

Whole Genome Sequencing of Infectious Bursal Disease Viruses Isolated from a Californian Outbreak Unravels the Underlying Virulence Markers and Highlights Positive Selection Incidence

Abstract
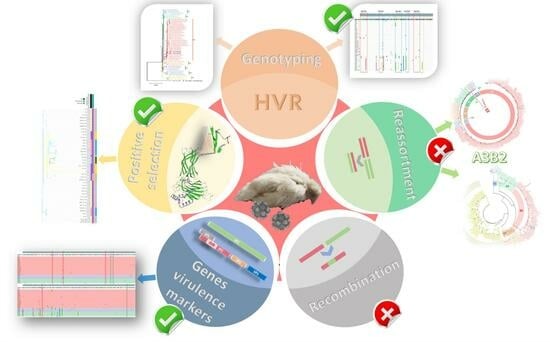
1. Introduction
2. Methods
2.1. Virus Isolation and Propagation
2.2. RNA Extraction and Reverse Transcription
2.3. IBDV Full-Length Genome Amplification and Cloning
2.4. IBDV Whole Genome Sequencing and Raw Data Processing
2.5. Phylogenetic Analyses
2.6. Detection of Recombination Incidence
2.7. Estimation of Selection Pressure
2.8. Secondary Structure Investigation of IBDV VP2
3. Results
3.1. Genomic Organization of Isolates rA and rB
3.2. rA and rB Isolates Belong to the Genogroup 3 Based on HVR Genotyping
3.3. HVR of rA and rB Contained the Virulence aa Hallmarks
3.4. rA and rB Do Not Show Evidence of Reassortment or Recombination
3.5. Positive Contribution of IBDV Genes Leading to Virulence of rA and rB
3.5.1. Gene-Wise Phylogenetic Analysis Showed Entire Clustering of rA and rB with vvIBDVs
3.5.2. Amino Acid Substitution Analysis Denoted the Virulence Signatures within rA and rB Genes
3.6. Selection Pressure Analysis Highlighted the Incidence of Both Purifying and Diversifying Selections
3.7. Secondary Structure Analysis Emphasized VP2 Protein Structure Fitness and Stability
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Edgar, S.; Cho, Y. Avian nephrosis (Gumboro disease) and its control by immunization. Poult. Sci. 1965, 44, 1366. [Google Scholar]
- Brown, M.D.; Skinner, M.A. Coding sequences of both genome segments of a European ‘very virulent’infectious bursal disease virus. Virus Res. 1996, 40, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Jackwood, D.J. Advances in vaccine research against economically important viral diseases of food animals: Infectious bursal disease virus. Vet. Microbiol. 2017, 206, 121–125. [Google Scholar] [CrossRef]
- Petit, S.P.; Lejal, N.; Huet, J.-C.; Delmas, B. Active residues and viral substrate cleavage sites of the protease of the birnavirus infectious pancreatic necrosis virus. J. Virol. 2000, 74, 2057–2066. [Google Scholar] [CrossRef]
- Aliyu, H.B.; Hair-Bejo, M.; Omar, A.R.; Ideris, A. Genetic diversity of recent infectious bursal disease viruses isolated from vaccinated poultry flocks in Malaysia. Front. Vet. Sci. 2021, 8, 643976. [Google Scholar] [CrossRef] [PubMed]
- Bayliss, C.; Spies, U.; Shaw, K.; Peters, R.; Papageorgiou, A.; Müller, H.; Boursnell, M. A comparison of the sequences of segment A of four infectious bursal disease virus strains and identification of a variable region in VP2. J. Gen. Virol. 1990, 71, 1303–1312. [Google Scholar] [CrossRef]
- Jackwood, D.H.; Saif, Y.M. Antigenic diversity of infectious bursal disease viruses. Avian Dis. 1987, 31, 766–770. [Google Scholar] [CrossRef] [PubMed]
- Vakharia, V.N.; He, J.; Ahamed, B.; Snyder, D.B. Molecular basis of antigenic variation in infectious bursal disease virus. Virus Res. 1994, 31, 265–273. [Google Scholar] [CrossRef]
- Letzel, T.; Coulibaly, F.; Rey, F.A.; Delmas, B.; Jagt, E.; Van Loon, A.A.; Mundt, E. Molecular and structural bases for the antigenicity of VP2 of infectious bursal disease virus. J. Virol. 2007, 81, 12827–12835. [Google Scholar] [CrossRef]
