. [Google Scholar] [CrossRef]
- Matsuda, M.; Tanaka, S.; Nagata, S.; Kojima, A.; Kurata, T.; Shibuya, M. Two species of human CRK cDNA encode proteins with distinct biological activities. Mol. Cell. Biol. 1992, 12, 3482–3489. [Google Scholar] [CrossRef] [PubMed]
- ten Hoeve, J.; Morris, C.; Heisterkamp, N.; Groffen, J. Isolation and chromosomal localization of CRKL, a human crk-like gene. Oncogene 1993, 8, 2469–2474. [Google Scholar] [PubMed]
- Hrincius, E.R.; Wixler, V.; Wolff, T.; Wagner, R.; Ludwig, S.; Ehrhardt, C. CRK adaptor protein expression is required for efficient replication of avian influenza A viruses and controls JNK-mediated apoptotic responses. Cell. Microbiol. 2010, 12, 831–843. [Google Scholar] [CrossRef] [PubMed]
- Ylösmäki, L.; Fagerlund, R.; Kuisma, I.; Julkunen, I.; Saksela, K. Nuclear Translocation of Crk Adaptor Proteins by the Influenza A Virus NS1 Protein. Viruses 2016, 8, 101. [Google Scholar] [CrossRef]
- Heikkinen, L.S.; Kazlauskas, A.; Melén, K.; Wagner, R.; Ziegler, T.; Julkunen, I.; Saksela, K. Avian and 1918 Spanish Influenza A Virus NS1 Proteins Bind to Crk/CrkL Src Homology 3 Domains to Activate Host Cell Signaling. J. Biol. Chem. 2008, 283, 5719–5727. [Google Scholar] [CrossRef]
- Li, S.S. Specificity and versatility of SH3 and other proline-recognition domains: Structural basis and implications for cellular signal transduction. Biochem. J. 2005, 390, 641–653. [Google Scholar] [CrossRef]
- Dolfi, F.; Garcia-Guzman, M.; Ojaniemi, M.; Nakamura, H.; Matsuda, M.; Vuori, K. The adaptor protein Crk connects multiple cellular stimuli to the JNK signaling pathway. Proc. Natl. Acad. Sci. USA 1998, 95, 15394–15399. [Google Scholar] [CrossRef]
- Ren, R.; Ye, Z.S.; Baltimore, D. Abl protein-tyrosine kinase selects the Crk adapter as a substrate using SH3-binding sites. Genes. Dev. 1994, 8, 783–795. [Google Scholar] [CrossRef]
- Bhatt, V.S.; Zeng, D.; Krieger, I.; Sacchettini, J.C.; Cho, J.H. Binding Mechanism of the N-Terminal SH3 Domain of CrkII and Proline-Rich Motifs in cAbl. Biophys. J. 2016, 110, 2630–2641. [Google Scholar] [CrossRef]
- Smith, J.J.; Richardson, D.A.; Kopf, J.; Yoshida, M.; Hollingsworth, R.E.; Kornbluth, S. Apoptotic Regulation by the Crk Adapter Protein Mediated by Interactions with Wee1 and Crm1/Exportin. Mol. Cell. Biol. 2002, 22, 1412–1423. [Google Scholar] [CrossRef]
- Ylösmäki, L.; Schmotz, C.; Ylösmäki, E.; Saksela, K. Reorganization of the host cell Crk(L)–PI3 kinase signaling complex by the influenza A virus NS1 protein. Virology 2015, 484, 146–152. [Google Scholar] [CrossRef][Green Version]
- Assémat, E.; Bazellières, E.; Pallesi-Pocachard, E.; Le Bivic, A.; Massey-Harroche, D. Polarity complex proteins. Biochim. Biophys. Acta (BBA) Biomembr. 2008, 1778, 614–630. [Google Scholar] [CrossRef]
- Massimi, P.; Narayan, N.; Thomas, M.; Gammoh, N.; Strand, S.; Strand, D.; Banks, L. Regulation of the hDlg/hScrib/Hugl-1 tumour suppressor complex. Exp. Cell Res. 2008, 314, 3306–3317. [Google Scholar] [CrossRef] [PubMed]
- Stephens, R.; Lim, K.; Portela, M.; Kvansakul, M.; Humbert, P.O.; Richardson, H.E. The Scribble Cell Polarity Module in the Regulation of Cell Signaling in Tissue Development and Tumorigenesis. J. Mol. Biol. 2018, 430, 3585–3612. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Golebiewski, L.; Dow, E.C.; Krug, R.M.; Javier, R.T.; Rice, A.P. The ESEV PDZ-binding motif of the avian influenza A virus NS1 protein protects infected cells from apoptosis by directly targeting Scribble. J. Virol. 2010, 84, 11164–11174. [Google Scholar] [CrossRef]
- Zhan, L.; Rosenberg, A.; Bergami, K.C.; Yu, M.; Xuan, Z.; Jaffe, A.B.; Allred, C.; Muthuswamy, S.K. Deregulation of scribble promotes mammary tumorigenesis and reveals a role for cell polarity in carcinoma. Cell 2008, 135, 865–878. [Google Scholar] [CrossRef]
- Lee, H.-J.; Zheng, J.J. PDZ domains and their binding partners: Structure, specificity, and modification. Cell Commun. Signal. 2010, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Javier, R.T.; Rice, A.P. Emerging Theme: Cellular PDZ Proteins as Common Targets of Pathogenic Viruses. J. Virol. 2011, 85, 11544–11556. [Google Scholar] [CrossRef]
- Obenauer, J.C.; Denson, J.; Mehta, P.K.; Su, X.; Mukatira, S.; Finkelstein, D.B.; Xu, X.; Wang, J.; Ma, J.; Fan, Y.; et al. Large-Scale Sequence Analysis of Avian Influenza Isolates. Science 2006, 311, 1576–1580. [Google Scholar] [CrossRef]
