
Sequence-Based Antigenic Analyses of H1 Swine Influenza A Viruses from Colombia (2008–2021) Reveals Temporal and Geographical Antigenic Variations
 , ,
, ,  ,
,
Abstract
1. Introduction
2. Materials and Methods
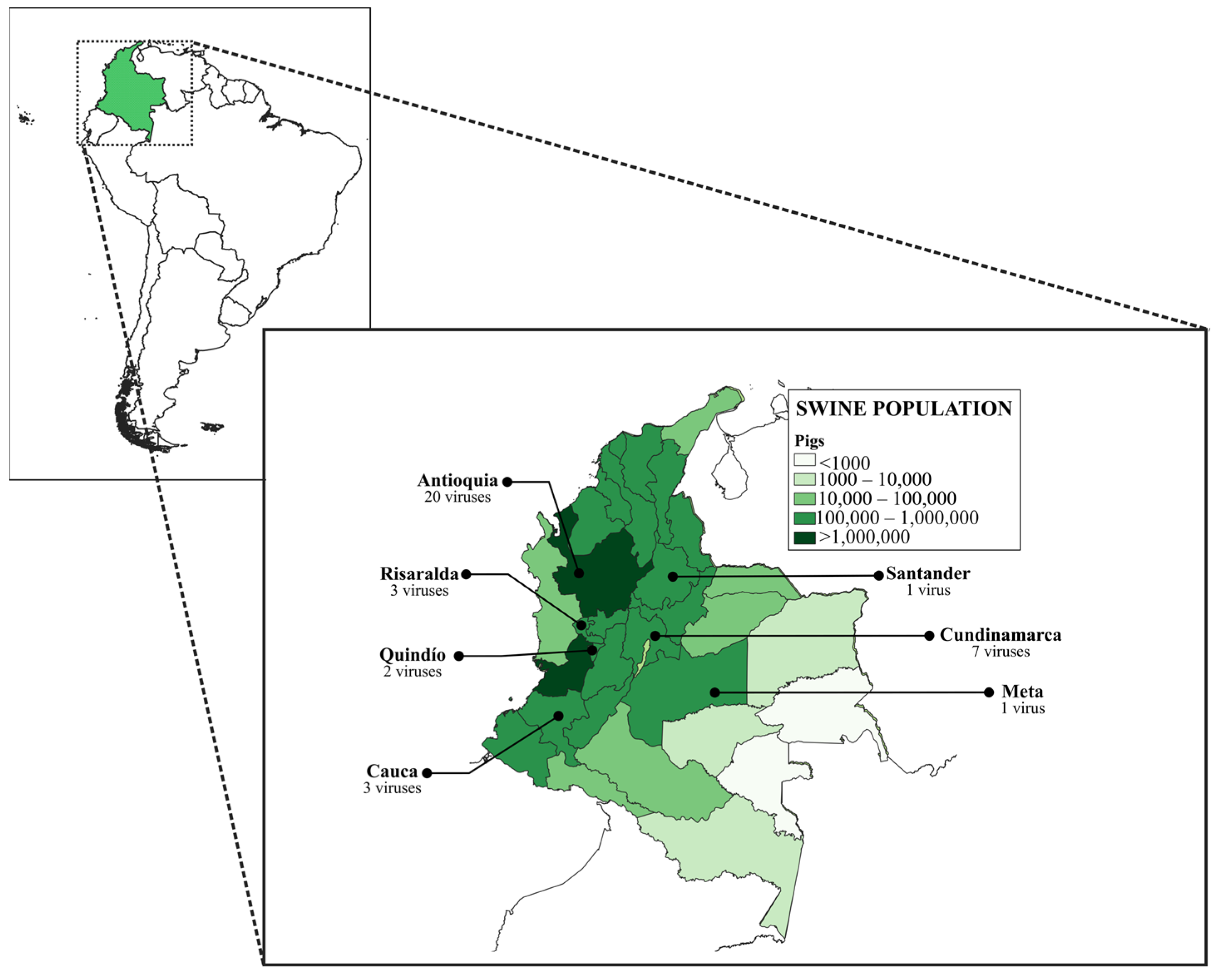
2.1. Viruses
2.2. Phylogenetic Characterization of the HA Glycoprotein
2.3. Antigenic Characterization
2.4. Epitope Analyses
2.5. N-Glycosylation Analyses
3. Results
