
Existing Evidence for Influenza B Virus Adaptations to Drive Replication in Humans as the Primary Host

Abstract
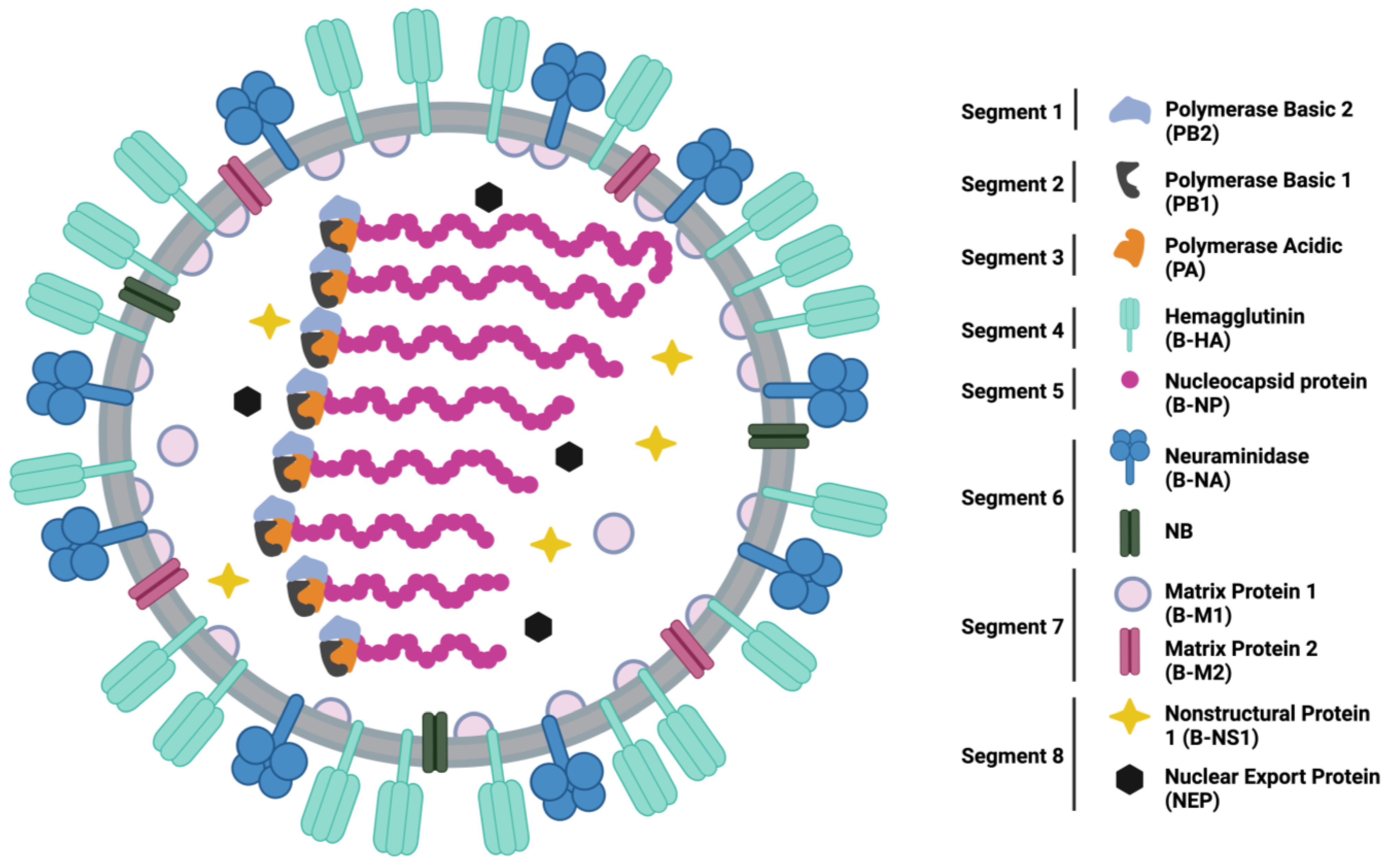
:1. Introduction
2. Hemagglutinin-Specific Adaptations
3. Nuclear Replication Adaptations
4. Immune Evasion
5. Summary and Future Research Directions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. Influenza (Seasonal). 6 November 2018. Available online: https://www.who.int/en/news-room/fact-sheets/detail/influenza-(seasonal) (accessed on 22 May 2023).
- Rota, P.A.; Wallis, T.R.; Harmon, M.W.; Rota, J.S.; Kendal, A.P.; Nerome, K. Cocirculation of two distinct evolutionary lineages of influenza type B virus since 1983. Virology 1990, 175, 59–68. [Google Scholar] [CrossRef]
- Rosu, M.E.; Lexmond, P.; Bestebroer, T.M.; Hauser, B.M.; Smith, D.J.; Herfst, S.; Fouchier, R.A.M. Substitutions near the HA receptor binding site explain the origin and major antigenic change of the B/Victoria and B/Yamagata lineages. Proc. Natl. Acad. Sci. USA 2022, 119, e2211616119. [Google Scholar] [CrossRef]
- Shaw, M.W.; Xu, X.; Li, Y.; Normand, S.; Ueki, R.T.; Kunimoto, G.Y.; Hall, H.; Klimov, A.; Cox, N.J.; Subbarao, K. Reappearance and Global Spread of Variants of Influenza B/Victoria/2/87 Lineage Viruses in the 2000–2001 and 2001–2002 Seasons. Virology 2002, 303, 1–8. [Google Scholar] [CrossRef]
- Skowronski, D.M.; Masaro, C.; Kwindt, T.L.; Mak, A.; Petric, M.; Li, Y.; Sebastian, R.; Chong, M.; Tam, T.; De Serres, G. Estimating vaccine effectiveness against laboratory-confirmed influenza using a sentinel physician network: Results from the 2005–2006 season of dual A and B vaccine mismatch in Canada. Vaccine 2007, 25, 2842–2851. [Google Scholar] [CrossRef] [PubMed]
- Lo, Y.C.; Chuang, J.H.; Kuo, H.W.; Huang, W.T.; Hsu, Y.F.; Liu, M.T.; Chen, C.H.; Huang, H.H.; Chang, C.H.; Chou, J.H.; et al. Surveillance and vaccine effectiveness of an influenza epidemic predominated by vaccine-mismatched influenza B/Yamagata-lineage viruses in Taiwan, 2011–2012 season. PLoS ONE 2013, 8, e58222. [Google Scholar] [CrossRef] [PubMed]
- Shang, M.; Blanton, L.; Brammer, L.; Olsen, S.J.; Fry, A.M. Influenza-Associated Pediatric Deaths in the United States, 2010–2016. Pediatrics 2018, 141, e20172918. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, J.; Vestergaard, L.S.; Richter, L.; Schmid, D.; Bustos, N.; Asikainen, T.; Trebbien, R.; Denissov, G.; Innos, K.; Virtanen, M.J.; et al. European all-cause excess and influenza-attributable mortality in the 2017/18 season: Should the burden of influenza B be reconsidered? Clin. Microbiol. Infect. 2019, 25, 1266–1276. [Google Scholar] [CrossRef] [PubMed]
- Zaraket, H.; Hurt, A.C.; Clinch, B.; Barr, I.; Lee, N. Burden of influenza B virus infection and considerations for clinical management. Antiviral Res. 2021, 185, 104970. [Google Scholar] [CrossRef]
- Saunders-Hastings, P.R.; Krewski, D. Reviewing the History of Pandemic Influenza: Understanding Patterns of Emergence and Transmission. Pathogens 2016, 5, 66. [Google Scholar] [CrossRef]
- Reperant, L.A.; Moesker, F.M.; Osterhaus, A.D. Influenza: From zoonosis to pandemic. ERJ Open Res. 2016, 2, 00013-2016. [Google Scholar] [CrossRef]
- Long, J.S.; Mistry, B.; Haslam, S.M.; Barclay, W.S. Host and viral determinants of influenza A virus species specificity. Nat. Rev. Microbiol. 2019, 17, 67–81. [Google Scholar] [CrossRef] [PubMed]
- Osterhaus, A.D.; Rimmelzwaan, G.F.; Martina, B.E.; Bestebroer, T.M.; Fouchier, R.A. Influenza B virus in seals. Science 2000, 288, 1051–1053. [Google Scholar] [CrossRef] [PubMed]
- Cauldwell, A.V.; Long, J.S.; Moncorgé, O.; Barclay, W.S. Viral determinants of influenza A virus host range. J. Gen. Virol. 2014, 95, 1193–1210. [Google Scholar] [CrossRef]
- Xiong, X.; McCauley, J.W.; Steinhauer, D.A. Receptor binding properties of the influenza virus hemagglutinin as a determinant of host range. Curr. Top Microbiol. Immunol. 2014, 385, 63–91. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.J.; Paulson, J.C. Adaptation of influenza viruses to human airway receptors. J. Biol. Chem. 2021, 296, 100017. [Google Scholar] [CrossRef]