- Fan, S.; Macken, C.A.; Li, C.; Ozawa, M.; Goto, H.; Iswahyudi, N.F.N.; Nidom, C.A.; Chen, H.; Neumann, G.; Kawaoka, Y. Synergistic Effect of the PDZ and p85β-Binding Domains of the NS1 Protein on Virulence of an Avian H5N1 Influenza A Virus. J. Virol. 2013, 87, 4861–4871. [Google Scholar] [CrossRef]
- Javorsky, A.; Humbert, P.O.; Kvansakul, M. Structural Basis of the Avian Influenza NS1 Protein Interactions with the Cell Polarity Regulator Scribble. Viruses 2022, 14, 583. [Google Scholar] [CrossRef] [PubMed]
- Li, D.-Q.; Nair, S.S.; Kumar, R. The MORC family. Epigenetics 2013, 8, 685–693. [Google Scholar] [CrossRef]
- Kimura, Y.; Sakai, F.; Nakano, O.; Kisaki, O.; Sugimoto, H.; Sawamura, T.; Sadano, H.; Osumi, T. The Newly Identified Human Nuclear Protein NXP-2 Possesses Three Distinct Domains, the Nuclear Matrix-binding, RNA-binding, and Coiled-coil Domains. J. Biol. Chem. 2002, 277, 20611–20617. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yoshida, N.; Murakami, N.; Kawata, K.; Ishizaki, H.; Tanaka-Okamoto, M.; Miyoshi, J.; Zinn, A.R.; Shime, H.; Inoue, N. Dynamic Regulation of p53 Subnuclear Localization and Senescence by MORC3. Mol. Biol. Cell 2007, 18, 1701–1709. [Google Scholar] [CrossRef] [PubMed]
- Geoffroy, M.-C.; Chelbi-Alix, M.K. Role of Promyelocytic Leukemia Protein in Host Antiviral Defense. J. Interferon Cytokine Res. 2011, 31, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Klein, B.J.; Cox, K.L.; Bertulat, B.; Tencer, A.H.; Holden, M.R.; Wright, G.M.; Black, J.; Cardoso, M.C.; Poirier, M.G.; et al. Mechanism for autoinhibition and activation of the MORC3 ATPase. Proc. Natl. Acad. Sci. USA 2019, 116, 6111–6119. [Google Scholar] [CrossRef]
- Zhang, Y.; Ahn, J.; Green, K.J.; Vann, K.R.; Black, J.; Brooke, C.B.; Kutateladze, T.G. MORC3 Is a Target of the Influenza A Viral Protein NS1. Structure 2019, 27, 1029–1033.e1023. [Google Scholar] [CrossRef]
- Jorba, N.; Juarez, S.; Torreira, E.; Gastaminza, P.; Zamarreño, N.; Albar, J.P.; Ortín, J. Analysis of the interaction of influenza virus polymerase complex with human cell factors. Proteomics 2008, 8, 2077–2088. [Google Scholar] [CrossRef]
- Ver, L.S.; Marcos-Villar, L.; Landeras-Bueno, S.; Nieto, A.; Ortín, J. The Cellular Factor NXP2/MORC3 Is a Positive Regulator of Influenza Virus Multiplication. J. Virol. 2015, 89, 10023–10030. [Google Scholar] [CrossRef]
- Bortz, E.; Westera, L.; Maamary, J.; Steel, J.; Albrecht, R.A.; Manicassamy, B.; Chase, G.; Martínez-Sobrido, L.; Schwemmle, M.; García-Sastre, A. Host- and Strain-Specific Regulation of Influenza Virus Polymerase Activity by Interacting Cellular Proteins. mBio 2011, 2, 10-1128. [Google Scholar] [CrossRef]
- Chan, S.; Choi, E.A.; Shi, Y. Pre-mRNA 3′-end processing complex assembly and function. Wiley Interdiscip. Rev. RNA 2011, 2, 321–335. [Google Scholar] [CrossRef] [PubMed]
- Poon, L.L.; Pritlove, D.C.; Fodor, E.; Brownlee, G.G. Direct evidence that the poly(A) tail of influenza A virus mRNA is synthesized by reiterative copying of a U track in the virion RNA template. J. Virol. 1999, 73, 3473–3476. [Google Scholar] [CrossRef] [PubMed]
- DeDiego, M.L.; Nogales, A.; Lambert-Emo, K.; Martinez-Sobrido, L.; Topham, D.J. NS1 Protein Mutation I64T Affects Interferon Responses and Virulence of Circulating H3N2 Human Influenza A Viruses. J. Virol. 2016, 90, 9693–9711. [Google Scholar] [CrossRef] [PubMed]
- Ayllon, J.; Domingues, P.; Rajsbaum, R.; Miorin, L.; Schmolke, M.; Hale, B.G.; García-Sastre, A. A single amino acid substitution in the novel H7N9 influenza A virus NS1 protein increases CPSF30 binding and virulence. J. Virol. 2014, 88, 12146–12151. [Google Scholar] [CrossRef]
- Spesock, A.; Malur, M.; Hossain, M.J.; Chen, L.-M.; Njaa Bradley, L.; Davis Charles, T.; Lipatov Aleksandr, S.; York Ian, A.; Krug Robert, M.; Donis Ruben, O. The Virulence of 1997 H5N1 Influenza Viruses in the Mouse Model Is Increased by Correcting a Defect in Their NS1 Proteins. J. Virol. 2011, 85, 7048–7058. [Google Scholar] [CrossRef]
- Kuo, R.-L.; Krug, R.M. Influenza A Virus Polymerase Is an Integral Component of the CPSF30-NS1A Protein Complex in Infected Cells. J. Virol. 2009, 83, 1611–1616. [Google Scholar] [CrossRef]