- Qi, X.; Gao, X.; Lu, Z.; Zhang, L.; Wang, Y.; Gao, L.; Gao, Y.; Li, K.; Gao, H.; Liu, C. A single mutation in the P BC loop of VP2 is involved in the in vitro replication of infectious bursal disease virus. Sci. China Life Sci. 2016, 59, 717–723. [Google Scholar] [CrossRef] [PubMed]
- Jackwood, D.J.; Sommer-Wagner, S.E. Amino acids contributing to antigenic drift in the infectious bursal disease Birnavirus (IBDV). Virology 2011, 409, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Ismail, N.; Saif, Y.; Moorhead, P. Lack of pathogenicity of five serotype 2 infectious bursal disease viruses in chickens. Avian Dis. 1988, 32, 757–759. [Google Scholar] [CrossRef] [PubMed]
- Lukert, P.D. Infectious bursal disease. In Disease of Poultry; Calnek, B.W., Ed.; IIowa State University Press: Ames, IA, USA, 1991; pp. 648–663. [Google Scholar]
- Ismail, N.; Saif, Y. Immunogenicity of infectious bursal disease viruses in chickens. Avian Dis. 1991, 35, 460–469. [Google Scholar] [CrossRef]
- Van den Berg, T.; Morales, D.; Eterradossi, N.; Rivallan, G.; Toquin, D.; Raue, R.; Zierenberg, K.; Zhang, M.; Zhu, Y.; Wang, C. Assessment of genetic, antigenic and pathotypic criteria for the characterization of IBDV strains. Avian Pathol. 2004, 33, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Jackwood, D.; Cookson, K.; Sommer-Wagner, S.; Galludec, H.L.; De Wit, J. Molecular characteristics of infectious bursal disease viruses from asymptomatic broiler flocks in Europe. Avian Dis. 2006, 50, 532–536. [Google Scholar] [CrossRef] [PubMed]
- Azad, A.A.; Jagadish, M.N.; Brown, M.A.; Hudson, P.J. Deletion mapping and expression in Escherichia coli of the large genomic segment of a birnavirus. Virology 1987, 161, 145–152. [Google Scholar] [CrossRef]
- Heine, H.-G.; Haritou, M.; Failla, P.; Fahey, K.; Azad, A. Sequence analysis and expression of the host-protective immunogen VP2 of a variant strain of infectious bursal disease virus which can circumvent vaccination with standard type I strains. J. Gen. Virol. 1991, 72, 1835–1843. [Google Scholar] [CrossRef]
- Wang, W.; He, X.; Zhang, Y.; Qiao, Y.; Shi, J.; Chen, R.; Chen, J.; Xiang, Y.; Wang, Z.; Chen, G. Analysis of the global origin, evolution and transmission dynamics of the emerging novel variant IBDV (A2dB1b): The accumulation of critical aa-residue mutations and commercial trade contributes to the emergence and transmission of novel variants. Transbound. Emerg. Dis. 2022, 69, e2832–e2851. [Google Scholar] [CrossRef]
- Fan, L.; Wu, T.; Hussain, A.; Gao, Y.; Zeng, X.; Wang, Y.; Gao, L.; Li, K.; Wang, Y.; Liu, C. Novel variant strains of infectious bursal disease virus isolated in China. Vet. Microbiol. 2019, 230, 212–220. [Google Scholar] [CrossRef]
- Muniz, E.; Verdi, R.; Jackwood, D.; Kuchpel, D.; Resende, M.; Mattos, J.; Cookson, K. Molecular epidemiologic survey of infectious bursal disease viruses in broiler farms raised under different vaccination programs. J. Appl. Poult. Res. 2018, 27, 253–261. [Google Scholar] [CrossRef]
- Hussain, A.; Wu, T.; Li, H.; Fan, L.; Li, K.; Gao, L.; Wang, Y.; Gao, Y.; Liu, C.; Cui, H. Pathogenic characterization and full length genome sequence of a reassortant infectious bursal disease virus newly isolated in Pakistan. Virol. Sin. 2019, 34, 102–105. [Google Scholar] [CrossRef] [PubMed]
- Pikuła, A.; Śmietanka, K.; Perez, L.J. Emergence and expansion of novel pathogenic reassortant strains of infectious bursal disease virus causing acute outbreaks of the disease in Europe. Transbound. Emerg. Dis. 2020, 67, 1739–1744. [Google Scholar] [CrossRef]