- de Graaf, M.; Fouchier, R.A. Role of receptor binding specificity in influenza A virus transmission and pathogenesis. Embo J. 2014, 33, 823–841. [Google Scholar] [CrossRef]
- Velkov, T. The specificity of the influenza B virus hemagglutinin receptor binding pocket: What does it bind to? J. Mol. Recognit. 2013, 26, 439–449. [Google Scholar] [CrossRef]
- Velkov, T.; Thompson, P.E.; El-Kabbani, O.; Lindh, F.; Stambas, J.; Rockman, S. Agel-capture assay for characterizing the sialyl-glycan selectivity of influenza viruses. Acta Virol. 2011, 55, 131–137. [Google Scholar] [CrossRef]
- Wang, Y.F.; Chang, C.F.; Chi, C.Y.; Wang, H.C.; Wang, J.R.; Su, I.J. Characterization of glycan binding specificities of influenza B viruses with correlation with hemagglutinin genotypes and clinical features. J. Med. Virol. 2012, 84, 679–685. [Google Scholar] [CrossRef]
- Ni, F.; Mbawuike, I.N.; Kondrashkina, E.; Wang, Q. The roles of hemagglutinin Phe-95 in receptor binding and pathogenicity of influenza B virus. Virology 2014, 450, 71–83. [Google Scholar] [CrossRef]
- Krammer, F.; Smith, G.J.D.; Fouchier, R.A.M.; Peiris, M.; Kedzierska, K.; Doherty, P.C.; Palese, P.; Shaw, M.L.; Treanor, J.; Webster, R.G.; et al. Influenza. Nat. Rev. Dis. Primers 2018, 4, 3. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Yang, G.; DeBeauchamp, J.; Crumpton, J.C.; Kim, H.; Li, L.; Wan, X.F.; Kercher, L.; Bowman, A.S.; Webster, R.G.; et al. HA stabilization promotes replication and transmission of swine H1N1 gamma influenza viruses in ferrets. Elife 2020, 9, e56236. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Pu, J. Influence of Host Sialic Acid Receptors Structure on the Host Specificity of Influenza Viruses. Viruses 2022, 14, 2141. [Google Scholar] [CrossRef] [PubMed]
- Trebbien, R.; Larsen, L.E.; Viuff, B.M. Distribution of sialic acid receptors and influenza A virus of avian and swine origin in experimentally infected pigs. Virol. J. 2011, 8, 434. [Google Scholar] [CrossRef]
- Shinya, K.; Ebina, M.; Yamada, S.; Ono, M.; Kasai, N.; Kawaoka, Y. Avian flu: Influenza virus receptors in the human airway. Nature 2006, 440, 435–436. [Google Scholar] [CrossRef]
- Walther, T.; Karamanska, R.; Chan, R.W.; Chan, M.C.; Jia, N.; Air, G.; Hopton, C.; Wong, M.P.; Dell, A.; Malik Peiris, J.S.; et al. Glycomic analysis of human respiratory tract tissues and correlation with influenza virus infection. PLoS Pathog. 2013, 9, e1003223. [Google Scholar] [CrossRef]
- Kuchipudi, S.V.; Nelli, R.K.; Gontu, A.; Satyakumar, R.; Surendran Nair, M.; Subbiah, M. Sialic Acid Receptors: The Key to Solving the Enigma of Zoonotic Virus Spillover. Viruses 2021, 13, 262. [Google Scholar] [CrossRef]
- Caini, S.; Kusznierz, G.; Garate, V.V.; Wangchuk, S.; Thapa, B.; de Paula Júnior, F.J.; Ferreira de Almeida, W.A.; Njouom, R.; Fasce, R.A.; Bustos, P.; et al. The epidemiological signature of influenza B virus and its B/Victoria and B/Yamagata lineages in the 21st century. PLoS ONE 2019, 14, e0222381. [Google Scholar] [CrossRef]
- Read, J.M.; Zimmer, S.; Vukotich, C., Jr.; Schweizer, M.L.; Galloway, D.; Lingle, C.; Yearwood, G.; Calderone, P.; Noble, E.; Quadelacy, T.; et al. Influenza and other respiratory viral infections associated with absence from school among schoolchildren in Pittsburgh, Pennsylvania, USA: A cohort study. BMC Infect. Dis. 2021, 21, 291. [Google Scholar] [CrossRef]
- Russell, C.J. Acid-induced membrane fusion by the hemagglutinin protein and its role in influenza virus biology. Curr. Top. Microbiol. Immunol. 2014, 385, 93–116. [Google Scholar] [CrossRef]
- Castro, L.K.; Daugherty, M.D. Tripping the wire: Sensing of viral protease activity by CARD8 and NLRP1 inflammasomes. Curr. Opin. Immunol. 2023, 83, 102354. [Google Scholar] [CrossRef] [PubMed]