- Kochs, G.; García-Sastre, A.; Martínez-Sobrido, L. Multiple anti-interferon actions of the influenza A virus NS1 protein. J. Virol. 2007, 81, 7011–7021. [Google Scholar] [CrossRef]
- Twu, K.Y.; Kuo, R.L.; Marklund, J.; Krug, R.M. The H5N1 influenza virus NS genes selected after 1998 enhance virus replication in mammalian cells. J. Virol. 2007, 81, 8112–8121. [Google Scholar] [CrossRef]
- Li, J.; Zhang, K.; Chen, Q.; Zhang, X.; Sun, Y.; Bi, Y.; Zhang, S.; Gu, J.; Li, J.; Liu, D.; et al. Three amino acid substitutions in the NS1 protein change the virus replication of H5N1 influenza virus in human cells. Virology 2018, 519, 64–73. [Google Scholar] [CrossRef]
- Rodriguez, L.; Nogales, A.; Iqbal, M.; Perez, D.R.; Martinez-Sobrido, L. Identification of Amino Acid Residues Responsible for Inhibition of Host Gene Expression by Influenza A H9N2 NS1 Targeting of CPSF30. Front. Microbiol. 2018, 9, 2546. [Google Scholar] [CrossRef]
- Zhu, Q.; Yang, H.; Chen, W.; Cao, W.; Zhong, G.; Jiao, P.; Deng, G.; Yu, K.; Yang, C.; Bu, Z.; et al. A naturally occurring deletion in its NS gene contributes to the attenuation of an H5N1 swine influenza virus in chickens. J. Virol. 2008, 82, 220–228. [Google Scholar] [CrossRef]
- Stutz, F.; Izaurralde, E. The interplay of nuclear mRNP assembly, mRNA surveillance and export. Trends Cell Biol. 2003, 13, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Carmody, S.R.; Wente, S.R. mRNA nuclear export at a glance. J. Cell Sci. 2009, 122, 1933–1937. [Google Scholar] [CrossRef] [PubMed]
- Braun, I.C.; Herold, A.; Rode, M.; Conti, E.; Izaurralde, E. Overexpression of TAP/p15 Heterodimers Bypasses Nuclear Retention and Stimulates Nuclear mRNA Export. J. Biol. Chem. 2001, 276, 20536–20543. [Google Scholar] [CrossRef] [PubMed]
- Blevins, M.B.; Smith, A.M.; Phillips, E.M.; Powers, M.A. Complex Formation among the RNA Export Proteins Nup98, Rae1/Gle2, and TAP*. J. Biol. Chem. 2003, 278, 20979–20988. [Google Scholar] [CrossRef]
- Bachi, A.; Braun, I.C.; Rodrigues, J.P.; Panté, N.; Ribbeck, K.; von Kobbe, C.; Kutay, U.; Wilm, M.; Görlich, D.; Carmo-Fonseca, M.; et al. The C-terminal domain of TAP interacts with the nuclear pore complex and promotes export of specific CTE-bearing RNA substrates. RNA 2000, 6, 136–158. [Google Scholar] [CrossRef]
- Satterly, N.; Tsai, P.L.; van Deursen, J.; Nussenzveig, D.R.; Wang, Y.; Faria, P.A.; Levay, A.; Levy, D.E.; Fontoura, B.M. Influenza virus targets the mRNA export machinery and the nuclear pore complex. Proc. Natl. Acad. Sci. USA 2007, 104, 1853–1858. [Google Scholar] [CrossRef]
- Enninga, J.; Levy, D.E.; Blobel, G.; Fontoura, B.M.A. Role of Nucleoporin Induction in Releasing an mRNA Nuclear Export Block. Science 2002, 295, 1523–1525. [Google Scholar] [CrossRef]
- Faria, P.A.; Chakraborty, P.; Levay, A.; Barber, G.N.; Ezelle, H.J.; Enninga, J.; Arana, C.; van Deursen, J.; Fontoura, B.M.A. VSV Disrupts the Rae1/mrnp41 mRNA Nuclear Export Pathway. Mol. Cell 2005, 17, 93–102. [Google Scholar] [CrossRef]
- Zhang, K.; Xie, Y.; Muñoz-Moreno, R.; Wang, J.; Zhang, L.; Esparza, M.; García-Sastre, A.; Fontoura, B.M.A.; Ren, Y. Structural basis for influenza virus NS1 protein block of mRNA nuclear export. Nat. Microbiol. 2019, 4, 1671–1679. [Google Scholar] [CrossRef]
- Gong, D.; Kim, Y.H.; Xiao, Y.; Du, Y.; Xie, Y.; Lee, K.K.; Feng, J.; Farhat, N.; Zhao, D.; Shu, S.; et al. A Herpesvirus Protein Selectively Inhibits Cellular mRNA Nuclear Export. Cell Host Microbe 2016, 20, 642–653. [Google Scholar] [CrossRef] [PubMed]
- Kuss, S.K.; Mata, M.A.; Zhang, L.; Fontoura, B.M. Nuclear imprisonment: Viral strategies to arrest host mRNA nuclear export. Viruses 2013, 5, 1824–1849. [Google Scholar] [CrossRef]
- Fribourg, S.; Braun, I.C.; Izaurralde, E.; Conti, E. Structural basis for the recognition of a nucleoporin FG repeat by the NTF2-like domain of the TAP/p15 mRNA nuclear export factor. Mol. Cell 2001, 8, 645–656. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Li, X.; Wang, Y.; Li, B.; Li, H.; Li, Y.; Zhou, W.; Zhang, C.; Wang, Y.; Rao, Z.; et al. PDlim2 selectively interacts with the PDZ binding motif of highly pathogenic avian H5N1 influenza A virus NS1. PLoS ONE 2011, 6, e19511. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.S.; Qu, Z. PDLIM2: Signaling pathways and functions in cancer suppression and host immunity. Biochim. Biophys. Acta (BBA) Rev. Cancer 2021, 1876, 188630. [Google Scholar] [CrossRef]