- Hussain, A.; Wu, T.-T.; Fan, L.-J.; Wang, Y.-L.; Muhammad, F.K.; Jiang, N.; Li, G.; Kai, L.; Gao, Y.-l.; Liu, C.-J. The circulation of unique reassortment strains of infectious bursal disease virus in Pakistan. J. Integr. Agric. 2020, 19, 1867–1875. [Google Scholar] [CrossRef]
- Lupini, C.; Giovanardi, D.; Pesente, P.; Bonci, M.; Felice, V.; Rossi, G.; Morandini, E.; Cecchinato, M.; Catelli, E. A molecular epidemiology study based on VP2 gene sequences reveals that a new genotype of infectious bursal disease virus is dominantly prevalent in Italy. Avian Pathol. 2016, 45, 458–464. [Google Scholar] [CrossRef]
- Felice, V.; Franzo, G.; Catelli, E.; Di Francesco, A.; Bonci, M.; Cecchinato, M.; Mescolini, G.; Giovanardi, D.; Pesente, P.; Lupini, C. Genome sequence analysis of a distinctive Italian infectious bursal disease virus. Poult. Sci. 2017, 96, 4370–4377. [Google Scholar] [CrossRef]
- Stoute, S.T.; Jackwood, D.J.; Sommer-Wagner, S.E.; Cooper, G.L.; Anderson, M.L.; Woolcock, P.R.; Bickford, A.A.; Sentíes-Cué, C.G.; Charlton, B.R. The diagnosis of very virulent infectious bursal disease in California pullets. Avian Dis. 2009, 53, 321–326. [Google Scholar] [CrossRef]
- Jackwood, D.J.; Sommer-Wagner, S.E.; Stoute, S.T.; Woolcock, P.R.; Crossley, B.M.; Hietala, S.K.; Charlton, B.R. Characteristics of a very virulent infectious bursal disease virus from California. Avian Dis. 2009, 53, 592–600. [Google Scholar] [CrossRef]
- De Coster, W.; D’hert, S.; Schultz, D.T.; Cruts, M.; Van Broeckhoven, C. NanoPack: Visualizing and processing long-read sequencing data. Bioinformatics 2018, 34, 2666–2669. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Michel, L.O.; Jackwood, D.J. Classification of infectious bursal disease virus into genogroups. Arch. Virol. 2017, 162, 3661–3670. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-L.; Fan, L.-J.; Jiang, N.; Li, G.; Kai, L.; Gao, Y.-L.; Liu, C.-J.; Cui, H.-Y.; Qing, P.; Zhang, Y.-P. An improved scheme for infectious bursal disease virus genotype classification based on both genome-segments A and B. J. Integr. Agric. 2021, 20, 1372–1381. [Google Scholar] [CrossRef]
- Feng, X.; Zhu, N.; Cui, Y.; Hou, L.; Zhou, J.; Qiu, Y.; Yang, X.; Liu, C.; Wang, D.; Guo, J. Characterization and pathogenicity of a naturally reassortant and recombinant infectious bursal disease virus in China. Transbound. Emerg. Dis. 2022, 69, e746–e758. [Google Scholar] [CrossRef] [PubMed]
- Lachheb, J.; Jbenyeni, A.; Nsiri, J.; Larbi, I.; Ammouna, F.; Ghram, A. Full-length genome sequencing of a very virulent infectious bursal disease virus isolated in Tunisia. Poult. Sci. 2021, 100, 496–506. [Google Scholar] [CrossRef]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef]
- Martin, D.; Posada, D.; Crandall, K.; Williamson, C. A modified bootscan algorithm for automated identification of recombinant sequences and recombination breakpoints. Virol. Sin. 2005, 21, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef]
- Arowolo, O.; George, U.; Luka, P.; Maurice, N.; Atuman, Y.; Shallmizhili, J.; Shittu, I.; Oluwayelu, D. Infectious bursal disease in Nigeria: Continuous circulation of reassortant viruses. Trop. Anim. Health Prod. 2021, 53, 271. [Google Scholar] [CrossRef]