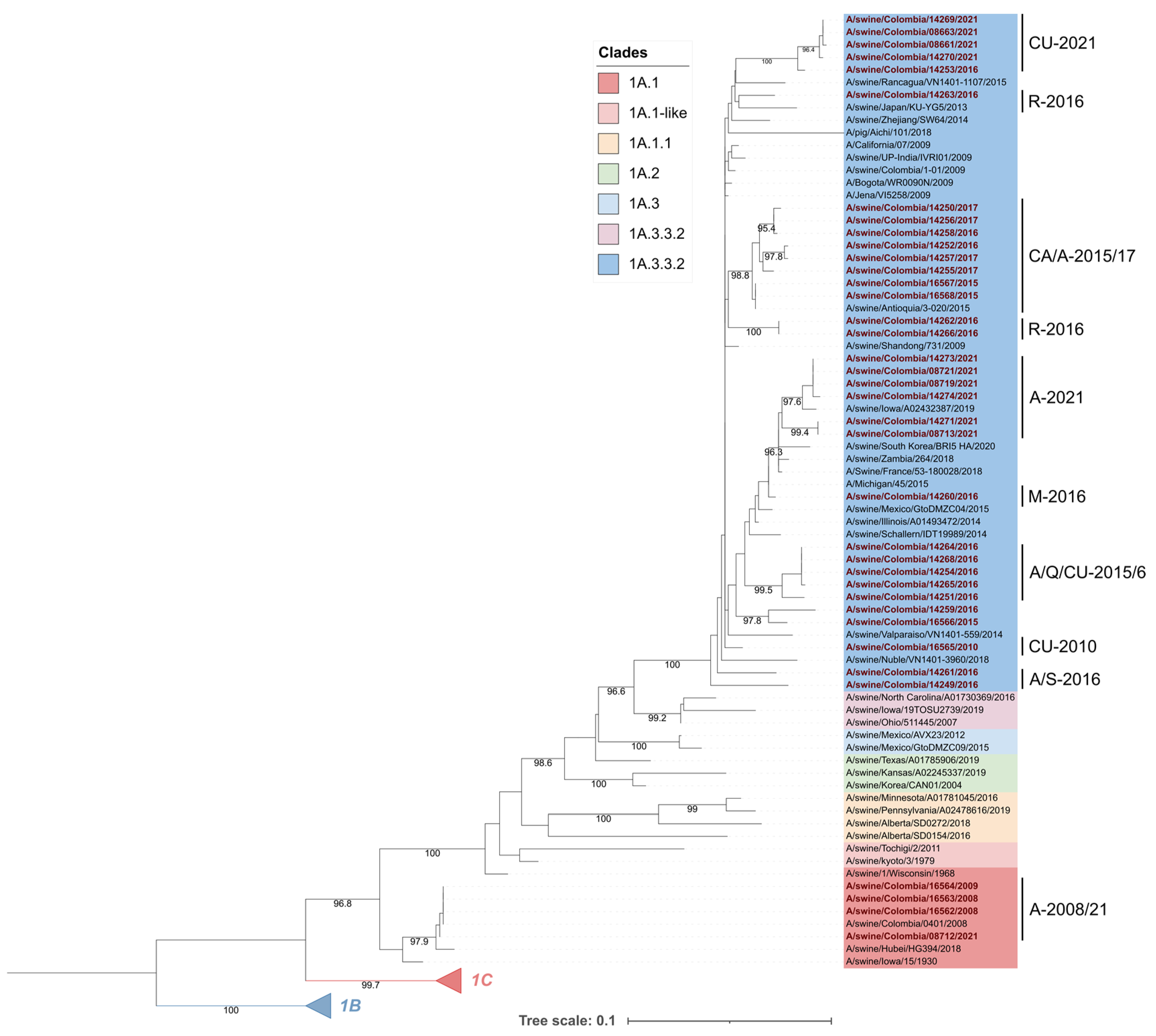
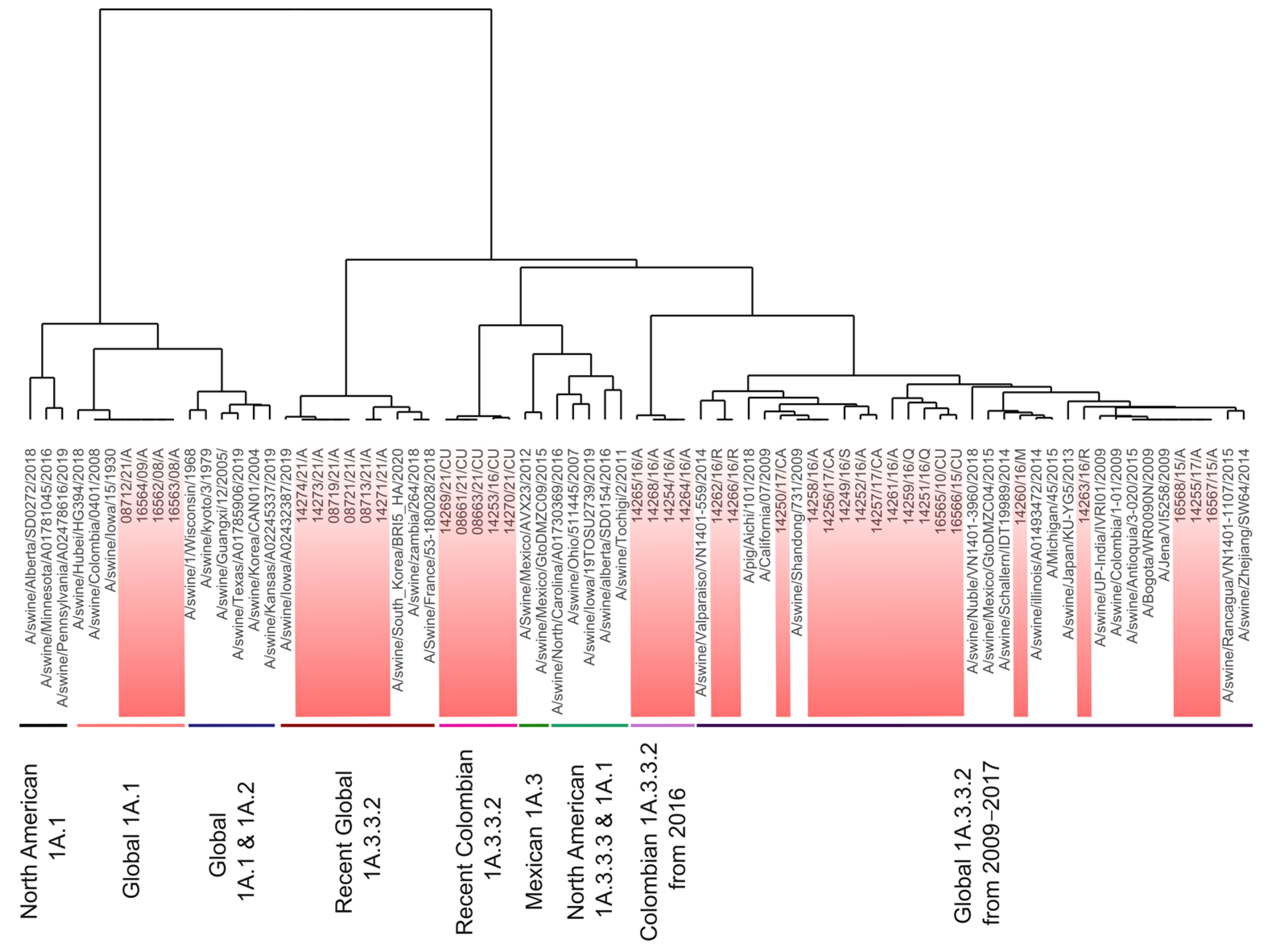
3.1. Colombian Swine H1 FLUAVs of the 1A.1 Clade Remain Genetically Stable, Whereas the 1A.3.3.2 Clade Shows Phylogeographic Divergence
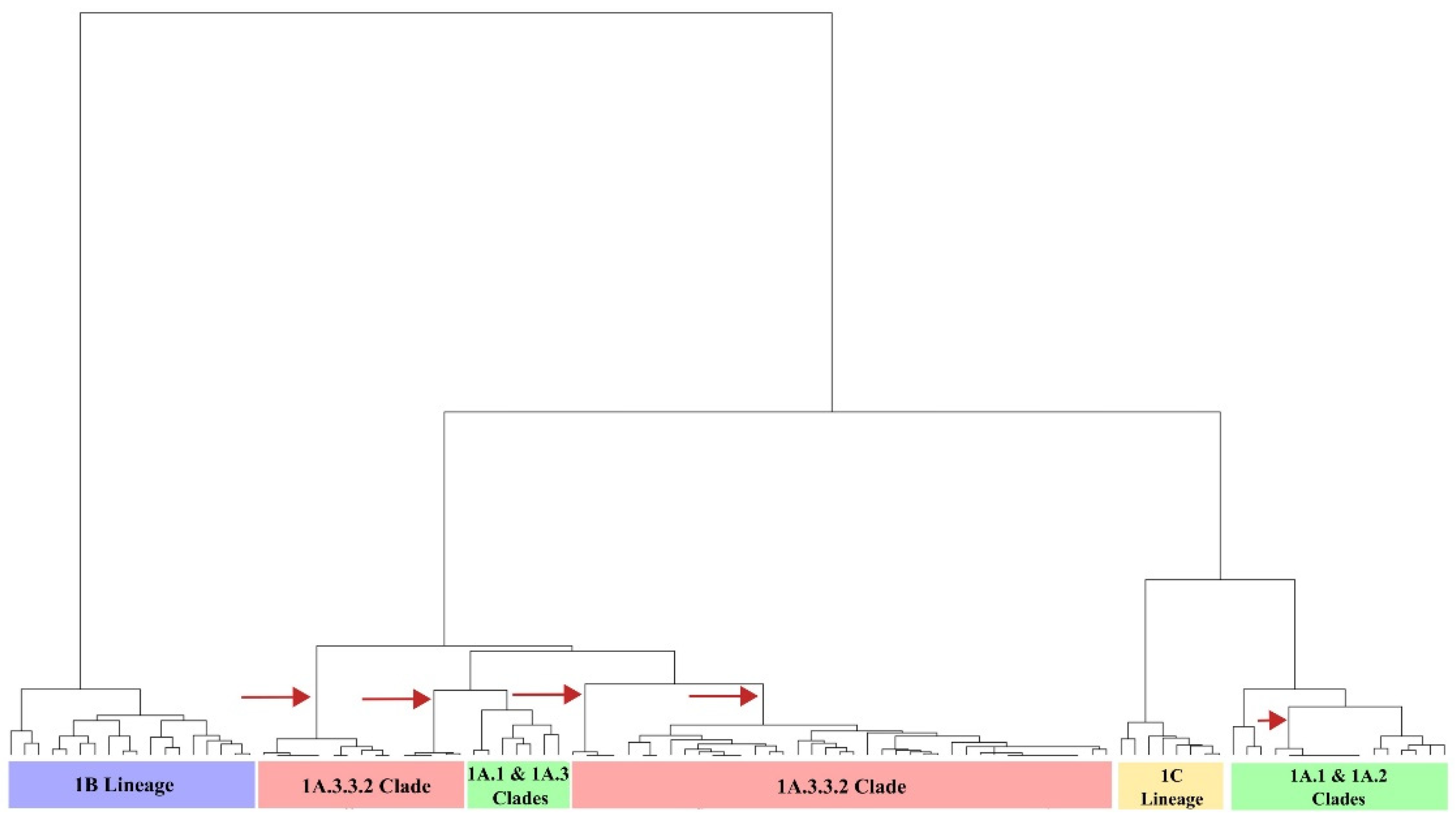
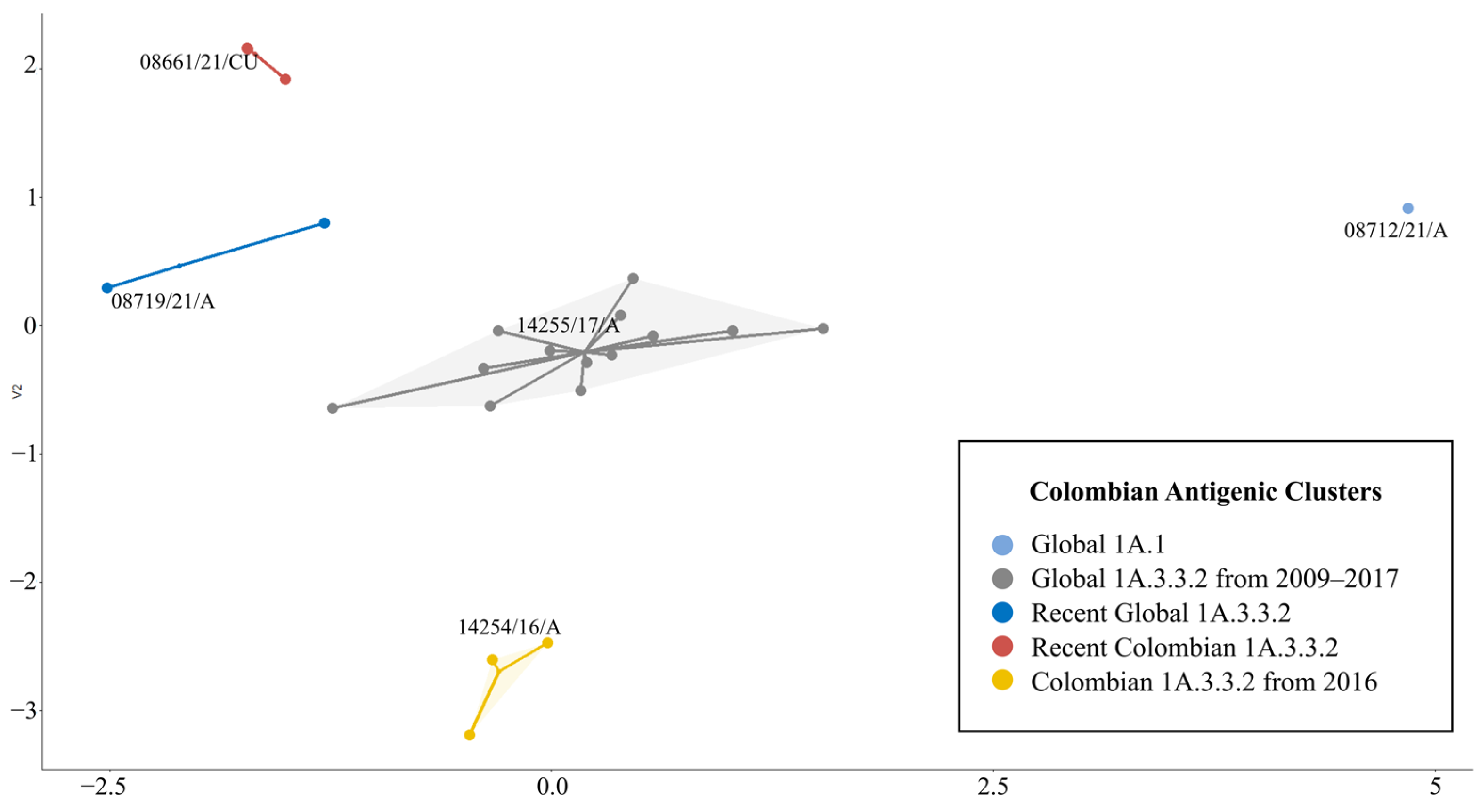
3.2. Sequence-Based Antigenic Cartography Shows the Relatedness with Phylogeny, Geographic Origin, and Temporal Factors
