
A Bioreactor-Based Yellow Fever Virus-like Particle Production Process with Integrated Process Analytical Technology Based on Transient Transfection

 ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Plasmid Production
2.1.1. pDNA Production under Standard Conditions
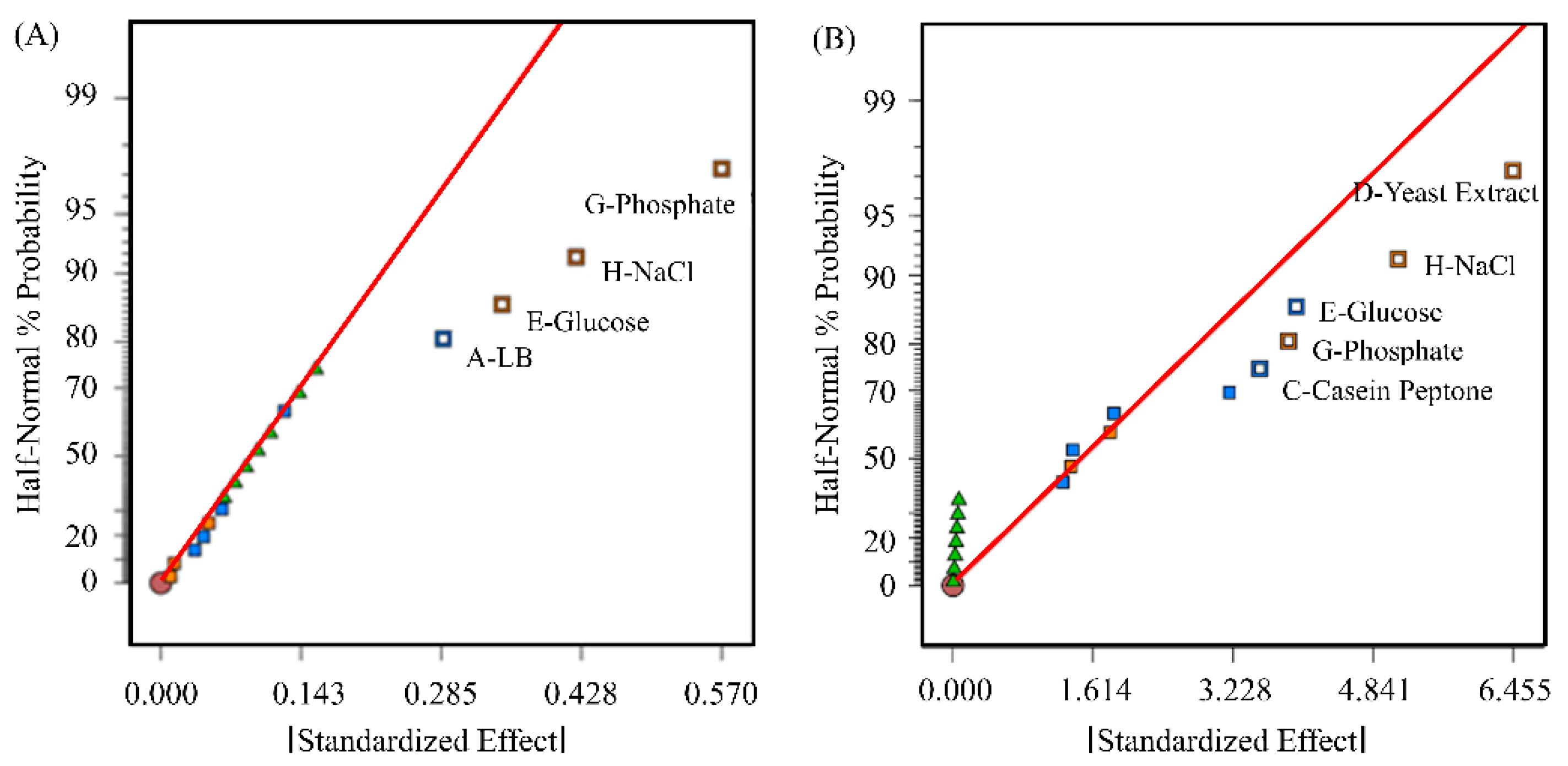
2.1.2. Optimization of pDNA Production Using a Plackett–Burman Design
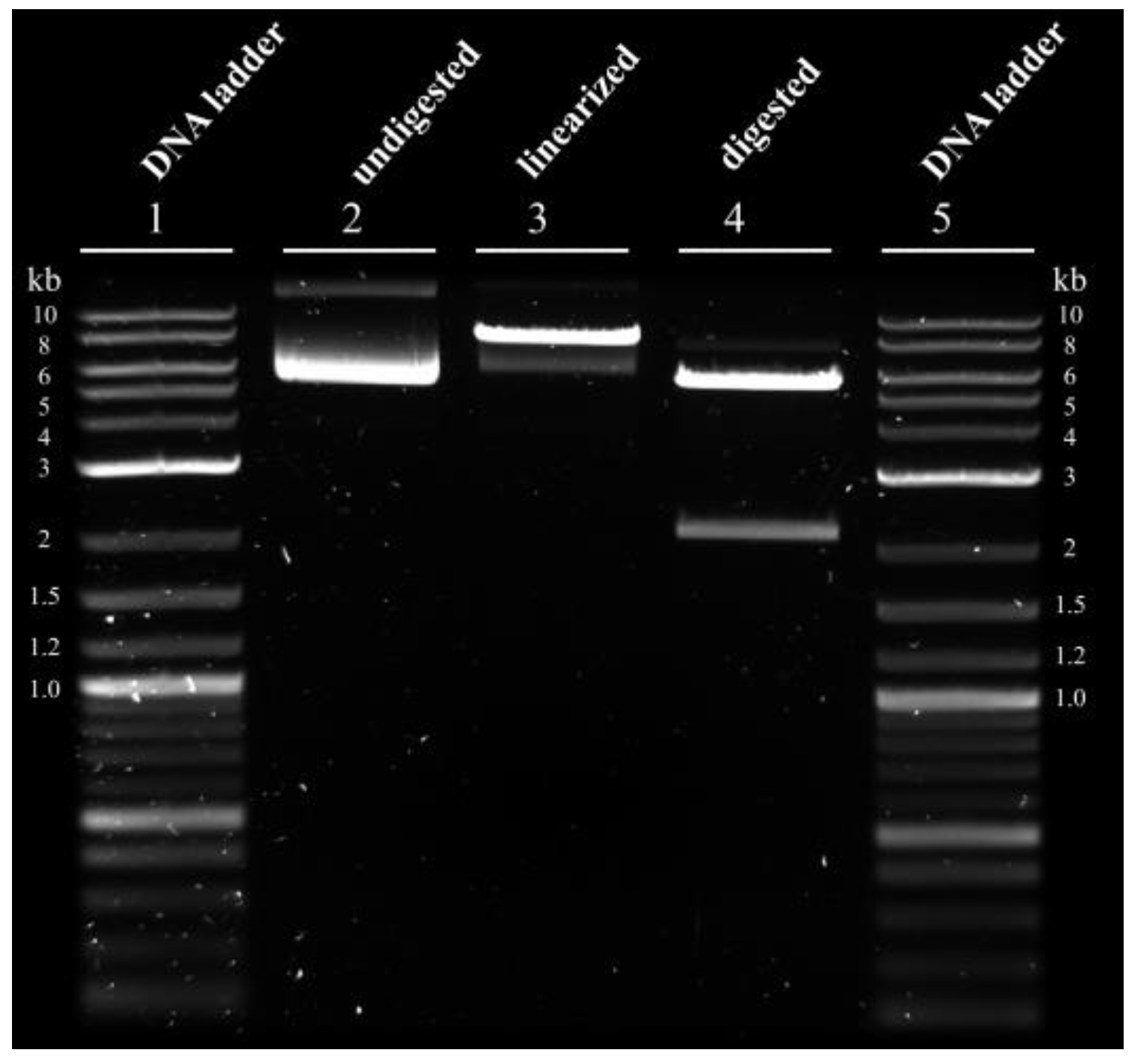
2.1.3. pDNA Purification
2.2. eGFP or YF-VLP Production
2.2.1. Cell Line and Medium
2.2.2. Transient Transfection in Shaking Flasks