- Brown, M.D.; Green, P.; Skinner, M.A. VP2 sequences of recent European ‘very virulent’ isolates of infectious bursal disease virus are closely related to each other but are distinct from those of ‘classical’ strains. J. Gen. Virol. 1994, 75, 675–680. [Google Scholar] [CrossRef]
- Hoque, M.M.; Omar, A.; Chong, L.; Hair-Bejo, M.; Aini, I. Pathogenicity of Ssp I-positive infectious bursal disease virus and molecular characterization of the VP2 hypervariable region. Avian Pathol. 2001, 30, 369–380. [Google Scholar] [CrossRef]
- Bao, K.; Qi, X.; Li, Y.; Gong, M.; Wang, X.; Zhu, P. Cryo-EM structures of infectious bursal disease viruses with different virulences provide insights into their assembly and invasion. Sci. Bull. 2022, 67, 646–654. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhou, J.; Kwang, J. Antigenic and molecular characterization of recent infectious bursal disease virus isolates in China. Virus Genes 2002, 24, 135–147. [Google Scholar] [CrossRef]
- Lim, B.-L.; Cao, Y.; Yu, T.; Mo, C.-W. Adaptation of very virulent infectious bursal disease virus to chicken embryonic fibroblasts by site-directed mutagenesis of residues 279 and 284 of viral coat protein VP2. J. Virol. 1999, 73, 2854–2862. [Google Scholar] [CrossRef] [PubMed]
- Shehata, A.A.; Sultan, H.; Halami, M.Y.; Talaat, S.; Vahlenkamp, T.W. Molecular characterization of very virulent infectious bursal disease virus strains circulating in Egypt from 2003 to 2014. Arch. Virol. 2017, 162, 3803–3815. [Google Scholar] [CrossRef] [PubMed]
- Jackwood, D.; Sreedevi, B.; LeFever, L.; Sommer-Wagner, S. Studies on naturally occurring infectious bursal disease viruses suggest that a single amino acid substitution at position 253 in VP2 increases pathogenicity. Virology 2008, 377, 110–116. [Google Scholar] [CrossRef]
- Qi, X.; Zhang, L.; Chen, Y.; Gao, L.; Wu, G.; Qin, L.; Wang, Y.; Ren, X.; Gao, Y.; Gao, H. Mutations of residues 249 and 256 in VP2 are involved in the replication and virulence of infectious bursal disease virus. PLoS ONE 2013, 8, e70982. [Google Scholar] [CrossRef] [PubMed]
- Dormitorio, T.; Giambrone, J.; Guo, K.; Jackwood, D. Molecular and phenotypic characterization of infectious bursal disease virus isolates. Avian Dis. 2007, 51, 597–600. [Google Scholar] [CrossRef]
- Lombardo, E.; Maraver, A.; Espinosa, I.; Fernández-Arias, A.; Rodriguez, J.F. VP5, the nonstructural polypeptide of infectious bursal disease virus, accumulates within the host plasma membrane and induces cell lysis. Virology 2000, 277, 345–357. [Google Scholar] [CrossRef]
- Wei, Y.; Li, J.; Zheng, J.; Xu, H.; Li, L.; Yu, L. Genetic reassortment of infectious bursal disease virus in nature. Biochem. Biophys. Res. Commun. 2006, 350, 277–287. [Google Scholar] [CrossRef]
- Legnardi, M.; Franzo, G.; Tucciarone, C.M.; Koutoulis, K.; Cecchinato, M. Infectious bursal disease virus in Western Europe: The rise of reassortant strains as the dominant field threat. Avian Pathol. 2023, 52, 25–35. [Google Scholar] [CrossRef]
- Mató, T.; Tatár-Kis, T.; Felföldi, B.; Jansson, D.S.; Homonnay, Z.; Bányai, K.; Palya, V. Occurrence and spread of a reassortant very virulent genotype of infectious bursal disease virus with altered VP2 amino acid profile and pathogenicity in some European countries. Vet. Microbiol. 2020, 245, 108663. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wang, X.; Gao, Y.; Qi, X. The Over-40-years-epidemic of infectious bursal disease virus in China. Viruses 2022, 14, 2253. [Google Scholar] [CrossRef] [PubMed]