- Limburg, H.; Harbig, A.; Bestle, D.; Stein, D.A.; Moulton, H.M.; Jaeger, J.; Janga, H.; Hardes, K.; Koepke, J.; Schulte, L.; et al. TMPRSS2 Is the Major Activating Protease of Influenza A Virus in Primary Human Airway Cells and Influenza B Virus in Human Type II Pneumocytes. J. Virol. 2019, 93, e00649-19. [Google Scholar] [CrossRef] [PubMed]
- Laporte, M.; Stevaert, A.; Raeymaekers, V.; Boogaerts, T.; Nehlmeier, I.; Chiu, W.; Benkheil, M.; Vanaudenaerde, B.; Pöhlmann, S.; Naesens, L. Hemagglutinin Cleavability, Acid Stability, and Temperature Dependence Optimize Influenza B Virus for Replication in Human Airways. J. Virol. 2019, 94, e01430-19. [Google Scholar] [CrossRef]
- Harbig, A.; Mernberger, M.; Bittel, L.; Pleschka, S.; Schughart, K.; Steinmetzer, T.; Stiewe, T.; Nist, A.; Böttcher-Friebertshäuser, E. Transcriptome profiling and protease inhibition experiments identify proteases that activate H3N2 influenza A and influenza B viruses in murine airways. J. Biol. Chem. 2020, 295, 11388–11407. [Google Scholar] [CrossRef] [PubMed]
- Abe, M.; Tahara, M.; Sakai, K.; Yamaguchi, H.; Kanou, K.; Shirato, K.; Kawase, M.; Noda, M.; Kimura, H.; Matsuyama, S.; et al. TMPRSS2 Is an Activating Protease for Respiratory Parainfluenza Viruses. J. Virol. 2013, 87, 11930–11935. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Itakura, Y.; Kishimoto, M.; Tabata, K.; Uemura, K.; Ito, N.; Sugiyama, M.; Wastika, C.E.; Orba, Y.; Sawa, H. Host serine proteases TMPRSS2 and TMPRSS11D mediate proteolytic activation and trypsin-independent infection in group A rotaviruses. J. Virol. 2021, 95, e00398-21. [Google Scholar] [CrossRef]
- Rahbar Saadat, Y.; Hosseiniyan Khatibi, S.M.; Zununi Vahed, S.; Ardalan, M. Host Serine Proteases: A Potential Targeted Therapy for COVID-19 and Influenza. Front. Mol. Biosci. 2021, 8, 725528. [Google Scholar] [CrossRef]
- Kido, H.; Okumura, Y.; Takahashi, E.; Pan, H.-Y.; Wang, S.; Yao, D.; Yao, M.; Chida, J.; Yano, M. Role of host cellular proteases in the pathogenesis of influenza and influenza-induced multiple organ failure. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2012, 1824, 186–194. [Google Scholar] [CrossRef]
- Bestle, D.; Limburg, H.; Kruhl, D.; Harbig, A.; Stein, D.A.; Moulton, H.; Matrosovich, M.; Abdelwhab, E.M.; Stech, J.; Böttcher-Friebertshäuser, E. Hemagglutinins of Avian Influenza Viruses Are Proteolytically Activated by TMPRSS2 in Human and Murine Airway Cells. J. Virol. 2021, 95, e0090621. [Google Scholar] [CrossRef]
- Bertram, S.; Glowacka, I.; Steffen, I.; Kühl, A.; Pöhlmann, S. Novel insights into proteolytic cleavage of influenza virus hemagglutinin. Rev. Med. Virol. 2010, 20, 298–310. [Google Scholar] [CrossRef]
- Sakai, K.; Ami, Y.; Nakajima, N.; Nakajima, K.; Kitazawa, M.; Anraku, M.; Takayama, I.; Sangsriratanakul, N.; Komura, M.; Sato, Y.; et al. TMPRSS2 Independency for Haemagglutinin Cleavage In Vivo Differentiates Influenza B Virus from Influenza A Virus. Sci. Rep. 2016, 6, 29430. [Google Scholar] [CrossRef] [PubMed]
- Virk, R.K.; Jayakumar, J.; Mendenhall, I.H.; Moorthy, M.; Lam, P.; Linster, M.; Lim, J.; Lin, C.; Oon, L.L.E.; Lee, H.K.; et al. Divergent evolutionary trajectories of influenza B viruses underlie their contemporaneous epidemic activity. Proc. Natl. Acad. Sci. USA 2020, 117, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Borchering, R.K.; Gunning, C.E.; Gokhale, D.V.; Weedop, K.B.; Saeidpour, A.; Brett, T.S.; Rohani, P. Anomalous influenza seasonality in the United States and the emergence of novel influenza B viruses. Proc. Natl. Acad. Sci. USA 2021, 118, e2012327118. [Google Scholar] [CrossRef] [PubMed]
- Kato-Miyashita, S.; Sakai-Tagawa, Y.; Yamashita, M.; Iwatsuki-Horimoto, K.; Ito, M.; Tokita, A.; Hagiwara, H.; Izumida, N.; Nishino, T.; Wada, N.; et al. Antigenic variants of influenza B viruses isolated in Japan during the 2017–2018 and 2018–2019 influenza seasons. Influenza Other Respir. Viruses 2020, 14, 311–319. [Google Scholar] [CrossRef]
- Tenforde, M.W.; Kondor, R.J.G.; Chung, J.R.; Zimmerman, R.K.; Nowalk, M.P.; Jackson, M.L.; Jackson, L.A.; Monto, A.S.; Martin, E.T.; Belongia, E.A.; et al. Effect of Antigenic Drift on Influenza Vaccine Effectiveness in the United States-2019–2020. Clin. Infect. Dis. 2021, 73, e4244–e4250. [Google Scholar] [CrossRef]