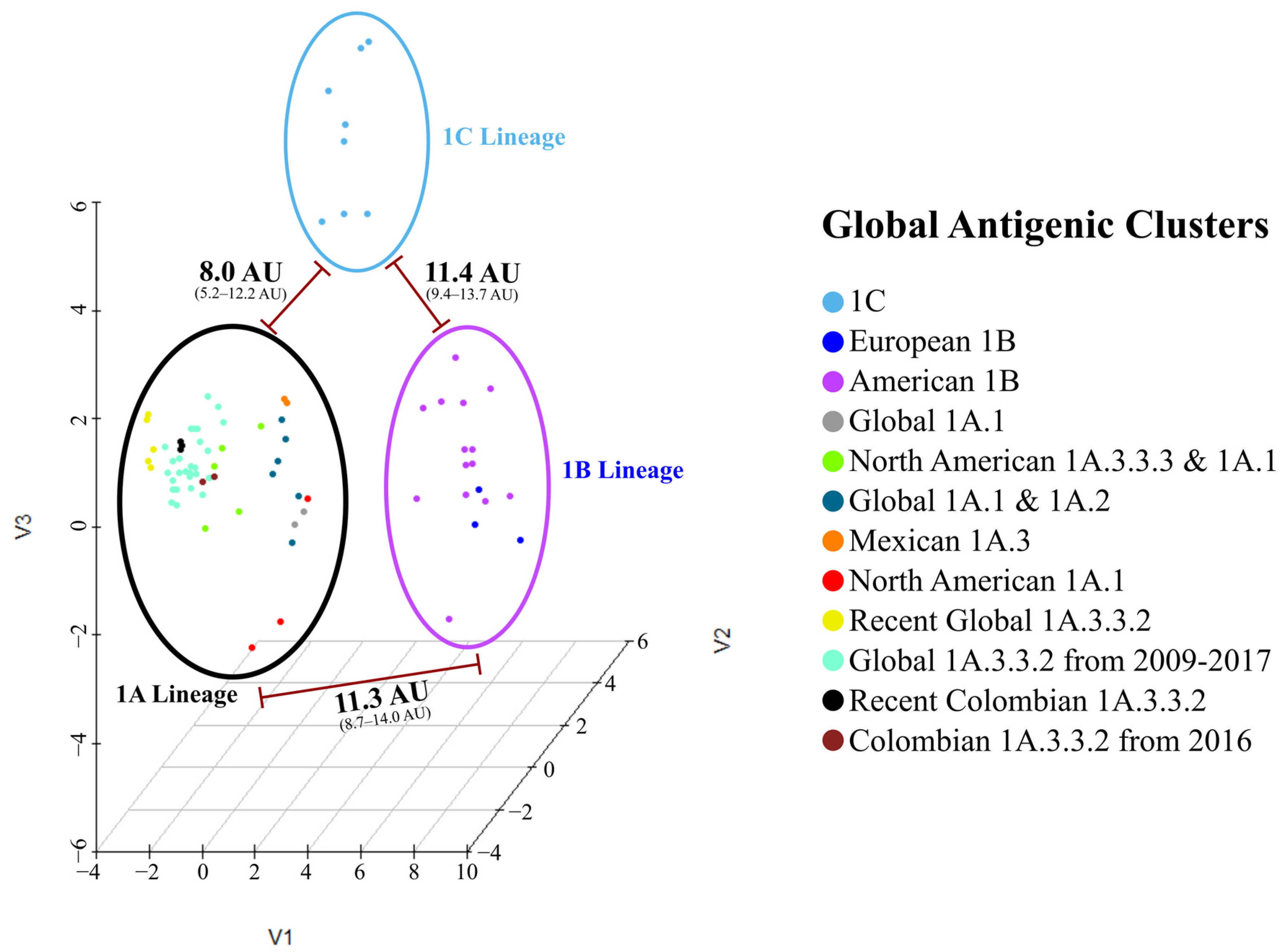
3.3. There Were at Least Five Antigenic Clusters Distributed in Different Regions of Colombia during Specific Years
3.4. Antigenic Characteristics of 1B and 1C Lineages Were Partially Influenced by Phylogeny and Geographic Factors
3.5. Predicted Antigenic Clusters in Colombia Carried Point Mutations whitin the Epitopes, with Ca2 Demonstrating Immunodominance