- Van den Berg, T.; Gonze, M.; Meulemans, G. Acute infectious bursal disease in poultry: Isolation and characterisation of a highly virulent strain. Avian Pathol. 1991, 20, 133–143. [Google Scholar] [CrossRef]
- Liu, M.; Vakharia, V.N. VP1 protein of infectious bursal disease virus modulates the virulence in vivo. Virology 2004, 330, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Wang, Y.; Li, H.; Fan, L.; Jiang, N.; Gao, L.; Li, K.; Gao, Y.; Liu, C.; Cui, H. Naturally occurring homologous recombination between novel variant infectious bursal disease virus and intermediate vaccine strain. Vet. Microbiol. 2020, 245, 108700. [Google Scholar] [CrossRef]
- Ganguly, B.; Rastogi, S.K. Structural and functional modeling of viral protein 5 of Infectious Bursal Disease Virus. Virus Res. 2018, 247, 55–60. [Google Scholar] [CrossRef]
- Qin, Y.; Zheng, S.J. Infectious bursal disease virus-host interactions: Multifunctional viral proteins that perform multiple and differing jobs. Int. J. Mol. Sci. 2017, 18, 161. [Google Scholar] [CrossRef]
- Hernández, M.; Villegas, P.; Hernández, D.; Banda, A.; Maya, L.; Romero, V.; Tomás, G.; Pérez, R. Sequence variability and evolution of the terminal overlapping VP5 gene of the infectious bursal disease virus. Virus Genes 2010, 41, 59–66. [Google Scholar] [CrossRef]
- Yao, K.; Goodwin, M.A.; Vakharia, V.N. Generation of a mutant infectious bursal disease virus that does not cause bursal lesions. J. Virol. 1998, 72, 2647–2654. [Google Scholar] [CrossRef]
- Pikuła, A.; Lisowska, A.; Jasik, A.; Śmietanka, K. Identification and assessment of virulence of a natural reassortant of infectious bursal disease virus. Vet. Res. 2018, 49, 89. [Google Scholar] [CrossRef]
- Brandt, M.; Yao, K.; Liu, M.; Heckert, R.A.; Vakharia, V.N. Molecular determinants of virulence, cell tropism, and pathogenic phenotype of infectious bursal disease virus. J. Virol. 2001, 75, 11974–11982. [Google Scholar] [CrossRef]
- Rudd, M.; Heine, H.; Sapats, S.; Parede, L.; Ignjatovic, J. Characterisation of an Indonesian very virulent strain of infectious bursal disease virus. Arch. Virol. 2002, 147, 1303–1322. [Google Scholar] [CrossRef]
- Wang, S.; Hu, B.; Si, W.; Jia, L.; Zheng, X.; Zhou, J. Avibirnavirus VP4 protein is a phosphoprotein and partially contributes to the cleavage of intermediate precursor VP4-VP3 polyprotein. PLoS ONE 2015, 10, e0128828. [Google Scholar] [CrossRef] [PubMed]
- Touzani, C.D.; Fellahi, S.; Fihri, O.F.; Gaboun, F.; Khayi, S.; Mentag, R.; Lico, C.; Baschieri, S.; El Houadfi, M.; Ducatez, M. Complete genome analysis and time scale evolution of very virulent infectious bursal disease viruses isolated from recent outbreaks in Morocco. Infect. Genet. Evol. 2020, 77, 104097. [Google Scholar] [CrossRef]
- Kong, L.L.; Omar, A.; Hair-Bejo, M.; Aini, I.; Seow, H.-F. Sequence analysis of both genome segments of two very virulent infectious bursal disease virus field isolates with distinct pathogenicity. Arch. Virol. 2004, 149, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zeng, X.; Gao, H.; Fu, C.; Wei, P. Changes in VP2 gene during the attenuation of very virulent infectious bursal disease virus strain Gx isolated in China. Avian Dis. 2004, 48, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Qi, X.; Kang, Z.; Yu, F.; Qin, L.; Gao, H.; Gao, Y.; Wang, X. A single amino acid in the C-terminus of VP3 protein influences the replication of attenuated infectious bursal disease virus in vitro and in vivo. Antivir. Res. 2010, 87, 223–229. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, H.; Gao, H.; Fu, C.; Gao, Y.; Ju, Y. Changes in VP3 and VP5 genes during the attenuation of the very virulent infectious bursal disease virus strain Gx isolated in China. Virus Genes 2007, 34, 67–73. [Google Scholar] [CrossRef]