- Te Velthuis, A.J.; Fodor, E. Influenza virus RNA polymerase: Insights into the mechanisms of viral RNA synthesis. Nat. Rev. Microbiol. 2016, 14, 479–493. [Google Scholar] [CrossRef]
- Walker, A.P.; Fodor, E. Interplay between Influenza Virus and the Host RNA Polymerase II Transcriptional Machinery. Trends Microbiol. 2019, 27, 398–407. [Google Scholar] [CrossRef]
- Bouloy, M.; Plotch, S.J.; Krug, R.M. Globin mRNAs are primers for the transcription of influenza viral RNA in vitro. Proc. Natl. Acad. Sci. USA 1978, 75, 4886–4890. [Google Scholar] [CrossRef]
- Reich, S.; Guilligay, D.; Cusack, S. An in vitro fluorescence based study of initiation of RNA synthesis by influenza B polymerase. Nucleic Acids Res. 2017, 45, 3353–3368. [Google Scholar] [CrossRef]
- Labaronne, A.; Milles, S.; Donchet, A.; Jensen, M.R.; Blackledge, M.; Bourhis, J.M.; Ruigrok, R.W.H.; Crépin, T. Structural analysis of the complex between influenza B nucleoprotein and human importin-α. Sci. Rep. 2017, 7, 17164. [Google Scholar] [CrossRef]
- Donchet, A.; Vassal-Stermann, E.; Gérard, F.C.A.; Ruigrok, R.W.H.; Crépin, T. Differential Behaviours and Preferential Bindings of Influenza Nucleoproteins on Importins-α. Viruses 2020, 12, 834. [Google Scholar] [CrossRef] [PubMed]
- Resa-Infante, P.; Gabriel, G. The nuclear import machinery is a determinant of influenza virus host adaptation. Bioessays 2013, 35, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhang, H.; Xu, L.; Guo, X.; Wang, W.; Ji, Y.; Lin, C.; Wang, Y.; Wang, X. Selective usage of ANP32 proteins by influenza B virus polymerase: Implications in determination of host range. PLoS Pathog. 2020, 16, e1008989. [Google Scholar] [CrossRef] [PubMed]
- Carrique, L.; Fan, H.; Walker, A.P.; Keown, J.R.; Sharps, J.; Staller, E.; Barclay, W.S.; Fodor, E.; Grimes, J.M. Host ANP32A mediates the assembly of the influenza virus replicase. Nature 2020, 587, 638–643. [Google Scholar] [CrossRef] [PubMed]
- Peacock, T.P.; Swann, O.C.; Salvesen, H.A.; Staller, E.; Leung, P.B.; Goldhill, D.H.; Zhou, H.; Lillico, S.G.; Whitelaw, C.B.A.; Long, J.S.; et al. Swine ANP32A Supports Avian Influenza Virus Polymerase. J. Virol. 2020, 94, e00132-20. [Google Scholar] [CrossRef]
- Yu, M.; Qu, Y.; Zhang, H.; Wang, X. Roles of ANP32 proteins in cell biology and viral replication. Anim. Dis. 2022, 2, 22. [Google Scholar] [CrossRef]
- Reilly, P.T.; Yu, Y.; Hamiche, A.; Wang, L. Cracking the ANP32 whips: Important functions, unequal requirement, and hints at disease implications. BioEssays 2014, 36, 1062–1071. [Google Scholar] [CrossRef]
- Conenello, G.M.; Zamarin, D.; Perrone, L.A.; Tumpey, T.; Palese, P. A single mutation in the PB1-F2 of H5N1 (HK/97) and 1918 influenza A viruses contributes to increased virulence. PLoS Pathog. 2007, 3, 1414–1421. [Google Scholar] [CrossRef]
- Jagger, B.W.; Wise, H.M.; Kash, J.C.; Walters, K.A.; Wills, N.M.; Xiao, Y.L.; Dunfee, R.L.; Schwartzman, L.M.; Ozinsky, A.; Bell, G.L.; et al. An overlapping protein-coding region in influenza A virus segment 3 modulates the host response. Science 2012, 337, 199–204. [Google Scholar] [CrossRef]
- Hayashi, T.; MacDonald, L.A.; Takimoto, T. Influenza A Virus Protein PA-X Contributes to Viral Growth and Suppression of the Host Antiviral and Immune Responses. J. Virol. 2015, 89, 6442–6452. [Google Scholar] [CrossRef]
- Klemm, C.; Boergeling, Y.; Ludwig, S.; Ehrhardt, C. Immunomodulatory Nonstructural Proteins of Influenza A Viruses. Trends Microbiol. 2018, 26, 624–636. [Google Scholar] [CrossRef] [PubMed]
- Nogales, A.; Martinez-Sobrido, L.; Topham, D.J.; DeDiego, M.L. Modulation of Innate Immune Responses by the Influenza A NS1 and PA-X Proteins. Viruses 2018, 10, 708. [Google Scholar] [CrossRef] [PubMed]
- James, J.; Howard, W.; Iqbal, M.; Nair, V.K.; Barclay, W.S.; Shelton, H. Influenza A virus PB1-F2 protein prolongs viral shedding in chickens lengthening the transmission window. J. Gen. Virol. 2016, 97, 2516–2527. [Google Scholar] [CrossRef] [PubMed]