- Karthikeyan, S.; Leung, T.; Ladias, J.A. Structural basis of the Na+/H+ exchanger regulatory factor PDZ1 interaction with the carboxyl-terminal region of the cystic fibrosis transmembrane conductance regulator. J. Biol. Chem. 2001, 276, 19683–19686. [Google Scholar] [CrossRef]
- Zhang, J.; Yan, X.; Shi, C.; Yang, X.; Guo, Y.; Tian, C.; Long, J.; Shen, Y. Structural basis of beta-catenin recognition by Tax-interacting protein-1. J. Mol. Biol. 2008, 384, 255–263. [Google Scholar] [CrossRef]
- Tanaka, T.; Grusby, M.J.; Kaisho, T. PDLIM2-mediated termination of transcription factor NF-kappaB activation by intranuclear sequestration and degradation of the p65 subunit. Nat. Immunol. 2007, 8, 584–591. [Google Scholar] [CrossRef]
- Wang, X.; Li, M.; Zheng, H.; Muster, T.; Palese, P.; Beg, A.A.; García-Sastre, A. Influenza A virus NS1 protein prevents activation of NF-kappaB and induction of alpha/beta interferon. J. Virol. 2000, 74, 11566–11573. [Google Scholar] [CrossRef]
- Tanaka, T.; Soriano, M.A.; Grusby, M.J. SLIM is a nuclear ubiquitin E3 ligase that negatively regulates STAT signaling. Immun. 2005, 22, 729–736. [Google Scholar] [CrossRef]
- Loughran, G.; Healy, N.C.; Kiely, P.A.; Huigsloot, M.; Kedersha, N.L.; O’Connor, R. Mystique is a new insulin-like growth factor-I-regulated PDZ-LIM domain protein that promotes cell attachment and migration and suppresses Anchorage-independent growth. Mol. Biol. Cell 2005, 16, 1811–1822. [Google Scholar] [CrossRef] [PubMed]
- Joyce, M.A.; Berry-Wynne, K.M.; dos Santos, T.; Addison, W.R.; McFarlane, N.; Hobman, T.; Tyrrell, D.L. HCV and flaviviruses hijack cellular mechanisms for nuclear STAT2 degradation: Up-regulation of PDLIM2 suppresses the innate immune response. PLoS Pathog. 2019, 15, e1007949. [Google Scholar] [CrossRef]
- Qu, Z.; Fu, J.; Ma, H.; Zhou, J.; Jin, M.; Mapara, M.Y.; Grusby, M.J.; Xiao, G. PDLIM2 restricts Th1 and Th17 differentiation and prevents autoimmune disease. Cell Biosci. 2012, 2, 23. [Google Scholar] [CrossRef] [PubMed]
- Cooray, S. The pivotal role of phosphatidylinositol 3-kinase–Akt signal transduction in virus survival. J. General. Virol. 2004, 85, 1065–1076. [Google Scholar] [CrossRef] [PubMed]
- Buchkovich, N.J.; Yu, Y.; Zampieri, C.A.; Alwine, J.C. The TORrid affairs of viruses: Effects of mammalian DNA viruses on the PI3K–Akt–mTOR signalling pathway. Nat. Rev. Microbiol. 2008, 6, 266–275. [Google Scholar] [CrossRef]
- Hale, B.G.; Jackson, D.; Chen, Y.H.; Lamb, R.A.; Randall, R.E. Influenza A virus NS1 protein binds p85beta and activates phosphatidylinositol-3-kinase signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 14194–14199. [Google Scholar] [CrossRef]
- Sirico, M.; D’Angelo, A.; Gianni, C.; Casadei, C.; Merloni, F.; De Giorgi, U. Current State and Future Challenges for PI3K Inhibitors in Cancer Therapy. Cancers 2023, 15, 703. [Google Scholar] [CrossRef]
- Carpenter, C.L.; Duckworth, B.C.; Auger, K.R.; Cohen, B.; Schaffhausen, B.S.; Cantley, L.C. Purification and characterization of phosphoinositide 3-kinase from rat liver. J. Biol. Chem. 1990, 265, 19704–19711. [Google Scholar] [CrossRef]
- Shin, Y.K.; Li, Y.; Liu, Q.; Anderson, D.H.; Babiuk, L.A.; Zhou, Y. SH3 binding motif 1 in influenza A virus NS1 protein is essential for PI3K/Akt signaling pathway activation. J. Virol. 2007, 81, 12730–12739. [Google Scholar] [CrossRef]
- Hale, B.G.; Batty, I.H.; Downes, C.P.; Randall, R.E. Binding of influenza A virus NS1 protein to the inter-SH2 domain of p85 suggests a novel mechanism for phosphoinositide 3-kinase activation. J. Biol. Chem. 2008, 283, 1372–1380. [Google Scholar] [CrossRef]
- Gallacher, M.; Brown, S.G.; Hale, B.G.; Fearns, R.; Olver, R.E.; Randall, R.E.; Wilson, S.M. Cation currents in human airway epithelial cells induced by infection with influenza A virus. J. Physiol. 2009, 587, 3159–3173. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.H.; Zhao, B.; Shi, J.; Savage, N.; Shen, Q.; Byrnes, J.; Yang, L.; Hwang, W.; Li, P. Molecular recognition of a host protein by NS1 of pandemic and seasonal influenza A viruses. Proc. Natl. Acad. Sci. USA 2020, 117, 6550–6558. [Google Scholar] [CrossRef] [PubMed]