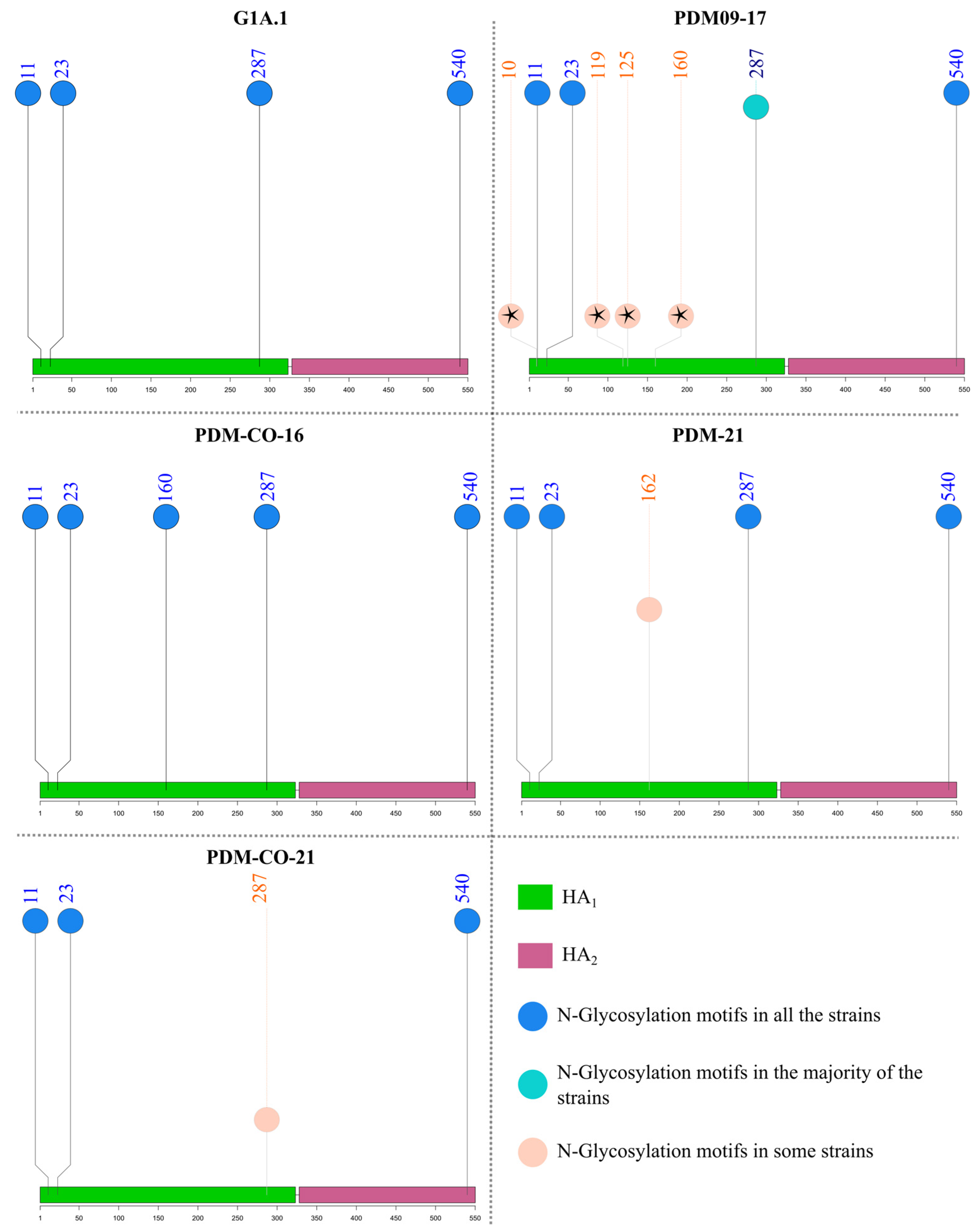
3.6. N-Glycosylation Motifs of Colombian Swine H1 FLUAVs Varied between Three and Five and Were Related to the Predicted Antigenic Cluster
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- ICTV ICTV 9th Report. 2011. Available online: https://talk.ictvonline.org/ictv-reports/ictv_9th_report/negative-sense-rna-viruses-2011/w/negrna_viruses/209/orthomyxoviridae (accessed on 29 March 2022).