- Lutz Iv, M.M.; Dunagan, M.M.; Kurebayashi, Y.; Takimoto, T. Key Role of the Influenza A Virus PA Gene Segment in the Emergence of Pandemic Viruses. Viruses 2020, 12, 365. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Liu, S.; Goraya, M.U.; Maarouf, M.; Huang, S.; Chen, J.L. Host Immune Response to Influenza A Virus Infection. Front. Immunol. 2018, 9, 320. [Google Scholar] [CrossRef]
- Iwasaki, A.; Pillai, P.S. Innate immunity to influenza virus infection. Nat. Rev. Immunol. 2014, 14, 315–328. [Google Scholar] [CrossRef]
- Jiang, J.; Li, J.; Fan, W.; Zheng, W.; Yu, M.; Chen, C.; Sun, L.; Bi, Y.; Ding, C.; Gao, G.F.; et al. Robust Lys63-Linked Ubiquitination of RIG-I Promotes Cytokine Eruption in Early Influenza B Virus Infection. J. Virol. 2016, 90, 6263–6275. [Google Scholar] [CrossRef]
- Jiao, P.; Fan, W.; Cao, Y.; Zhang, H.; Tian, L.; Sun, L.; Luo, T.; Liu, W.; Li, J. Robust induction of interferon and interferon-stimulated gene expression by influenza B/Yamagata lineage virus infection of A549 cells. PLoS ONE 2020, 15, e0231039. [Google Scholar] [CrossRef]
- Dissanayake, T.K.; Schäuble, S.; Mirhakkak, M.H.; Wu, W.L.; Ng, A.C.; Yip, C.C.Y.; López, A.G.; Wolf, T.; Yeung, M.L.; Chan, K.H.; et al. Comparative Transcriptomic Analysis of Rhinovirus and Influenza Virus Infection. Front. Microbiol. 2020, 11, 1580. [Google Scholar] [CrossRef]
- Österlund, P.; Strengell, M.; Sarin, L.P.; Poranen, M.M.; Fagerlund, R.; Melén, K.; Julkunen, I. Incoming Influenza A Virus Evades Early Host Recognition, while Influenza B Virus Induces Interferon Expression Directly upon Entry. J. Virol. 2012, 86, 11183–11193. [Google Scholar] [CrossRef]
- Mäkelä, S.M.; Österlund, P.; Westenius, V.; Latvala, S.; Diamond, M.S.; Gale, M., Jr.; Julkunen, I. RIG-I Signaling Is Essential for Influenza B Virus-Induced Rapid Interferon Gene Expression. J. Virol. 2015, 89, 12014–12025. [Google Scholar] [CrossRef]
- Nogales, A.; Aydillo, T.; Ávila-Pérez, G.; Escalera, A.; Chiem, K.; Cadagan, R.; DeDiego, M.L.; Li, F.; García-Sastre, A.; Martínez-Sobrido, L. Functional Characterization and Direct Comparison of Influenza A, B, C, and D NS1 Proteins in vitro and in vivo. Front. Microbiol. 2019, 10, 2862. [Google Scholar] [CrossRef]
- Hensen, L.; Kedzierska, K.; Koutsakos, M. Innate and adaptive immunity toward influenza B viruses. Future Microbiol. 2020, 15, 1045–1058. [Google Scholar] [CrossRef] [PubMed]
- Donelan, N.R.; Dauber, B.; Wang, X.; Basler, C.F.; Wolff, T.; García-Sastre, A. The N- and C-terminal domains of the NS1 protein of influenza B virus can independently inhibit IRF-3 and beta interferon promoter activation. J. Virol. 2004, 78, 11574–11582. [Google Scholar] [CrossRef] [PubMed]
- Yuan, W.; Krug, R.M. Influenza B virus NS1 protein inhibits conjugation of the interferon (IFN)-induced ubiquitin-like ISG15 protein. Embo J. 2001, 20, 362–371. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Denison, C.; Huibregtse, J.M.; Gygi, S.; Krug, R.M. Human ISG15 conjugation targets both IFN-induced and constitutively expressed proteins functioning in diverse cellular pathways. Proc. Natl. Acad. Sci. USA 2005, 102, 10200–10205. [Google Scholar] [CrossRef]
- Versteeg, G.A.; Hale, B.G.; van Boheemen, S.; Wolff, T.; Lenschow, D.J.; García-Sastre, A. Species-specific antagonism of host ISGylation by the influenza B virus NS1 protein. J. Virol. 2010, 84, 5423–5430. [Google Scholar] [CrossRef]
- Sridharan, H.; Zhao, C.; Krug, R.M. Species specificity of the NS1 protein of influenza B virus: NS1 binds only human and non-human primate ubiquitin-like ISG15 proteins. J. Biol. Chem. 2010, 285, 7852–7856. [Google Scholar] [CrossRef]
- Guan, R.; Ma, L.-C.; Leonard, P.G.; Amer, B.R.; Sridharan, H.; Zhao, C.; Krug, R.M.; Montelione, G.T. Structural basis for the sequence-specific recognition of human ISG15 by the NS1 protein of influenza B virus. Proc. Natl. Acad. Sci. USA 2011, 108, 13468–13473. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Sridharan, H.; Chen, R.; Baker, D.P.; Wang, S.; Krug, R.M. Influenza B virus non-structural protein 1 counteracts ISG15 antiviral activity by sequestering ISGylated viral proteins. Nat. Commun. 2016, 7, 12754. [Google Scholar] [CrossRef]