- Ayllon, J.; Hale, B.G.; García-Sastre, A. Strain-specific contribution of NS1-activated phosphoinositide 3-kinase signaling to influenza A virus replication and virulence. J. Virol. 2012, 86, 5366–5370. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Anderson, D.H.; Liu, Q.; Zhou, Y. Mechanism of influenza A virus NS1 protein interaction with the p85beta, but not the p85alpha, subunit of phosphatidylinositol 3-kinase (PI3K) and up-regulation of PI3K activity. J. Biol. Chem. 2008, 283, 23397–23409. [Google Scholar] [CrossRef] [PubMed]
- Dubrow, A.; Kim, I.; Topo, E.; Cho, J.H. Understanding the Binding Transition State After the Conformational Selection Step: The Second Half of the Molecular Recognition Process Between NS1 of the 1918 Influenza Virus and Host p85β. Front. Mol. Biosci. 2021, 8, 716477. [Google Scholar] [CrossRef]
- Dubrow, A.; Lin, S.; Savage, N.; Shen, Q.; Cho, J.H. Molecular Basis of the Ternary Interaction between NS1 of the 1918 Influenza A Virus, PI3K, and CRK. Viruses 2020, 12, 338. [Google Scholar] [CrossRef]
- Yoneyama, M.; Onomoto, K.; Jogi, M.; Akaboshi, T.; Fujita, T. Viral RNA detection by RIG-I-like receptors. Curr. Opin. Immunol. 2015, 32, 48–53. [Google Scholar] [CrossRef]
- Saito, T.; Hirai, R.; Loo, Y.M.; Owen, D.; Johnson, C.L.; Sinha, S.C.; Akira, S.; Fujita, T.; Gale, M., Jr. Regulation of innate antiviral defenses through a shared repressor domain in RIG-I and LGP2. Proc. Natl. Acad. Sci. USA 2007, 104, 582–587. [Google Scholar] [CrossRef]
- Beckham, S.A.; Brouwer, J.; Roth, A.; Wang, D.; Sadler, A.J.; John, M.; Jahn-Hofmann, K.; Williams, B.R.; Wilce, J.A.; Wilce, M.C. Conformational rearrangements of RIG-I receptor on formation of a multiprotein:dsRNA assembly. Nucleic Acids Res. 2013, 41, 3436–3445. [Google Scholar] [CrossRef]
- Gack, M.U.; Shin, Y.C.; Joo, C.H.; Urano, T.; Liang, C.; Sun, L.; Takeuchi, O.; Akira, S.; Chen, Z.; Inoue, S.; et al. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 2007, 446, 916–920. [Google Scholar] [CrossRef]
- Jiang, X.; Kinch, L.N.; Brautigam, C.A.; Chen, X.; Du, F.; Grishin, N.V.; Chen, Z.J. Ubiquitin-induced oligomerization of the RNA sensors RIG-I and MDA5 activates antiviral innate immune response. Immunity 2012, 36, 959–973. [Google Scholar] [CrossRef] [PubMed]
- Fang, R.; Jiang, Q.; Zhou, X.; Wang, C.; Guan, Y.; Tao, J.; Xi, J.; Feng, J.M.; Jiang, Z. MAVS activates TBK1 and IKKε through TRAFs in NEMO dependent and independent manner. PLoS Pathog. 2017, 13, e1006720. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Taniguchi, T. IRFs: Master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat. Rev. Immunol. 2006, 6, 644–658. [Google Scholar] [CrossRef] [PubMed]
- Fensterl, V.; Grotheer, D.; Berk, I.; Schlemminger, S.; Vallbracht, A.; Dotzauer, A. Hepatitis A Virus Suppresses RIG-I-Mediated IRF-3 Activation To Block Induction of Beta Interferon. J. Virol. 2005, 79, 10968–10977. [Google Scholar] [CrossRef]
- Haller, O.; Kochs, G.; Weber, F. The interferon response circuit: Induction and suppression by pathogenic viruses. Virology 2006, 344, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Miranda, E.; Forafonov, F.; Tavassoli, A. Deciphering interactions used by the influenza virus NS1 protein to silence the host antiviral sensor protein RIG-I using a bacterial reverse two-hybrid system. Mol. Biosyst. 2011, 7, 1042–1045. [Google Scholar] [CrossRef] [PubMed]
- Pothlichet, J.; Meunier, I.; Davis, B.K.; Ting, J.P.; Skamene, E.; von Messling, V.; Vidal, S.M. Type I IFN triggers RIG-I/TLR3/NLRP3-dependent inflammasome activation in influenza A virus infected cells. PLoS Pathog. 2013, 9, e1003256. [Google Scholar] [CrossRef]
- Esposito, D.; Koliopoulos, M.G.; Rittinger, K. Structural determinants of TRIM protein function. Biochem. Soc. Trans. 2017, 45, 183–191. [Google Scholar] [CrossRef]
- D’Cruz, A.A.; Kershaw, N.J.; Chiang, J.J.; Wang, M.K.; Nicola, N.A.; Babon, J.J.; Gack, M.U.; Nicholson, S.E. Crystal structure of the TRIM25 B30.2 (PRYSPRY) domain: A key component of antiviral signalling. Biochem. J. 2013, 456, 231–240. [Google Scholar] [CrossRef]
- Gack, M.U.; Albrecht, R.A.; Urano, T.; Inn, K.S.; Huang, I.C.; Carnero, E.; Farzan, M.; Inoue, S.; Jung, J.U.; García-Sastre, A. Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe 2009, 5, 439–449. [Google Scholar] [CrossRef]
- Rajsbaum, R.; Albrecht, R.A.; Wang, M.K.; Maharaj, N.P.; Versteeg, G.A.; Nistal-Villán, E.; García-Sastre, A.; Gack, M.U. Species-specific inhibition of RIG-I ubiquitination and IFN induction by the influenza A virus NS1 protein. PLoS Pathog. 2012, 8, e10030592012. [Google Scholar] [CrossRef] [PubMed]