- Tong, S.; Zhu, X.; Li, Y.; Shi, M.; Zhang, J. New World Bats Harbor Diverse Influenza A Viruses. PLoS Pathog. 2013, 9, 1003657. [Google Scholar] [CrossRef]
- Zhuang, Q.; Wang, S.; Liu, S.; Hou, G.; Li, J.; Jiang, W.; Wang, K.; Peng, C.; Liu, D.; Guo, A.; et al. Diversity and Distribution of Type A Influenza Viruses: An Updated Panorama Analysis Based on Protein Sequences. Virol. J. 2019, 16, 85. [Google Scholar] [CrossRef]
- Kandeil, A.; Gomaa, M.R.; Shehata, M.M.; El Taweel, A.N.; Mahmoud, S.H.; Bagato, O.; Moatasim, Y.; Kutkat, O.; Kayed, A.S.; Dawson, P.; et al. Isolation and Characterization of a Distinct Influenza A Virus from Egyptian Bats. J. Virol. 2019, 93, e01059-18. [Google Scholar] [CrossRef]
- Ha, Y.; Stevens, D.J.; Skehel, J.J.; Wiley, D.C. X-ray Structure of the Hemagglutinin of a Potential H3 Avian Progenitor of the 1968 Hong Kong Pandemic Influenza Virus☆. Virology 2003, 309, 209–218. [Google Scholar] [CrossRef]
- Cheng, C.; Holyoak, M.; Xu, L.; Li, J.; Liu, W.; Stenseth, N.C.; Zhang, Z. Host and Geographic Barriers Shape the Competition, Coexistence, and Extinction Patterns of Influenza A (H1N1) Viruses. Ecol. Evol. 2022, 12, 8732. [Google Scholar] [CrossRef]
- Lewis, N.S.; Russell, C.A.; Langat, P.; Anderson, T.K.; Berger, K.; Bielejec, F.; Burke, D.F.; Dudas, G.; Fonville, J.M.; Fouchier, R.A.M.; et al. The Global Antigenic Diversity of Swine Influenza A Viruses. Elife 2016, 5, e12217. [Google Scholar] [CrossRef]
- Anderson, T.K.; Macken, C.A.; Lewis, N.S.; Scheuermann, R.H.; Van Reeth, K.; Brown, I.H.; Swenson, S.L.; Simon, G.; Saito, T.; Berhane, Y.; et al. A Phylogeny-Based Global Nomenclature System and Automated Annotation Tool for H1 Hemagglutinin Genes from Swine Influenza A Viruses. mSphere 2016, 1, e00275-16. [Google Scholar] [CrossRef]
- Anderson, T.K.; Chang, J.; Arendsee, Z.W.; Venkatesh, D.; Souza, C.K.; Kimble, J.B.; Lewis, N.S.; Davis, C.T.; Vincent, A.L. Swine Influenza A Viruses and the Tangled Relationship with Humans. Cold Spring Harb. Perspect. Med. 2021, 11, a038737. [Google Scholar] [CrossRef]
- Zhang, H.; Li, H.; Wang, W.; Wang, Y.; Han, G.Z.; Chen, H.; Wang, X. A Unique Feature of Swine ANP32A Provides Susceptibility to Avian Influenza Virus Infection in Pigs. PLoS Pathog. 2020, 16, e1008330. [Google Scholar] [CrossRef]
- Nelli, R.K.; Kuchipudi, S.V.; White, G.A.; Baquero Perez, B.; Dunham, S.P.; Chang, K.-C. Comparative Distribution of Human and Avian Type Sialic Acid Influenza Receptors in the Pig. BMC Vet. Res. 2010, 6, 4. [Google Scholar] [CrossRef]
- Trebbien, R.; Larsen, L.E.; Viuff, B.M. Distribution of Sialic Acid Receptors and Influenza A Virus of Avian and Swine Origin in Experimentally Infected Pigs. Virol. J. 2011, 8, 434. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.J.D.; Vijaykrishna, D.; Bahl, J.; Lycett, S.J.; Worobey, M.; Pybus, O.G.; Ma, S.K.; Cheung, C.L.; Raghwani, J.; Bhatt, S.; et al. Origins and Evolutionary Genomics of the 2009 Swine-Origin H1N1 Influenza A Epidemic. Nature 2009, 459, 1122–1125. [Google Scholar] [CrossRef] [PubMed]
- Ryt-Hansen, P.; Pedersen, A.G.; Larsen, I.; Kristensen, C.S.; Krog, J.S.; Wacheck, S.; Larsen, L.E. Substantial Antigenic Drift in the Hemagglutinin Protein of Swine Influenza A Viruses. Viruses 2020, 12, 248. [Google Scholar] [CrossRef]
- Retamal, M.; Abed, Y.; Rh, C.; Baz, M.; Boivin, G. In Vitro and in Vivo Evidence of a Potential A(H1N1)Pdm09 Antigenic Drift Mediated by Escape Mutations in the Haemagglutinin Sa Antigenic Site. J. Gen. Virol. 2012, 98, 1224–1231. [Google Scholar] [CrossRef] [PubMed]
- López-Valiñas, Á.; Sisteré-Oró, M.; López-Serrano, S.; Baioni, L.; Darji, A.; Chiapponi, C.; Segalés, J.; Ganges, L.; Núñez, J.I. Identification and Characterization of Swine Influenza Virus H1n1 Variants Generated in Vaccinated and Nonvaccinated, Challenged Pigs. Viruses 2021, 13, 2087. [Google Scholar] [CrossRef]
- Caton, A.J.; Brownlee, G.G.; Yewdell, J.W.; Gerhard, W. The Antigenic Structure of the Influenza Virus A/PR/8/34 Hemagglutinin (H1 Subtype). Cell 1982, 31, 417–427. [Google Scholar] [CrossRef]
- Kilbourne, E.D.; Gerhard, W.; Whitaker, C.W. Monoclonal Antibodies to the Hemagglutinin Sa Antigenic Site of A/PR/8/34 Influenza Virus Distinguish Biologic Mutants of Swine Influenza Virus. Proc. Natl. Acad. Sci. USA 1983, 80, 6399–6402. [Google Scholar] [CrossRef]
- Wiley, D.C.; Wilson, I.A.; Skehel, J.J. Structural Identification of the Antibody-Binding Sites of Hong Kong Influenza Haemagglutinin and Their Involvement in Antigenic Variation. Nature 1981, 289, 373–378. [Google Scholar] [CrossRef]
- Furuse, Y.; Shimabukuro, K.; Odagiri, T.; Sawayama, R.; Okada, T.; Khandaker, I.; Suzuki, A.; Oshitani, H. Comparison of Selection Pressures on the HA Gene of Pandemic (2009) and Seasonal Human and Swine Influenza A H1 Subtype Viruses. Virology 2010, 405, 314–321. [Google Scholar] [CrossRef]
- Henritzi, D.; Petric, P.P.; Lewis, N.S.; Graaf, A.; Pessia, A.; Starick, E.; Breithaupt, A.; Strebelow, G.; Luttermann, C.; Parker, L.M.K.; et al. Surveillance of European Domestic Pig Populations Identifies an Emerging Reservoir of Potentially Zoonotic Swine Influenza A Viruses. Cell Host Microbe 2020, 28, 614–627.e6. [Google Scholar] [CrossRef]
- Venkatesh, D.; Anderson, T.K.; Kimble, J.B.; Chang, J.; Lopes, S.; Souza, C.K.; Pekosz, A.; Shaw-Saliba, K.; Rothman, R.E.; Chen, K.-F.; et al. Antigenic Characterization and Pandemic Risk Assessment of North American H1 Influenza A Viruses Circulating in Swine. Microbiol. Spectr. 2022, 10, e01781-22. [Google Scholar] [CrossRef] [PubMed]