- Koutsakos, M.; McWilliam, H.E.G.; Aktepe, T.E.; Fritzlar, S.; Illing, P.T.; Mifsud, N.A.; Purcell, A.W.; Rockman, S.; Reading, P.C.; Vivian, J.P.; et al. Downregulation of MHC Class I Expression by Influenza A and B Viruses. Front. Immunol. 2019, 10, 1158. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Xiao, W.; Wu, Y.; Fan, W.; Li, L.; Yue, C.; Zhang, Q.; Zhang, D.; Yuan, X.; Yao, S.; et al. Parallel T Cell Immunogenic Regions in Influenza B and A Viruses with Distinct Nuclear Export Signal Functions: The Balance between Viral Life Cycle and Immune Escape. J. Immunol. 2023, 210, 1074–1085. [Google Scholar] [CrossRef] [PubMed]
- Griffin, B.D.; Verweij, M.C.; Wiertz, E.J. Herpesviruses and immunity: The art of evasion. Vet. Microbiol. 2010, 143, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Quinn, L.L.; Williams, L.R.; White, C.; Forrest, C.; Zuo, J.; Rowe, M. The Missing Link in Epstein-Barr Virus Immune Evasion: The BDLF3 Gene Induces Ubiquitination and Downregulation of Major Histocompatibility Complex Class I (MHC-I) and MHC-II. J. Virol. 2016, 90, 356–367. [Google Scholar] [CrossRef]
- Vinjamuri, S.; Li, L.; Bouvier, M. SARS-CoV-2 ORF8: One protein, seemingly one structure, and many functions. Front. Immunol. 2022, 13, 1035559. [Google Scholar] [CrossRef] [PubMed]
- Dauber, B.; Schneider, J.; Wolff, T. Double-stranded RNA binding of influenza B virus nonstructural NS1 protein inhibits protein kinase R but is not essential to antagonize production of alpha/beta interferon. J. Virol. 2006, 80, 11667–11677. [Google Scholar] [CrossRef]
- Yin, C.; Khan, J.A.; Swapna, G.V.; Ertekin, A.; Krug, R.M.; Tong, L.; Montelione, G.T. Conserved surface features form the double-stranded RNA binding site of non-structural protein 1 (NS1) from influenza A and B viruses. J. Biol. Chem. 2007, 282, 20584–20592. [Google Scholar] [CrossRef]
- Schreiber, A.; Liedmann, S.; Brunotte, L.; Anhlan, D.; Ehrhardt, C.; Ludwig, S. Type I interferon antagonistic properties of influenza B virus polymerase proteins. Cell Microbiol. 2020, 22, e13143. [Google Scholar] [CrossRef]
- Gabriel, G.; Herwig, A.; Klenk, H.D. Interaction of polymerase subunit PB2 and NP with importin alpha1 is a determinant of host range of influenza A virus. PLoS Pathog. 2008, 4, e11. [Google Scholar] [CrossRef]
- Resa-Infante, P.; Jorba, N.; Zamarreño, N.; Fernández, Y.; Juárez, S.; Ortín, J. The host-dependent interaction of alpha-importins with influenza PB2 polymerase subunit is required for virus RNA replication. PLoS ONE 2008, 3, e3904. [Google Scholar] [CrossRef]
- Gabriel, G.; Klingel, K.; Otte, A.; Thiele, S.; Hudjetz, B.; Arman-Kalcek, G.; Sauter, M.; Shmidt, T.; Rother, F.; Baumgarte, S.; et al. Differential use of importin-α isoforms governs cell tropism and host adaptation of influenza virus. Nat. Commun. 2011, 2, 156. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Liu, M.; Bai, S.; Zhao, C.; Li, Z.; Xu, J.; Zhang, X. Influenza A Virus-Host Specificity: An Ongoing Cross-Talk Between Viral and Host Factors. Front. Microbiol. 2021, 12, 777885. [Google Scholar] [CrossRef] [PubMed]
- Gilbertson, B.; Duncan, M.; Subbarao, K. Role of the viral polymerase during adaptation of influenza A viruses to new hosts. Curr. Opin. Virol. 2023, 62, 101363. [Google Scholar] [CrossRef]
- Bisset, A.T.; Hoyne, G.F. Evolution and Adaptation of the Avian H7N9 Virus into the Human Host. Microorganisms 2020, 8, 778. [Google Scholar] [CrossRef]
- Ciminski, K.; Ran, W.; Gorka, M.; Lee, J.; Malmlov, A.; Schinköthe, J.; Eckley, M.; Murrieta, R.A.; Aboellail, T.A.; Campbell, C.L.; et al. Bat influenza viruses transmit among bats but are poorly adapted to non-bat species. Nat. Microbiol. 2019, 4, 2298–2309. [Google Scholar] [CrossRef] [PubMed]