2.2.3. Transient Transfection in a Stirred-Tank Bioreactor
2.3. Analysis
2.3.1. pDNA Analysis
2.3.2. Analysis of E. coli Growth
2.3.3. Offline Determination of Cell Concentration
2.3.4. Metabolite Analysis
2.3.5. SDS-PAGE
2.3.6. Western Blot
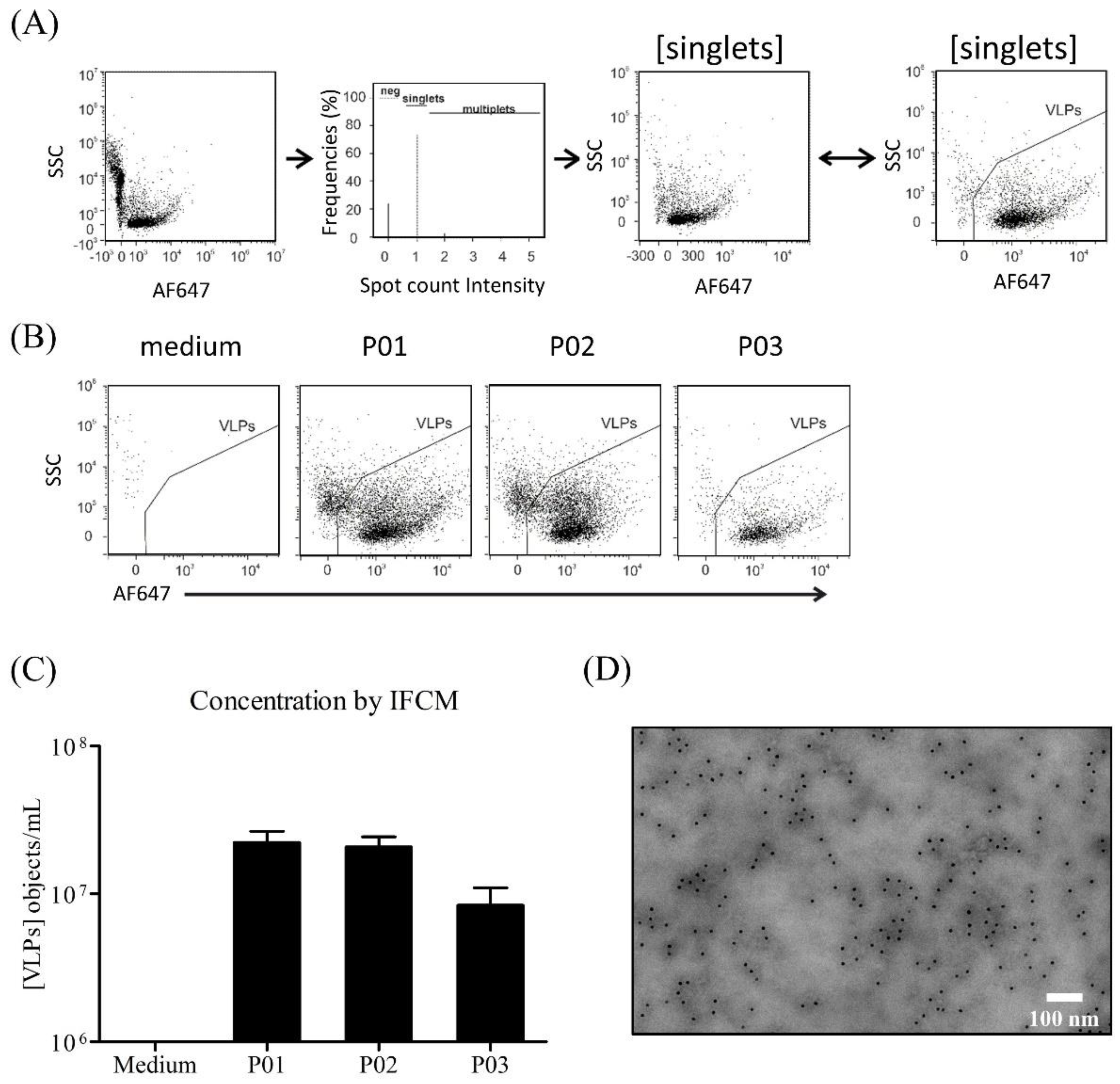
2.3.7. Imaging Flow Cytometry Measurement (IFCM)