- Souza, C.K.; Anderson, T.K.; Chang, J.; Venkatesh, D.; Lewis, N.S.; Pekosz, A.; Shaw-Saliba, K.; Rothman, R.E.; Chen, K.-F.; Vincent, A.L. Antigenic Distance between North American Swine and Human Seasonal H3N2 Influenza A Viruses as an Indication of Zoonotic Risk to Humans. J. Virol. 2022, 96, e01374-21. [Google Scholar] [CrossRef] [PubMed]
- Tapia, R.; Torremorell, M.; Culhane, M.; Medina, R.A.; Neira, V. Antigenic Characterization of Novel H1 Influenza A Viruses in Swine. Sci. Rep. 2020, 10, 4510. [Google Scholar] [CrossRef] [PubMed]
- Hanssen, H.; Hincapié, O.; López, J.H. Influenza En Porcinos de Antioquia, Colombia. Pan Am. J. Public Health 1977, 8, 35–43. [Google Scholar]
- Osorio-Zambrano, W.F.; Ospina-Jimenez, A.F.; Alvarez-Munoz, S.; Gomez, A.P.; Ramirez-Nieto, G.C. Zooming in on the Molecular Characteristics of Swine Influenza Virus Circulating in Colombia before and after the H1N1pdm09 Virus. Front. Vet. Sci. 2022, 9, 983304. [Google Scholar] [CrossRef]
- Ramirez-Nieto, G.C.; Diaz Rojas, C.A.; Vera Alfonso, V.J.; Correa, J.J.; Mogollon Galvis, J.D. First Isolation and Identification of H1N1 Swine Influenza Viruses in Colombian Pig Farms. Health 2012, 4, 983–990. [Google Scholar] [CrossRef]
- Ciuoderis, K.A.; Perez, L.S.; Cardona, A.; Hernandez-Ortíz, J.P.; Osorio, J.E. Use of Oral Fluids for Efficient Monitoring of Influenza Viruses in Swine Herds in Colombia. Rev. Colomb. Cienc. Pecu. 2019, 35, 141–152. [Google Scholar] [CrossRef]
- Flórez Ramos, J.; Vera, V.; Lora, Á.; Ramírez-Nieto, G. Molecular Evaluation of Influenza A Virus in Swine at Slaughterhouses in Colombia. Rev. MVZ Cordoba 2018, 23, 7013–7024. [Google Scholar] [CrossRef]
- Edgar, R.C. Muscle5: High-Accuracy Alignment Ensembles Enable Unbiased Assessments of Sequence Homology and Phylogeny. Nat. Commun. 2022, 13, 6968. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; Von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Anderson, C.S.; McCall, P.R.; Stern, H.A.; Yang, H.; Topham, D.J. Antigenic Cartography of H1N1 Influenza Viruses Using Sequence-Based Antigenic Distance Calculation. BMC Bioinform. 2018, 19, 51. [Google Scholar] [CrossRef]
- Burke, D.F.; Smith, D.J. A Recommended Numbering Scheme for Influenza A HA Subtypes. PLoS ONE 2014, 9, e112302. [Google Scholar] [CrossRef]
- Zhu, W.; Yang, S.; Guo, Y.; Yang, L.; Bai, T.; Yu, Z.; Li, X.; Li, M.; Guo, J.; Wang, D.; et al. Imported Pigs May Have Introduced the First Classical Swine Influenza Viruses into Mainland China. Infect. Genet. Evol. 2013, 17, 142–146. [Google Scholar] [CrossRef][Green Version]
- Shope, R.E. The Etiology of Swine Influenza. Science (1979) 1931, 73, 214–215. [Google Scholar] [CrossRef]
- Diaz, A.; Perez, A.; Sreevatsan, S.; Davies, P.; Culhane, M.; Torremorell, M. Association between Influenza A Virus Infection and Pigs Subpopulations in Endemically Infected Breeding Herds. PLoS ONE 2015, 10, e0129213. [Google Scholar] [CrossRef]
- Diaz, A.; Marthaler, D.; Culhane, M.; Sreevatsan, S.; Alkhamis, M.; Torremorell, M. Complete Genome Sequencing of Influenza A Viruses within Swine Farrow-to-Wean Farms Reveals the Emergence, Persistence, and Subsidence of Diverse Viral Genotypes. J. Virol. 2017, 91, e00745-17. [Google Scholar] [CrossRef]
- Ryt-Hansen, P.; Nielsen, H.G.; Sørensen, S.S.; Larsen, I.; Kristensen, C.S.; Larsen, L.E. The Role of Gilts in Transmission Dynamics of Swine Influenza Virus and Impacts of Vaccination Strategies and Quarantine Management. Porc. Health Manag. 2022, 8, 19. [Google Scholar] [CrossRef]
- Chamba Pardo, F.O.; Schelkopf, A.; Allerson, M.; Morrison, R.; Culhane, M.; Perez, A.; Torremorell, M. Breed-to-Wean Farm Factors Associated with Influenza A Virus Infection in Piglets at Weaning. Prev. Vet. Med. 2018, 161, 33–40. [Google Scholar] [CrossRef]
- Markin, A.; Zanella, G.C.; Arendsee, Z.W.; Zhang, J.; Krueger, K.M.; Gauger, P.C.; Baker, A.L.V.; Anderson, T.K. Reverse-Zoonoses of 2009 H1N1 Pandemic Influenza A Viruses and Evolution in United States Swine Results in Viruses with Zoonotic Potential. bioRxiv 2022. [Google Scholar] [CrossRef]
- Nelson, M.I.; Gramer, M.R.; Vincent, A.L.; Holmes, E.C. Global Transmission of Influenza Viruses from Humans to Swine. J. Gen. Virol. 2012, 93, 2195. [Google Scholar] [CrossRef] [PubMed]
- Charoenvisal, N.; Keawcharoen, J.; Sreta, D.; Chaiyawong, S.; Nonthabenjawan, N.; Tantawet, S.; Jittimanee, S.; Arunorat, J.; Amonsin, A.; Thanawongnuwech, R. Genetic Characterization of Thai Swine Influenza Viruses after the Introduction of Pandemic H1N1 2009. Virus Genes 2013, 47, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Chastagner, A.; Hervé, S.; Bonin, E.; Quéguiner, S.; Hirchaud, E.; Henritzi, D.; Béven, V.; Gorin, S.; Barbier, N.; Blanchard, Y.; et al. Spatiotemporal Distribution and Evolution of the A/H1N1 2009 Pandemic Influenza Virus in Pigs in France from 2009 to 2017: Identification of a Potential Swine-Specific Lineage. J. Virol. 2018, 92, 24. [Google Scholar] [CrossRef] [PubMed]
- Grøntvedt, C.A.; Er, C.; Gjerset, B.; Hauge, A.G.; Brun, E.; Jørgensen, A.; Lium, B.; Framstad, T. Influenza A(H1N1)Pdm09 Virus Infection in Norwegian Swine Herds 2009/10: The Risk of Human to Swine Transmission. Prev. Vet. Med. 2013, 110, 429. [Google Scholar] [CrossRef] [PubMed]
- Pereda, A.; Cappuccio, J.; Quiroga, M.A.; Baumeister, E.; Insarralde, L.; Ibar, M.; Sanguinetti, R.; Cannilla, M.L.; Franzese, D.; Escobar Cabrera, O.E.; et al. Pandemic (H1N1) 2009 Outbreak on Pig Farm, Argentina. Emerg. Infect. Dis. 2010, 16, 307. [Google Scholar] [CrossRef]