- Matrosovich, M.; Zhou, N.; Kawaoka, Y.; Webster, R. The surface glycoproteins of H5 influenza viruses isolated from humans, chickens, and wild aquatic birds have distinguishable properties. J. Virol. 1999, 73, 1146–1155. [Google Scholar] [CrossRef]
- Webby, R.J.; Woolcock, P.R.; Krauss, S.L.; Webster, R.G. Reassortment and interspecies transmission of North American H6N2 influenza viruses. Virology 2002, 295, 44–53. [Google Scholar] [CrossRef]
- Li, J.; Zu Dohna, H.; Cardona, C.J.; Miller, J.; Carpenter, T.E. Emergence and genetic variation of neuraminidase stalk deletions in avian influenza viruses. PLoS ONE 2011, 6, e14722. [Google Scholar] [CrossRef]
- Sederdahl, B.K.; Williams, J.V. Epidemiology and Clinical Characteristics of Influenza C Virus. Viruses 2020, 12, 89. [Google Scholar] [CrossRef]
- Hause, B.M.; Ducatez, M.; Collin, E.A.; Ran, Z.; Liu, R.; Sheng, Z.; Armien, A.; Kaplan, B.; Chakravarty, S.; Hoppe, A.D.; et al. Isolation of a novel swine influenza virus from Oklahoma in 2011 which is distantly related to human influenza C viruses. PLoS Pathog. 2013, 9, e1003176. [Google Scholar] [CrossRef]
- Herrler, G.; Dürkop, I.; Becht, H.; Klenk, H.D. The glycoprotein of influenza C virus is the haemagglutinin, esterase and fusion factor. J. Gen. Virol. 1988, 69 Pt 4, 839–846. [Google Scholar] [CrossRef] [PubMed]
- Rogers, G.N.; Herrler, G.; Paulson, J.C.; Klenk, H.D. Influenza C virus uses 9-O-acetyl-N-acetylneuraminic acid as a high affinity receptor determinant for attachment to cells. J. Biol. Chem. 1986, 261, 5947–5951. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Veit, M. Hemagglutinin-esterase-fusion (HEF) protein of influenza C virus. Protein Cell 2016, 7, 28–45. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Sreenivasan, C.; Yu, H.; Sheng, Z.; Newkirk, S.J.; An, W.; Smith, D.F.; Chen, X.; Wang, D.; Li, F. Influenza D virus diverges from its related influenza C virus in the recognition of 9-O-acetylated N-acetyl- or N-glycolyl-neuraminic acid-containing glycan receptors. Virology 2020, 545, 16–23. [Google Scholar] [CrossRef]
- Sreenivasan, C.C.; Sheng, Z.; Wang, D.; Li, F. Host Range, Biology, and Species Specificity of Seven-Segmented Influenza Viruses-A Comparative Review on Influenza C and D. Pathogens 2021, 10, 1583. [Google Scholar] [CrossRef]
- Sato, K.; Hayashi, H.; Shimotai, Y.; Yamaya, M.; Hongo, S.; Kawakami, K.; Matsuzaki, Y.; Nishimura, H. TMPRSS2 Activates Hemagglutinin-Esterase Glycoprotein of Influenza C Virus. J. Virol. 2021, 95, e0129621. [Google Scholar] [CrossRef]
- Pachler, K.; Vlasak, R. Influenza C virus NS1 protein counteracts RIG-I-mediated IFN signalling. Virol. J. 2011, 8, 48. [Google Scholar] [CrossRef]
- Chastagner, A.; Enouf, V.; Peroz, D.; Hervé, S.; Lucas, P.; Quéguiner, S.; Gorin, S.; Beven, V.; Behillil, S.; Leneveu, P.; et al. Bidirectional Human-Swine Transmission of Seasonal Influenza A(H1N1)pdm09 Virus in Pig Herd, France, 2018. Emerg. Infect. Dis. 2019, 25, 1940–1943. [Google Scholar] [CrossRef]
- Markin, A.; Ciacci Zanella, G.; Arendsee, Z.W.; Zhang, J.; Krueger, K.M.; Gauger, P.C.; Vincent Baker, A.L.; Anderson, T.K. Reverse-zoonoses of 2009 H1N1 pandemic influenza A viruses and evolution in United States swine results in viruses with zoonotic potential. PLoS Pathog. 2023, 19, e1011476. [Google Scholar] [CrossRef]
- Zeller, M.A.; Ma, J.; Wong, F.Y.; Tum, S.; Hidano, A.; Holt, H.; Chhay, T.; Sorn, S.; Koeut, D.; Seng, B.; et al. The genomic landscape of swine influenza A viruses in Southeast Asia. Proc. Natl. Acad. Sci. USA 2023, 120, e2301926120. [Google Scholar] [CrossRef]
- Rajao, D.S.; Vincent, A.L.; Perez, D.R. Adaptation of Human Influenza Viruses to Swine. Front. Vet. Sci. 2018, 5, 347. [Google Scholar] [CrossRef] [PubMed]
- Munir, K.; Ashraf, S.; Munir, I.; Khalid, H.; Muneer, M.A.; Mukhtar, N.; Amin, S.; Ashraf, S.; Imran, M.A.; Chaudhry, U.; et al. Zoonotic and reverse zoonotic events of SARS-CoV-2 and their impact on global health. Emerg. Microbes Infect. 2020, 9, 2222–2235. [Google Scholar] [CrossRef] [PubMed]