- Meyerson, N.R.; Zhou, L.; Guo, Y.R.; Zhao, C.; Tao, Y.J.; Krug, R.M.; Sawyer, S.L. Nuclear TRIM25 Specifically Targets Influenza Virus Ribonucleoproteins to Block the Onset of RNA Chain Elongation. Cell Host Microbe 2017, 22, 627–638.e627. [Google Scholar] [CrossRef] [PubMed]
- Smelkinson, M.G.; Guichard, A.; Teijaro, J.R.; Malur, M.; Loureiro, M.E.; Jain, P.; Ganesan, S.; Zuniga, E.I.; Krug, R.M.; Oldstone, M.B.; et al. Influenza NS1 directly modulates Hedgehog signaling during infection. PLoS Pathog. 2017, 13, e1006588. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, M.; Nishihara, H.; Hasegawa, H.; Tashiro, M.; Wang, L.; Kimura, T.; Tanino, M.; Tsuda, M.; Tanaka, S. NS1-binding protein abrogates the elevation of cell viability by the influenza A virus NS1 protein in association with CRKL. Biochem. Biophys. Res. Commun. 2013, 441, 953–957. [Google Scholar] [CrossRef] [PubMed]
- Cruz, A.; Joseph, S. Interaction of the Influenza A Virus NS1 Protein with the 5′-m7G-mRNA.eIF4E.eIF4G1 Complex. Biochemistry 2022, 61, 1485–1494. [Google Scholar] [CrossRef]
- Burgui, I.; Aragon, T.; Ortin, J.; Nieto, A. PABP1 and eIF4GI associate with influenza virus NS1 protein in viral mRNA translation initiation complexes. J. Gen. Virol. 2003, 84, 3263–3274. [Google Scholar] [CrossRef]
- Aragon, T.; de la Luna, S.; Novoa, I.; Carrasco, L.; Ortin, J.; Nieto, A. Eukaryotic translation initiation factor 4GI is a cellular target for NS1 protein, a translational activator of influenza virus. Mol. Cell. Biol. 2000, 20, 6259–6268. [Google Scholar] [CrossRef]
- Yang, H.; Winkler, W.; Wu, X. Interferon Inducer IFI35 regulates RIG-I-mediated innate antiviral response through mutual antagonism with Influenza protein NS1. J. Virol. 2021, 95. [Google Scholar] [CrossRef]
- Kumar, M.; Liu, H.; Rice, A.P. Regulation of interferon-beta by MAGI-1 and its interaction with influenza A virus NS1 protein with ESEV PBM. PLoS ONE 2012, 7, e41251. [Google Scholar] [CrossRef]
- Wolff, T.; O’Neill, R.E.; Palese, P. NS1-Binding protein (NS1-BP): A novel human protein that interacts with the influenza A virus nonstructural NS1 protein is relocalized in the nuclei of infected cells. J. Virol. 1998, 72, 7170–7180. [Google Scholar] [CrossRef]
- Yan, Y.; Du, Y.; Zheng, H.; Wang, G.; Li, R.; Chen, J.; Li, K. NS1 of H7N9 Influenza A Virus Induces NO-Mediated Cellular Senescence in Neuro2a Cells. Cell Physiol. Biochem. 2017, 43, 1369–1380. [Google Scholar] [CrossRef] [PubMed]
- Melen, K.; Tynell, J.; Fagerlund, R.; Roussel, P.; Hernandez-Verdun, D.; Julkunen, I. Influenza A H3N2 subtype virus NS1 protein targets into the nucleus and binds primarily via its C-terminal NLS2/NoLS to nucleolin and fibrillarin. Virol. J. 2012, 9, 167. [Google Scholar] [CrossRef] [PubMed]
- Murayama, R.; Harada, Y.; Shibata, T.; Kuroda, K.; Hayakawa, S.; Shimizu, K.; Tanaka, T. Influenza A virus non-structural protein 1 (NS1) interacts with cellular multifunctional protein nucleolin during infection. Biochem. Biophys. Res. Commun. 2007, 362, 880–885. [Google Scholar] [CrossRef]
- de Rozieres, C.M.; Joseph, S. Influenza A Virus NS1 Protein Binds as a Dimer to RNA-Free PABP1 but Not to the PABP1.Poly(A) RNA Complex. Biochemistry 2020, 59, 4439–4448. [Google Scholar] [CrossRef] [PubMed]
- Arias-Mireles, B.H.; de Rozieres, C.M.; Ly, K.; Joseph, S. RNA Modulates the Interaction between Influenza A Virus NS1 and Human PABP1. Biochemistry 2018, 57, 3590–3598. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Li, Y.; Krug, R.M. Influenza A virus NS1 protein targets poly(A)-binding protein II of the cellular 3′-end processing machinery. EMBO J. 1999, 18, 2273–2283. [Google Scholar] [CrossRef]
- Tawaratsumida, K.; Phan, V.; Hrincius, E.R.; High, A.A.; Webby, R.; Redecke, V.; Hacker, H. Quantitative proteomic analysis of the influenza A virus nonstructural proteins NS1 and NS2 during natural cell infection identifies PACT as an NS1 target protein and antiviral host factor. J. Virol. 2014, 88, 9038–9048. [Google Scholar] [CrossRef]