- Escaffre, O.; Le Nouën, C.; Amelot, M.; Ambroggio, X.; Ogden, K.M.; Guionie, O.; Toquin, D.; Müller, H.; Islam, M.R.; Eterradossi, N. Both genome segments contribute to the pathogenicity of very virulent infectious bursal disease virus. J. Virol. 2013, 87, 2767–2780. [Google Scholar] [CrossRef]
- Cui, P.; Ma, S.-J.; Zhang, Y.-G.; Li, X.-S.; Gao, X.-Y.; Cui, B.-A.; Chen, H.-Y. Genomic sequence analysis of a new reassortant infectious bursal disease virus from commercial broiler flocks in Central China. Arch. Virol. 2013, 158, 1973–1978. [Google Scholar] [CrossRef]
- Pan, J.; Vakharia, V.N.; Tao, Y.J. The structure of a birnavirus polymerase reveals a distinct active site topology. Proc. Natl. Acad. Sci. USA 2007, 104, 7385–7390. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Li, K.; Qi, X.; Gao, H.; Gao, Y.; Qin, L.; Wang, Y.; Shen, N.; Kong, X.; Wang, X. Triplet amino acids located at positions 145/146/147 of the RNA polymerase of very virulent infectious bursal disease virus contribute to viral virulence. J. Gen. Virol. 2014, 95, 888–897. [Google Scholar] [CrossRef] [PubMed]
- Jackwood, D.J.; Sommer, S.E. Identification of infectious bursal disease virus quasispecies in commercial vaccines and field isolates of this double-stranded RNA virus. Virology 2002, 304, 105–113. [Google Scholar] [CrossRef][Green Version]
- Domingo, E.; Sheldon, J.; Perales, C. Viral quasispecies evolution. Microbiol. Mol. Biol. Rev. 2012, 76, 159–216. [Google Scholar] [CrossRef] [PubMed]
- Legnardi, M.; Franzo, G.; Tucciarone, C.M.; Koutoulis, K.; Duarte, I.; Silva, M.; Le Tallec, B.; Cecchinato, M. Detection and molecular characterization of a new genotype of infectious bursal disease virus in Portugal. Avian Pathol. 2022, 51, 97–105. [Google Scholar] [CrossRef]
- Coulibaly, F.; Chevalier, C.; Gutsche, I.; Pous, J.; Navaza, J.; Bressanelli, S.; Delmas, B.; Rey, F.A. The birnavirus crystal structure reveals structural relationships among icosahedral viruses. Cell 2005, 120, 761–772. [Google Scholar] [CrossRef]
- Durairaj, V.; Sellers, H.S.; Linnemann, E.G.; Icard, A.H.; Mundt, E. Investigation of the antigenic evolution of field isolates using the reverse genetics system of infectious bursal disease virus (IBDV). Arch. Virol. 2011, 156, 1717–1728. [Google Scholar] [CrossRef]
- Lojkić, I.; Bidin, Z.; Pokrić, B. Sequence analysis of both genome segments of three Croatian infectious bursal disease field viruses. Avian Dis. 2008, 52, 513–519. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Segment A | |||||||
| 5′UTR | VP5 | VP2 | VP4 | VP3 | 3′UTR | ||
| Whole | Mature | HVR * | |||||
| 1–84 | 85–534 | 131–1666 | 131–1453 | 761–1180 | 1667–2395 | 2396–3169 | 3170–3261 |
| Segment B | |||||||
| 5′UTR | VP1 | 3′UTR | |||||
| 1–111 | 112–2751 | 2752–2827 | |||||
| Strain | Phenotype | VP5 | Mature VP2 (Excluding HVR) | VP4 | VP3 | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 18 | 80 | 95 | 59 | 80 | 163 | 173 | 178 | 426 | 535 (23) ¥ | 604 (92) | 642 (130) | 667 (155) | 745 (233) | 773 (18) | 774 (19) | 970 (215) | 990 (235) | ||