- Goraichuk, I.V.; Arefiev, V.; Stegniy, B.T.; Gerilovych, A.P. Zoonotic and Reverse Zoonotic Transmissibility of SARS-CoV-2. Virus Res. 2021, 302, 198473. [Google Scholar] [CrossRef]
- Olsen, S.J.; Azziz-Baumgartner, E.; Budd, A.P.; Brammer, L.; Sullivan, S.; Pineda, R.F.; Cohen, C.; Fry, A.M. Decreased Influenza Activity during the COVID-19 Pandemic—United States, Australia, Chile, and South Africa, 2020. MMWR Morb. Mortal. Wkly. Rep. 2020, 69, 1305–1309. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Shaman, J.; Pei, S. Quantifying the Impact of COVID-19 Nonpharmaceutical Interventions on Influenza Transmission in the United States. J. Infect. Dis. 2021, 224, 1500–1508. [Google Scholar] [CrossRef]
- Takeuchi, H.; Kawashima, R. Disappearance and Re-Emergence of Influenza during the COVID-19 Pandemic: Association with Infection Control Measures. Viruses 2023, 15, 223. [Google Scholar] [CrossRef] [PubMed]
- Koutsakos, M.; Wheatley, A.K.; Laurie, K.; Kent, S.J.; Rockman, S. Influenza lineage extinction during the COVID-19 pandemic? Nat. Rev. Microbiol. 2021, 19, 741–742. [Google Scholar] [CrossRef]
- Paget, J.; Caini, S.; Del Riccio, M.; van Waarden, W.; Meijer, A. Has influenza B/Yamagata become extinct and what implications might this have for quadrivalent influenza vaccines? Eurosurveillance 2022, 27, 2200753. [Google Scholar] [CrossRef]
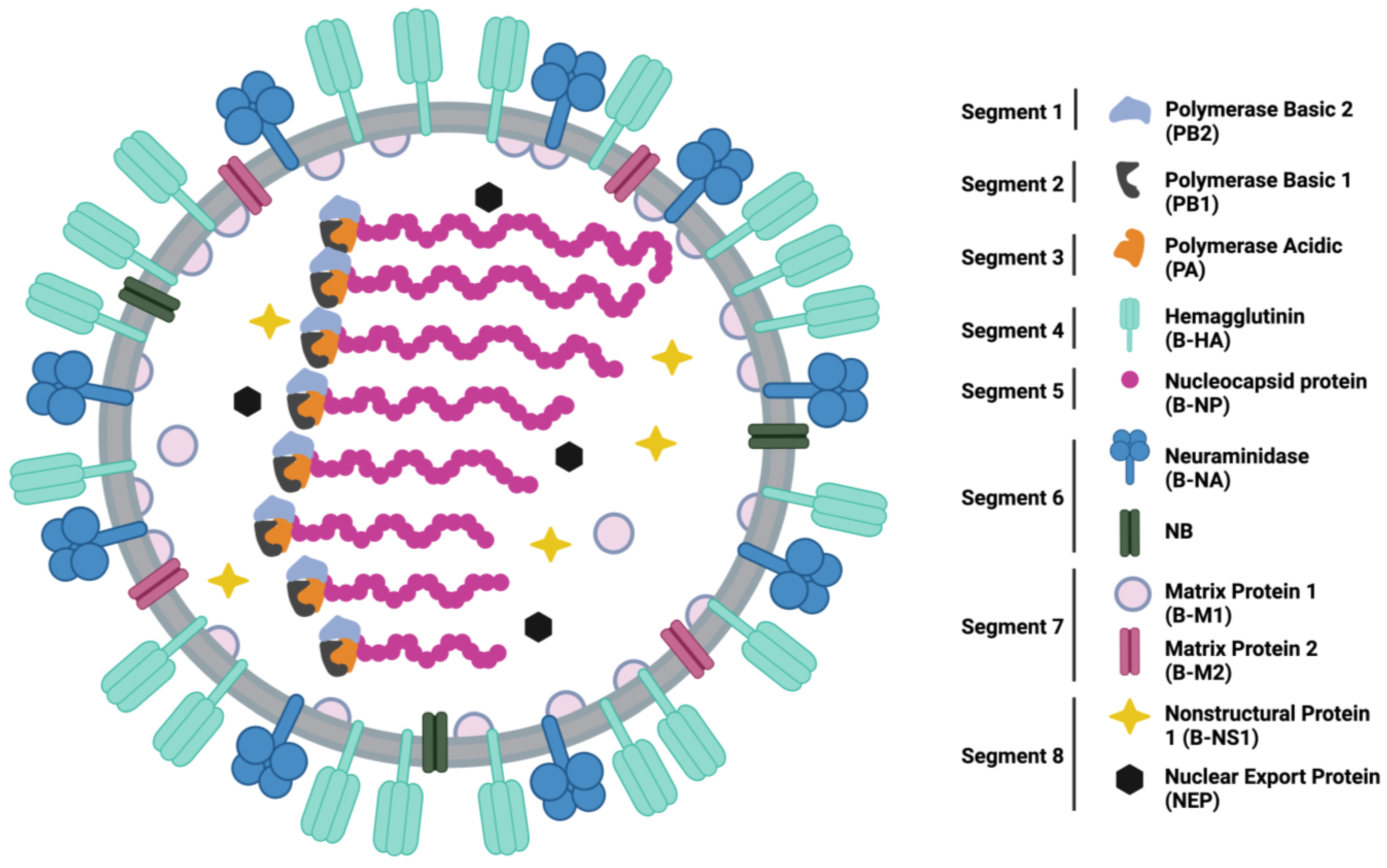

{kind=link}
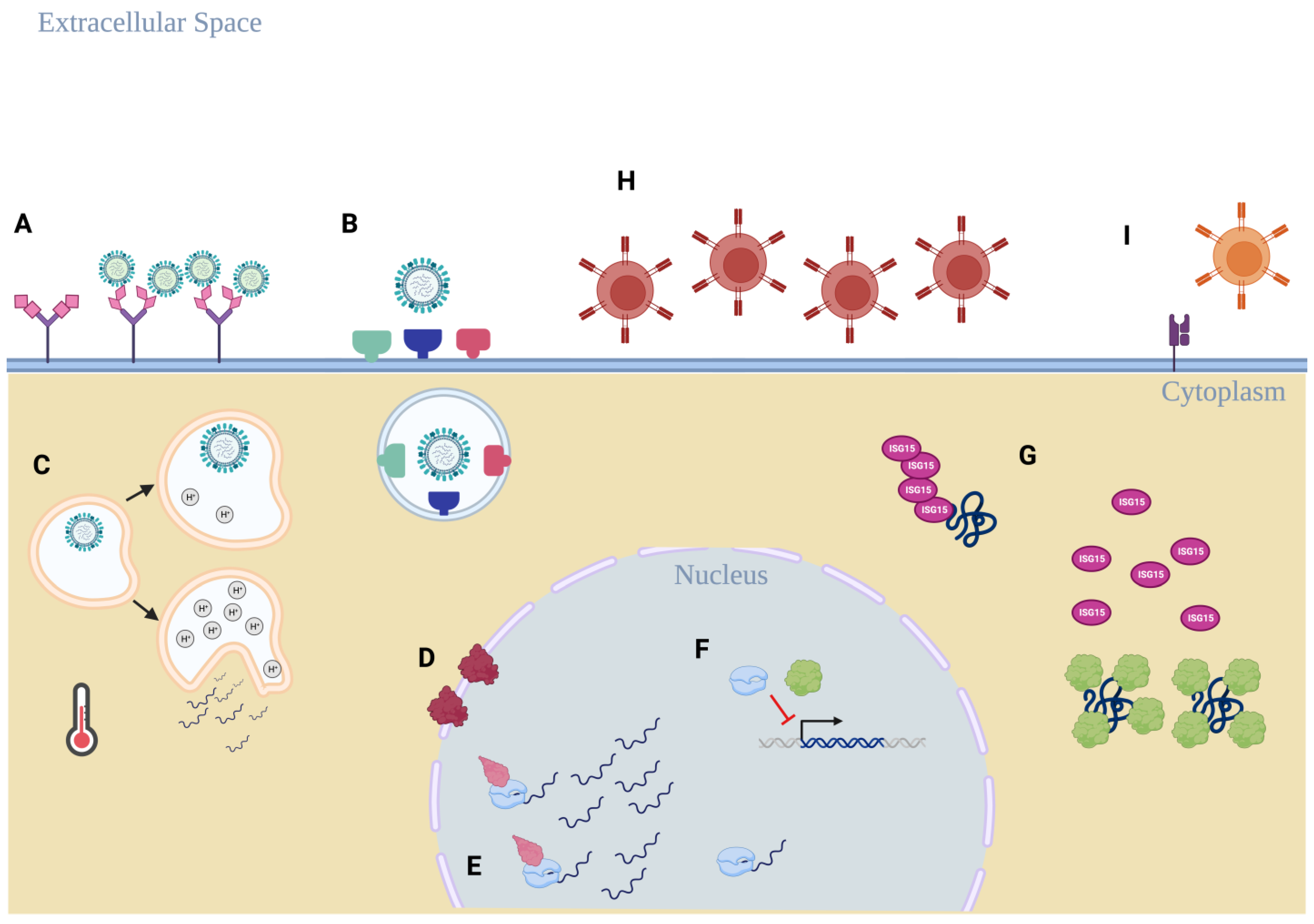
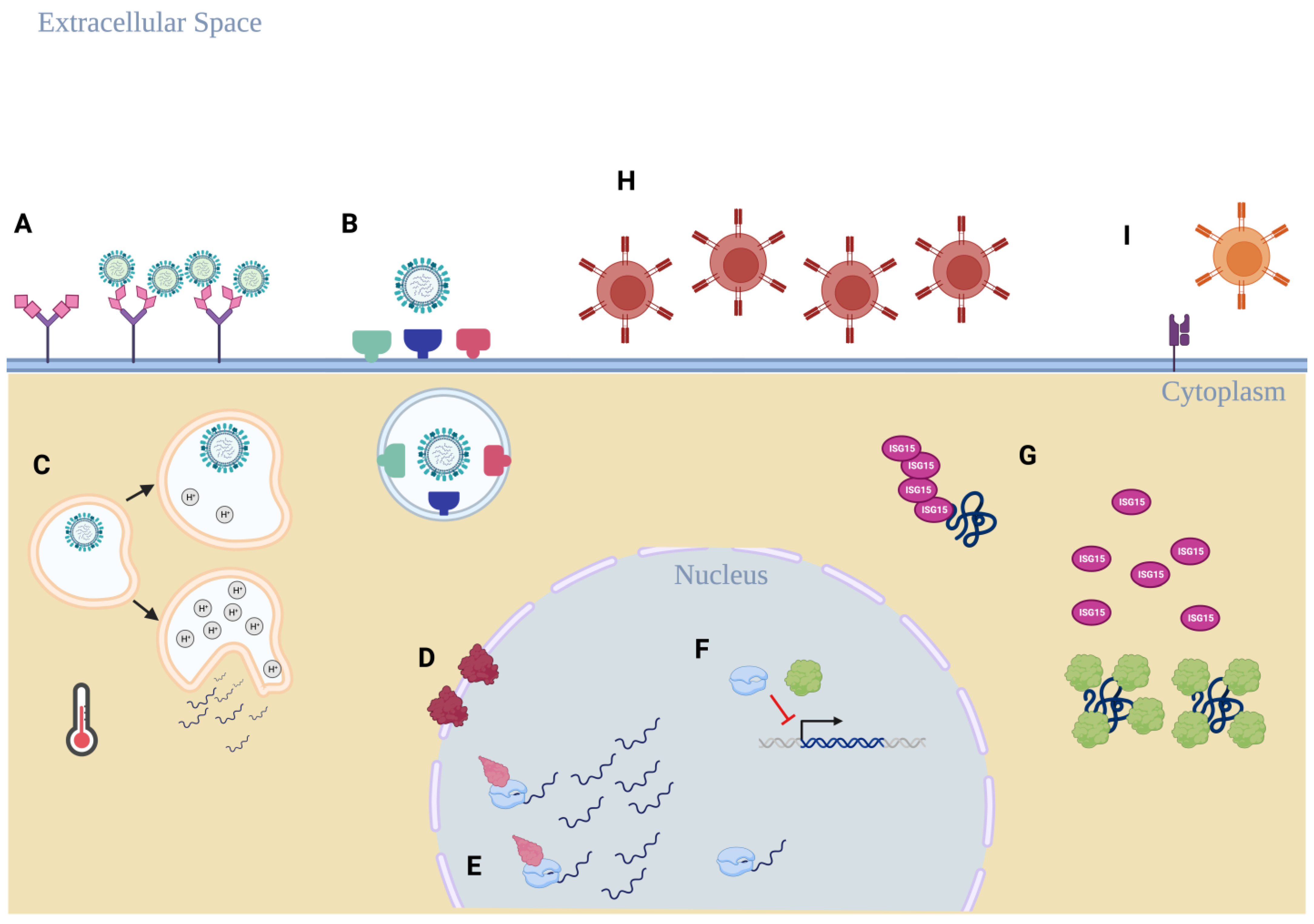{kind=link}
| B-HA Role in Virus Infection | B-HA Function | Optimal Characteristic Found in Human Respiratory Tract | References |
|---|---|---|---|
| Receptor Binding | B-HA binds α2,6 linked sialic acid residues more efficiently than α2,3 linked sialic acid residues | Abundance and distribution of α2,6 linked sialic acid residues in the upper respiratory tract | [19,20,21,26,27] |
| Activation | B-HA must be enzymatically activated from its precursor form into its active subunits | A broad range of enzymes capable of activating B-HA are found in the upper respiratory tract | [34,35,42] |
| Membrane Fusion | B-HA undergoes pH-induced conformational change to fuse viral envelope and host endosomal membrane | Human endosomal pH and temperature of upper respiratory tract | [22,34] |
| Host Protein Family | Role in IBV Replication | Questions to Be Answered about Possible Host Restriction | References |
|---|---|---|---|
| Respiratory proteases | Activation of B-HA | Do homologs of human proteases known to activate B-HA similarly activate the glycoprotein? | [34,35,42] |
| Importin-α | Transport of vRNPs from cytoplasm into nucleus | Does the IBV vRNP interact with importin-α from non-human species? Can non-human importin-α proteins support efficient IBV replication? | [51,52] |
| ANP32 | Support B-Pol complex activity | How well can non-human mammalian ANP32 proteins support IBV replication? | [54,56,57] |
| Innate immune sensors | Sensing of intracellular viral infection | Does B-NS1 interact with non-human homologs of innate immune sensing proteins? Can B-NS1 interaction lead to evasion of immune response in non-human hosts? | [73,74,75,87,88,89] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pekarek, M.J.; Weaver, E.A. Existing Evidence for Influenza B Virus Adaptations to Drive Replication in Humans as the Primary Host. Viruses 2023, 15, 2032. https://doi.org/10.3390/v15102032
Pekarek MJ, Weaver EA. Existing Evidence for Influenza B Virus Adaptations to Drive Replication in Humans as the Primary Host. Viruses. 2023; 15(10):2032. https://doi.org/10.3390/v15102032
Chicago/Turabian StylePekarek, Matthew J., and Eric A. Weaver. 2023. "Existing Evidence for Influenza B Virus Adaptations to Drive Replication in Humans as the Primary Host" Viruses 15, no. 10: 2032. https://doi.org/10.3390/v15102032
APA StylePekarek, M. J., & Weaver, E. A. (2023). Existing Evidence for Influenza B Virus Adaptations to Drive Replication in Humans as the Primary Host. Viruses, 15(10), 2032. https://doi.org/10.3390/v15102032