- Schierhorn, K.L.; Jolmes, F.; Bespalowa, J.; Saenger, S.; Peteranderl, C.; Dzieciolowski, J.; Mielke, M.; Budt, M.; Pleschka, S.; Herrmann, A.; et al. Influenza A Virus Virulence Depends on Two Amino Acids in the N-Terminal Domain of Its NS1 Protein To Facilitate Inhibition of the RNA-Dependent Protein Kinase PKR. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Tan, S.L.; Katze, M.G. Biochemical and genetic evidence for complex formation between the influenza A virus NS1 protein and the interferon-induced PKR protein kinase. J. Interferon Cytokine Res. 1998, 18, 757–766. [Google Scholar] [CrossRef]
- Zhang, H.; Li, W.; Wang, G.; Su, Y.; Zhang, C.; Chen, X.; Xu, Y.; Li, K. The distinct binding properties between avian/human influenza A virus NS1 and Postsynaptic density protein-95 (PSD-95), and inhibition of nitric oxide production. Virol. J. 2011, 8, 298. [Google Scholar] [CrossRef]
- Bavagnoli, L.; Dundon, W.G.; Garbelli, A.; Zecchin, B.; Milani, A.; Parakkal, G.; Baldanti, F.; Paolucci, S.; Volmer, R.; Tu, Y.; et al. The PDZ-ligand and Src-homology type 3 domains of epidemic avian influenza virus NS1 protein modulate human Src kinase activity during viral infection. PLoS ONE 2011, 6, e27789. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Ahn, S.H.; Kim, K.M.; Kim, Y.K. Non-structural protein 1 of influenza viruses inhibits rapid mRNAdegradation mediated by double-stranded RNA-binding protein, staufen1. FEBS Lett. 2013, 587, 2118–2124. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Oh, J.Y.; Pascua, P.N.; Kim, E.G.; Choi, Y.K.; Kim, H.K. Impairment of the Staufen1-NS1 interaction reduces influenza viral replication. Biochem. Biophys. Res. Commun. 2011, 414, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Falcon, A.M.; Fortes, P.; Marion, R.M.; Beloso, A.; Ortin, J. Interaction of influenza virus NS1 protein and the human homologue of Staufen in vivo and in vitro. Nucleic Acids Res. 1999, 27, 2241–2247. [Google Scholar] [CrossRef]
- Liu, Y.C.; Mok, B.W.; Wang, P.; Kuo, R.L.; Chen, H.; Shih, S.R. Cellular 5′-3′ mRNA Exoribonuclease XRN1 Inhibits Interferon Beta Activation and Facilitates Influenza A Virus Replication. mBio 2021, 12, e0094521. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhang, X.; Lei, X.; Wang, H.; Jiang, J.; Wang, Y.; Bi, K.; Diao, H. Influenza A virus NS1 protein hijacks YAP/TAZ to suppress TLR3-mediated innate immune response. PLoS Pathog. 2022, 18, e1010505. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Host Protein Function | Results of Interaction | Methods | PDB ID | Figure | Citations |
|---|---|---|---|---|---|---|
| CHD1 | chromatin remodeling | downregulation of host genes | IP*, ITC, X-ray | 4NW2 4O42 | 3 | Marazzi (2012) [87] Qin (2014) [115] |
| Ci/Gli1 | transcriptional mediator in Hedgehog pathway | modulates Hedgehog signaling | FRET-FLIM | Smelkinson (2017) [215] | ||
| CPSF30 | mRNA processing | downregulation of host genes | co-IP, Confocal, Pulldown, X-ray, Y2H | 2RHK | 7 | Das (2008) [40] Nemeroff (1998) [41] Dankar (2013) [62] Twu (2006) [68] Kuo (2018) [77] DeDiego (2016) [155] Kuo (2009) [158] Kochs (2007) [159] Twu (2007) [160] Li (2018) [161] Rodriguez (2018) [162] Zhu (2008) [163] |
| CrkII | cellular signaling through binding to JNK1 | inhibits innate immune response | Confocal, NMR Relaxation Dispersion, Pulldown, X-ray | 5UL6 | 4 | Shen (2017) [116] Ylösmäki (2016) [126] |
| CrkL | PI3K signaling | inhibits apoptosis | Binding assays *, co-IP | Hrincius (2010) [125] Heikinnen (2008) [127] Ylösmäki (2015) [133] Miyazaki (2013) [216] | ||
| Dlg | tight junction formation | disrupts tight junction formation to enhance viral spread | co-IP, Confocal, Pulldown * | Golebiewski (2011) [88] Thomas (2011) [89] Liu (2010) [137] | ||
| eIF4E | translation initiation | stimulates translation of viral mRNA | FA | Cruz (2022) [217] | ||
| eIF4G1 | translation initiation | enhances viral translation | co-IP, Pulldown * | Burgui (2003) [218] Aragon (2000) [219] | ||
| hPAF1C | transcription elongation | downregulation of host genes | IP * | Marazzi (2012) [87] | ||
| IFI35 | RIG-I regulation through degradation | inhibits type I interferon production to inhibit innate immune system | co-IP, Confocal, Pulldown * | Yang (2021) [220] | ||
| MAGI-1 | tight junction formation | disables tight junction formation to enhance viral spread | co-IP, Confocal, Pulldown * | Thomas (2011) [89] Liu (2010) [137] Kumar (2012) [221] | ||
| MORC3 | ATPase with antiviral activity | prevents MORC3 histone binding and autoinhibition | NMR, X-ray | 6O5W | 6 | Zhang (2019) [149] |