| D6948 | Very virulent | E | W | L | F | Y | G | Y | V | F | I | S | K | V | N | E | A | M | A |
| rA | K | G | P | S | H | E | * | * | * | T | F | N | A | S | V | V | * | V | |
| rB | K | G | * | * | * | * | C | A | S | * | * | N | * | S | * | V | V | V | |
| SH | K | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | V | |
| ks | K | * | * | * | * | * | * | * | * | * | * | N | * | S | * | * | * | V | |
| mb | K | * | * | * | * | * | * | * | * | * | * | N | * | S | * | * | * | V | |
| DD1 | K | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | V | |
| 3529 | * | * | * | * | * | * | * | * | * | * | * | N | * | * | * | * | * | V | |
| UK661 | K | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | V | |
| 89163 | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | |
| HuB-1 | K | * | * | * | * | * | * | * | * | * | * | * | * | * | * | V | * | V | |
| GZ/96 | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | V | |
| Hairbin-1 | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | V | |
| TASIK | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | V | |
| Chinju | K | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | V | |
| Cro-Ig | K | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | |
| T09 | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | V | |
| TN46/19 | K | G | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | |
| 150124/1.1 | K | G | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | |
| 150133/3.2 | K | G | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | |
| 150144/5.1 | K | G | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | |
| 150128/4.1 | K | G | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | |
| CAFHS-785 | K | * | * | * | * | * | * | * | * | * | * | N | * | * | * | * | * | V | |
| 7741-SEGA | K | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | V | |
| KZC-104 | K | * | * | * | * | * | * | * | * | * | * | * | * | S | * | * | * | V | |
| Bpop-03 | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | |
| HHN81 | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | |
| HHN79 | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | |
| Ventri | * | * | * | * | * | * | * | * | * | * | * | N | * | S | * | * | * | V | |
| IBDV78/ABIC | K | * | * | * | * | * | * | * | * | * | * | * | * | S | * | * | * | V | |
| VarE | Variant | K | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * |
| 9109 | K | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | |
| F52/70 | Classical | K | * | * | * | * | * | * | * | * | * | * | R | * | * | * | * | * | * |
| STC | K | * | * | * | L | * | * | * | * | * | * | * | * | * | * | * | * | * | |
| Cu-1wt | K | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | |
| B87 | Attenuated | K | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * |
| JD1 | K | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | |
| HZ2 | K | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | |
| 903/78 | K | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | |
| CEF94 | K | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | |
| Gt | K | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | |
| dIBDV | Distinct | K | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * |
| Strain | Phenotype | VP1 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 4 | 111 | 186 | 378 | 424 | 494 | 513 | 545 | 607 | 637 | ||