- Ciacci-Zanella, J.R.; Schaefer, R.; Gava, D.; Haach, V.; Cantão, M.E.; Coldebella, A. Influenza A Virus Infection in Brazilian Swine Herds Following the Introduction of Pandemic 2009 H1N1. Vet. Microbiol. 2015, 180, 118–122. [Google Scholar] [CrossRef]
- Nelson, M.; Spiro, D.; Wentworth, D.; Fan, J.; Beck, E.; St. George, K.; Ghedin, E.; Halpin, R.; Bera, J.; Hine, E.; et al. The Early Diversification of Influenza A/H1N1pdm. PLoS Curr. 2009, 1, RRN1126. [Google Scholar] [CrossRef]
- Castelán-Vega, J.A.; Magaña-Hernández, A.; Jiménez-Alberto, A.; Ribas-Aparicio, R.M. The Hemagglutinin of the Influenza A(H1N1)Pdm09 Is Mutating towards Stability. Adv. Appl. Bioinform. Chem. 2014, 7, 37–44. [Google Scholar] [CrossRef]
- Klein, E.Y.; Serohijos, A.W.R.; Choi, J.M.; Shakhnovich, E.I.; Pekosz, A. Influenza A H1N1 Pandemic Strain Evolution—Divergence and the Potential for Antigenic Drift Variants. PLoS ONE 2014, 9, e93632. [Google Scholar] [CrossRef]
- Kim, P.; Jang, Y.H.; Kwon, S.B.; Lee, C.M.; Han, G.; Seong, B.L. Glycosylation of Hemagglutinin and Neuraminidase of Influenza A Virus as Signature for Ecological Spillover and Adaptation among Influenza Reservoirs. Viruses 2018, 10, 183. [Google Scholar] [CrossRef]
- Sun, S.; Wang, Q.; Zhao, F.; Chen, W.; Li, Z. Glycosylation Site Alteration in the Evolution of Influenza A (H1N1) Viruses. PLoS ONE 2011, 6, 22844. [Google Scholar] [CrossRef] [PubMed]
- Decker, C.H.; Rapier-Sharman, N.; Pickett, B.E. Mutation in Hemagglutinin Antigenic Sites in Influenza A PH1N1 Viruses from 2015–2019 in the United States Mountain West, Europe, and the Northern Hemisphere. Genes 2022, 13, 909. [Google Scholar] [CrossRef] [PubMed]
- Guldemir, D.; Coskun-Ari, F.F.; Altas, A.B.; Bakkaloglu, Z.; Unaldi, O.; Bayraktar, F.; Korukluoglu, G.; Aktas, A.R.; Durmaz, R. Molecular Characterization of the Influenza A(H1N1)Pdm09 Isolates Collected in the 2015-2016 Season and Comparison of HA Mutations Detected in Turkey since 2009. J. Med. Virol. 2019, 91, 2074–2082. [Google Scholar] [CrossRef]
- Liu, B.; Wang, Y.; Liu, Y.; Chen, Y.; Liu, Y.; Cong, X.; Ji, Y.; Gao, Y. Molecular Evolution and Characterization of Hemagglutinin and Neuraminidase of Influenza A(H1N1)Pdm09 Viruses Isolated in Beijing, China, during the 2017–2018 and 2018–2019 Influenza Seasons. Arch. Virol. 2021, 166, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Potdar, V.; Vijay, N.; Gupta, N.; Arunkumar, G.; Borkakoty, B.; Malhotra, B.; Rabha, D.; Hinge, D.; Kaur, H.; Chadha, M. Molecular Characterization of Influenza A(H1N1)Pdm09 Viruses Circulating at Various Geographical Locations in India, 2017. Indian J. Med. Res. 2019, 149, 783–789. [Google Scholar] [CrossRef]
- Zolotarova, O.; Mironenko, A.; Budzanivska, I.; Leibenko, L.; Radchenko, L. Antigenic Site Variation in the Hemagglutinin of Pandemic Influenza A(H1N1)Pdm09 Viruses between 2009–2017 in Ukraine. Pathogens 2019, 8, 194. [Google Scholar] [CrossRef]
- Altman, M.O.; Angel, M.; Košík, I.; Trovão, N.S.; Zost, S.J.; Gibbs, J.S.; Casalino, L.; Amaro, R.E.; Hensley, S.E.; Nelson, M.I.; et al. Human Influenza a Virus Hemagglutinin Glycan Evolution Follows a Temporal Pattern to a Glycan Limit. mBio 2019, 10, e00204-19. [Google Scholar] [CrossRef]
- Gibbs, A.J.; Armstrong, J.S.; Downie, J.C. From Where Did the 2009 “swine-Origin” Influenza A Virus (H1N1) Emerge? Virol. J. 2009, 6, 207. [Google Scholar] [CrossRef]
- Takemae, N.; Nguyen, T.; Ngo, L.T.; Hiromoto, Y.; Uchida, Y.; Pham, V.P.; Kageyama, T.; Kasuo, S.; Shimada, S.; Yamashita, Y.; et al. Antigenic Variation of H1N1, H1N2 and H3N2 Swine Influenza Viruses in Japan and Vietnam. Arch. Virol. 2013, 158, 859–876. [Google Scholar] [CrossRef]
- Xu, C.; Zhang, N.; Yang, Y.; Liang, W.; Zhang, Y.; Wang, J.; Suzuki, Y.; Wu, Y.; Chen, Y.; Yang, H.; et al. Immune Escape Adaptive Mutations in Hemagglutinin Are Responsible for the Antigenic Drift of Eurasian Avian-Like H1N1 Swine Influenza Viruses. J. Virol. 2022, 96, e00971-22. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, Y.; Chen, H.; Meng, F.; Tao, S.; Ma, S.; Qiao, C.; Chen, H.; Yang, H. A Single Amino Acid at Position 158 in Haemagglutinin Affects the Antigenic Property of Eurasian Avian-like H1N1 Swine Influenza Viruses. Transbound. Emerg. Dis. 2021, 69, e236–e243. [Google Scholar] [CrossRef] [PubMed]
- Dunham, E.J.; Dugan, V.G.; Kaser, E.K.; Perkins, S.E.; Brown, I.H.; Holmes, E.C.; Taubenberger, J.K. Different Evolutionary Trajectories of European Avian-Like and Classical Swine H1N1 Influenza A Viruses. J. Virol. 2009, 83, 5485–5494. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus Name | Virus ID | Year | Subtype | HA Lineage | Accession Number |
|---|---|---|---|---|---|
| A/swine/Colombia/16562/2008 | 16562/08/A | 2008 | H1N1 | 1A.1 | EPI2719368 |
| A/swine/Colombia/16563/2008 | 16563/08/A | 2008 | H1N1 | 1A.1 | EPI2721678 |
| A/swine/Colombia/16564/2009 | 16564/09/A | 2009 | H1N1 | 1A.1 | EPI2721686 |
| A/swine/Colombia/16567/2015 | 16567/15/A | 2015 | H1N1 | 1A.3.3.2 | EPI2721687 |
| A/swine/Colombia/16568/2015 | 16568/15/A | 2015 | H1N1 | 1A.3.3.2 | EPI2721688 |
| A/swine/Colombia/14254/2016 | 14254/16/A | 2016 | H1N1 | 1A.3.3.2 | EPI2721689 |
| A/swine/Colombia/14258/2016 | 14258/16/A | 2016 | H1N1 | 1A.3.3.2 | EPI2721690 |
| A/swine/Colombia/14264/2016 | 14264/16/A | 2016 | H1N1 | 1A.3.3.2 | EPI2721691 |
| A/swine/Colombia/14268/2016 | 14268/16/A | 2016 | H1N1 | 1A.3.3.2 | EPI2721692 |
| A/swine/Colombia/14252/2016 | 14252/16/A | 2016 | H1N1 | 1A.3.3.2 | EPI2721693 |