| NS1-BP | inhibits splicing, inhibits some NS1 activities | regulates cell viability through blocking NS1 stimulation of ERK pathway | IP *, Pulldown, Y2H | Miyazaki (2013) [216] Wolff (1998) [222] | ||
| Nucleolin and Fibrillarin | rRNA production | represses ribosome production | co-IP *, Confocal, Pulldown * | Yan (2017) [223] Melen (2012) [224] Murayama (2007) [225] | ||
| NXF1-NXT1 | RNA nuclear export | inhibits host mRNA translation | IP, Pulldown *, X-ray | 6E5U | 8 | Satterly (2007) [169] Zhang (2019) [172] |
| PABP1 | translation initiation | enhances viral translation | co-IP, EMSA, FA, FRET, Pulldown * | Burgui (2003) [218] de Rozieres (2020) [226] Arias-Mireles (2018) [227] | ||
| PABPII | mRNA processing | downregulation of host genes | IP * | Chen (1999) [228] | ||
| PACT | stimulates RIG-I-induced interferon production | inhibits interferon production | co-IP, Pulldown *, SILAC | Li (2006) [47] Tawaratsumida (2014) [229] | ||
| PDlim2 | scaffold protein that recruits NF-kB to ubiquitin ligase | unknown | BiFC, Pulldown *, M2H, X-ray | 3PDV | 9 | Yu (2011) [176] |
| PI3K | apoptosis regulation | inhibits apoptosis | BLI, co-IP, Confocal, ITC, MD, NMR, Pulldown, X-ray | 6U28 6OX7 | 10 | Hale (2010) [49] Ehrhardt (2007) [50] Ylösmäki (2015) [133] Hale (2006) [188] Shin (2007) [191] Hale (2008) [192] Cho (2020) [194] Li (2008) [196] Dubrow (2021) [197] Dubrow (2020) [198] |
| PKR | antiviral activity through translational shutdown | continued viral mRNA translation | co-IP, FRET, Pulldown *, Y2H | Li (2006) [47] Min (2007) [48] Schierhorn (2017) [230] Tan (1998) [231] | ||
| PSD-95 | regulates NO production | reduces NO production | IP *, Pulldown *, Y2H | Zhang (2011) [232] | ||
| RIG-I | interferon induction | inhibits interferon production | co-IP, Confocal, Crosslinking IP, NMR, Pulldown, Y2H, | Mibayashi (2007) [44] Dankar (2013) [62] Jureka (2015) [95] Jureka (2020) [109] Pothlichet (2013) [209] | ||
| RIL and Src Kinase | kinase cascade for cell signaling | alters cell signaling | Protein arrays, Pulldown * | Bavagnoli (2011) [233] | ||
| RuvBL2 | ATPase and helicase with multiple regulatory functions | inhibits infection-induced apoptosis by maintaining RuvBL2 protein abundance | 2DE-MS, co-IP, Pulldown * | Wang (2021) [79] | ||
| Scribble | cell polarity regulator, tight junction formation | disrupts tight junction formation to enhance viral spread | CD, co-IP, Confocal, ITC, Pulldown *, X-ray | 7QTO 7QTP 7QTU | 5 | Golebiewski (2011) [88] Thomas (2011) [89] Liu (2010) [137] Javorsky (2022) [143] |
| Staufen | RNA translocation and regulation | inhibits viral mRNA degradation | Confocal, IP *, Tethering assay, Y2H | Cho (2013) [234] Lee (2011) [235] Falcon (1999) [236] | ||
| TRIM25 | RIG-I activation | inhibits innate immune response | BLI, co-IP *, Confocal, NMR, SECMALS, X-ray | 5NT1 5NT2 | 11 | Koliopoulos (2018) [114] Gack (2009) [212] Rajsbaum (2012) [213] |
| WDR5 | coactivator involved in gene regulation | downregulation of host genes | ITC, X-ray | 4O45 | 1 | Qin (2014) [115] |
| XRN1 | mRNA decay factor in P-bodies | inhibits degradation of viral mRNAs | co-IP, Confocal, Pulldown * | Liu (2021) [237] | ||
| YAP/TAZ | Hippo pathway activation | downregulates cytokines and the innate immune system | co-IP with exogenous Protein | Zhang (2022) [238] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blake, M.E.; Kleinpeter, A.B.; Jureka, A.S.; Petit, C.M. Structural Investigations of Interactions between the Influenza a Virus NS1 and Host Cellular Proteins. Viruses 2023, 15, 2063. https://doi.org/10.3390/v15102063
Blake ME, Kleinpeter AB, Jureka AS, Petit CM. Structural Investigations of Interactions between the Influenza a Virus NS1 and Host Cellular Proteins. Viruses. 2023; 15(10):2063. https://doi.org/10.3390/v15102063
Chicago/Turabian StyleBlake, Morgan E., Alex B. Kleinpeter, Alexander S. Jureka, and Chad M. Petit. 2023. "Structural Investigations of Interactions between the Influenza a Virus NS1 and Host Cellular Proteins" Viruses 15, no. 10: 2063. https://doi.org/10.3390/v15102063
APA StyleBlake, M. E., Kleinpeter, A. B., Jureka, A. S., & Petit, C. M. (2023). Structural Investigations of Interactions between the Influenza a Virus NS1 and Host Cellular Proteins. Viruses, 15(10), 2063. https://doi.org/10.3390/v15102063