| D6948 | Very virulent | V | I | R | F | C | H | D | V | S | L |
| rA | I | * | * | * | * | * | * | * | * | * | |
| rB | I | F | G | V | R | L | G | A | G | P | |
| ks | * | * | * | * | * | * | * | * | * | * | |
| mb | * | * | * | * | * | * | * | * | * | * | |
| DD1 | * | * | * | * | *s | * | * | * | * | * | |
| UK661 | * | * | * | * | * | * | * | * | * | * | |
| 89163 | * | * | * | * | * | * | * | * | * | * | |
| HuB-1 | * | * | * | * | * | * | * | * | * | * | |
| GZ/96 | * | * | * | * | * | * | * | * | * | * | |
| Hairbin-1 | * | * | * | * | * | * | H | * | * | * | |
| TASIK | * | * | * | * | * | * | * | * | * | * | |
| Cro-Ig | * | * | * | * | * | * | * | * | * | * | |
| T09 | * | * | * | * | * | * | * | * | * | * | |
| TN46/19 | I | * | * | * | * | * | * | * | * | * | |
| 150124/1.1 | * | * | * | * | * | * | * | * | * | * | |
| 150133/3.2 | I | * | * | * | * | * | * | * | * | * | |
| 150144/5.1 | * | * | * | * | * | * | * | * | * | * | |
| 150128/4.1 | I | * | * | * | * | * | * | * | * | * | |
| VarE | Variant | I | * | * | * | * | * | * | * | * | * |
| 9109 | I | * | * | * | * | * | * | * | * | * | |
| F52/70 | Classical | I | * | * | * | * | * | * | * | * | * |
| JD1 | Attenuated | I | * | * | * | * | * | * | * | * | * |
| HZ2 | I | * | * | * | * | * | * | * | * | * | |
| CEF94 | I | * | * | * | * | * | * | * | * | * | |
| dIBDV | Distinct | I | * | * | * | * | * | * | * | * | * |
| Protein | Tajima’s D | Z-Test | dN/dS | Positively Selected Sites | Negatively Selected Sites | |||
|---|---|---|---|---|---|---|---|---|
| SLAC (Codon) | FUBAR (Codon) | SLAC | FUBAR | |||||
| VP1 | 0.72408 | NS | 0.0473 | 0 | 0 | 133 | 445 | |
| VP5 | −1.35217 | NS | 1.62 | 0 | 5 (44, 78, 104, 116, 138) | 2 | 2 | |
| PP | VP2 | −1.02634 | SS | 0.111 | 0 | 1 (222) | 66 | 171 |
| VP4 | −0.85195 | NS | 0.0938 | 0 | 1 (168 [680 *]) | 26 | 65 | |
| VP3 | −1.35183 | NS | 0.0983 | 0 | 0 | 37 | 74 | |
| HVR | −0.47423 | NS | 0.161 ¥/0.162 § | 0 ¥/1 § (13[222] *) | 1 (13[222] *) | 16 ¥/49 § | 30 ¥/65 § | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nour, I.; Blakey, J.R.; Alvarez-Narvaez, S.; Mohanty, S.K. Whole Genome Sequencing of Infectious Bursal Disease Viruses Isolated from a Californian Outbreak Unravels the Underlying Virulence Markers and Highlights Positive Selection Incidence. Viruses 2023, 15, 2044. https://doi.org/10.3390/v15102044
Nour I, Blakey JR, Alvarez-Narvaez S, Mohanty SK. Whole Genome Sequencing of Infectious Bursal Disease Viruses Isolated from a Californian Outbreak Unravels the Underlying Virulence Markers and Highlights Positive Selection Incidence. Viruses. 2023; 15(10):2044. https://doi.org/10.3390/v15102044
Chicago/Turabian StyleNour, Islam, Julia R. Blakey, Sonsiray Alvarez-Narvaez, and Sujit K. Mohanty. 2023. "Whole Genome Sequencing of Infectious Bursal Disease Viruses Isolated from a Californian Outbreak Unravels the Underlying Virulence Markers and Highlights Positive Selection Incidence" Viruses 15, no. 10: 2044. https://doi.org/10.3390/v15102044
APA StyleNour, I., Blakey, J. R., Alvarez-Narvaez, S., & Mohanty, S. K. (2023). Whole Genome Sequencing of Infectious Bursal Disease Viruses Isolated from a Californian Outbreak Unravels the Underlying Virulence Markers and Highlights Positive Selection Incidence. Viruses, 15(10), 2044. https://doi.org/10.3390/v15102044