| A/swine/Colombia/14265/2016 | 14265/16/A | 2016 | H1N1 | 1A.3.3.2 | EPI2721694 |
| A/swine/Colombia/14261/2016 | 14261/16/A | 2016 | H1N2 | 1A.3.3.2 | EPI2721695 |
| A/swine/Colombia/14255/2017 | 14255/17/A | 2017 | H1N1 | 1A.3.3.2 | EPI2721696 |
| A/swine/Colombia/08712/2021 | 08712/21/A | 2021 | H1N1 | 1A.1 | EPI2721697 |
| A/swine/Colombia/08713/2021 | 08713/21/A | 2021 | H1N1 | 1A.3.3.2 | EPI2721698 |
| A/swine/Colombia/14271/2021 | 14271/21/A | 2021 | H1N1 | 1A.3.3.2 | EPI2721699 |
| A/swine/Colombia/08719/2021 | 08719/21/A | 2021 | H1N1 | 1A.3.3.2 | EPI2721700 |
| A/swine/Colombia/08721/2021 | 08721/21/A | 2021 | H1N1 | 1A.3.3.2 | EPI2721701 |
| A/swine/Colombia/14273/2021 | 14273/21/A | 2021 | H1N1 | 1A.3.3.2 | EPI2721702 |
| A/swine/Colombia/14274/2021 | 14274/21/A | 2021 | H1N1 | 1A.3.3.2 | EPI2721703 |
| A/swine/Colombia/14250/2017 | 14250/17/CA | 2017 | H1N1 | 1A.3.3.2 | EPI2721704 |
| A/swine/Colombia/14256/2017 | 14256/17/CA | 2017 | H1N1 | 1A.3.3.2 | EPI2721705 |
| A/swine/Colombia/14257/2017 | 14257/17/CA | 2017 | H1N1 | 1A.3.3.2 | EPI2721706 |
| A/swine/Colombia/16565/2010 | 16565/10/CU | 2010 | H1N1 | 1A.3.3.2 | EPI2721707 |
| A/swine/Colombia/16566/2015 | 16566/15/CU | 2015 | H1N1 | 1A.3.3.2 | EPI2721708 |
| A/swine/Colombia/14253/2016 | 14253/16/CU | 2016 | H1N1 | 1A.3.3.2 | EPI2721709 |
| A/swine/Colombia/08661/2021 | 08661/21/CU | 2021 | H1N1 | 1A.3.3.2 | EPI2721710 |
| A/swine/Colombia/08663/2021 | 08663/21/CU | 2021 | H1N1 | 1A.3.3.2 | EPI2721711 |
| A/swine/Colombia/14269/2021 | 14269/21/CU | 2021 | H1N1 | 1A.3.3.2 | EPI2721712 |
| A/swine/Colombia/14270/2021 | 14270/21/CU | 2021 | H1N1 | 1A.3.3.2 | EPI2721713 |
| A/swine/Colombia/14260/2016 | 14260/16/M | 2016 | H1N2 | 1A.3.3.2 | EPI2721714 |
| A/swine/Colombia/14259/2016 | 14259/16/Q | 2016 | H1N1 | 1A.3.3.2 | EPI2721715 |
| A/swine/Colombia/14251/2016 | 14251/16/Q | 2016 | H1N1 | 1A.3.3.2 | EPI2721716 |
| A/swine/Colombia/14262/2016 | 14262/16/R | 2016 | H1N1 | 1A.3.3.2 | EPI2721717 |
| A/swine/Colombia/14266/2016 | 14266/16/R | 2016 | H1N1 | 1A.3.3.2 | EPI2721718 |
| A/swine/Colombia/14263/2016 | 14263/16/R | 2016 | H1N1 | 1A.3.3.2 | EPI2721719 |
| A/swine/Colombia/14249/2016 | 14249/16/S | 2016 | H1N1 | 1A.3.3.2 | EPI2721720 |
| G1A.1 | N1A.1 | G1A.1–2 | N1A.1–3 | M1A.3 | PDM-CO-16 | PDM09-17 | PDM-CO-21 | PDM-21 | |
|---|---|---|---|---|---|---|---|---|---|
| G1A.1 | 0.3 * | ||||||||
| N1A.1 | 5.2 | 4.7 * | |||||||
| G1A.1–2 | 2.8 | 6.8 | 2.6 * | ||||||
| N1A.1–3 | 6.2 | 8.5 | 6.1 | 5.0 * | |||||
| M1A.3 | 4.8 | 8.7 | 5.4 | 6.3 | 1.3 * | ||||
| PDM-CO-16 | 6.5 | 8.2 | 6.0 | 6.0 | 5.1 | 0.3 * | |||
| PDM09-17 | 5.4 | 7.7 | 5.0 | 4.8 | 5.8 | 3.6 | 2.0 * | ||
| PDM-CO-21 | 6.9 | 8.3 | 7.3 | 4.9 | 6.3 | 5.1 | 3.9 | 0.2 * | |
| PDM-21 | 7.2 | 8.1 | 6.8 | 5.8 | 7.3 | 4.5 | 3.1 | 3.5 | 0.9 * |
| Chilean 1B.2-Other | Mexican 1B.2-Other | Asian 1B.2-Other | North America 1B.2.1 | North America 1B.2.2 | Human | |
|---|---|---|---|---|---|---|
| Chilean 1B.2-other | 3.1 * | |||||
| Mexican 1B.2-other | 6.3 | 0.0 * | ||||
| Asian 1B.2-other | 4.0 | 4.5 | 0.0 * | |||
| North America 1B.2.1 | 4.2 | 5.7 | 3.8 | 2.9 * | ||
| North America 1B.2.2 | 5.0 | 6.3 | 4.5 | 4.9 | 4.0 * | |
| Human | 4.0 | 3.5 | 2.1 | 4.0 | 4.5 | 0.2 * |
| Epitope | Conservation | Conserved Amino Acids | Mutations | Associated Region | Associated Year |
|---|---|---|---|---|---|
| Sa | 38.5% | P124, K153, K154, S157, and P159 | G155E | Cundinamarca | 2021 |
| K160M | Antioquia | 2021 | |||
| K160N | Antioquia | 2016 | |||
| S162N | Antioquia | 2021 | |||
| Cundinamarca | |||||
| S162Y | Antioquia | 2021 | |||
| K163Q | Antioquia | 2021 | |||
| K163I | Cundinamarca | 2021 | |||
| Sb | 50% | D187, Q188, Q189, L191, Y192, and N194 | A186T | Cundinamarca | 2021 |
| S190H | Cundinamarca | 2021 | |||
| Ca1 | 63.3% | N167, K169, G170, S204, and G237 | E235D | Antioquia | 2021 |
| Ca2 | 12.5% | G140 | P137S | Cundinamarca | 2021 |
| Antioquia | 2016 | ||||
| A141T | Cundinamarca | 2021 | |||
| Cb | 50% | L70, T72, and G75 | S71F | Antioquia | 2016 |
| A73T | Antioquia | 2016 | |||
| S74R | Antioquia | 2021 | |||
| Cundinamarca |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ospina-Jimenez, A.F.; Gomez, A.P.; Rincon-Monroy, M.A.; Ortiz, L.; Perez, D.R.; Peña, M.; Ramirez-Nieto, G. Sequence-Based Antigenic Analyses of H1 Swine Influenza A Viruses from Colombia (2008–2021) Reveals Temporal and Geographical Antigenic Variations. Viruses 2023, 15, 2030. https://doi.org/10.3390/v15102030
Ospina-Jimenez AF, Gomez AP, Rincon-Monroy MA, Ortiz L, Perez DR, Peña M, Ramirez-Nieto G. Sequence-Based Antigenic Analyses of H1 Swine Influenza A Viruses from Colombia (2008–2021) Reveals Temporal and Geographical Antigenic Variations. Viruses. 2023; 15(10):2030. https://doi.org/10.3390/v15102030
Chicago/Turabian StyleOspina-Jimenez, Andres F., Arlen P. Gomez, Maria A. Rincon-Monroy, Lucia Ortiz, Daniel R. Perez, Mario Peña, and Gloria Ramirez-Nieto. 2023. "Sequence-Based Antigenic Analyses of H1 Swine Influenza A Viruses from Colombia (2008–2021) Reveals Temporal and Geographical Antigenic Variations" Viruses 15, no. 10: 2030. https://doi.org/10.3390/v15102030
APA StyleOspina-Jimenez, A. F., Gomez, A. P., Rincon-Monroy, M. A., Ortiz, L., Perez, D. R., Peña, M., & Ramirez-Nieto, G. (2023). Sequence-Based Antigenic Analyses of H1 Swine Influenza A Viruses from Colombia (2008–2021) Reveals Temporal and Geographical Antigenic Variations. Viruses, 15(10), 2030. https://doi.org/10.3390/v15102030