2.3.8. Transmission Electron Microscopy (TEM)
2.3.9. Statistical Analysis
3. Results
3.1. Optimization of Transfection-Grade pDNA Production
3.2. Optimization of Process Parameters for a Transient VLP Production in a Stirred-Tank Bioreactor
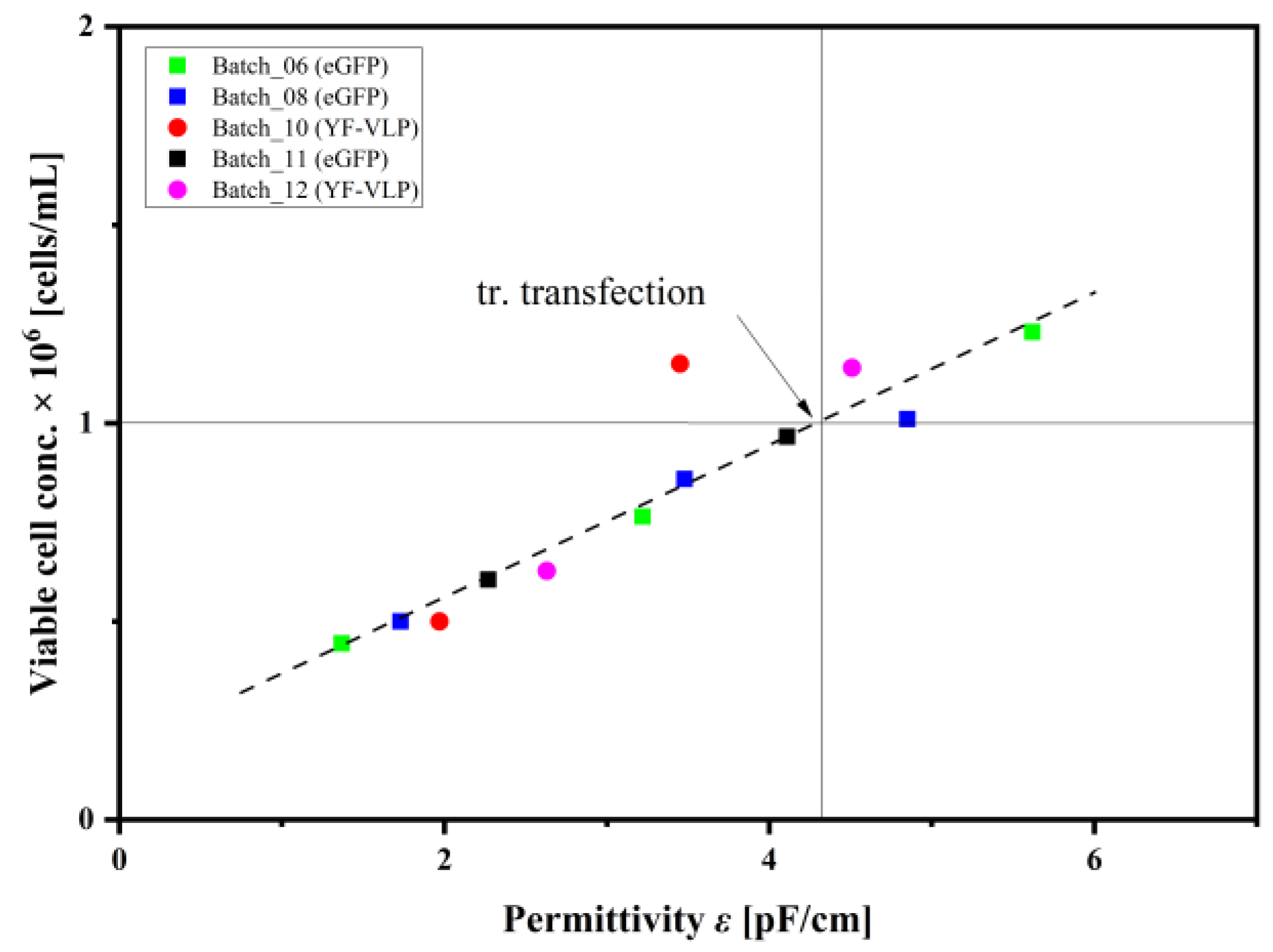
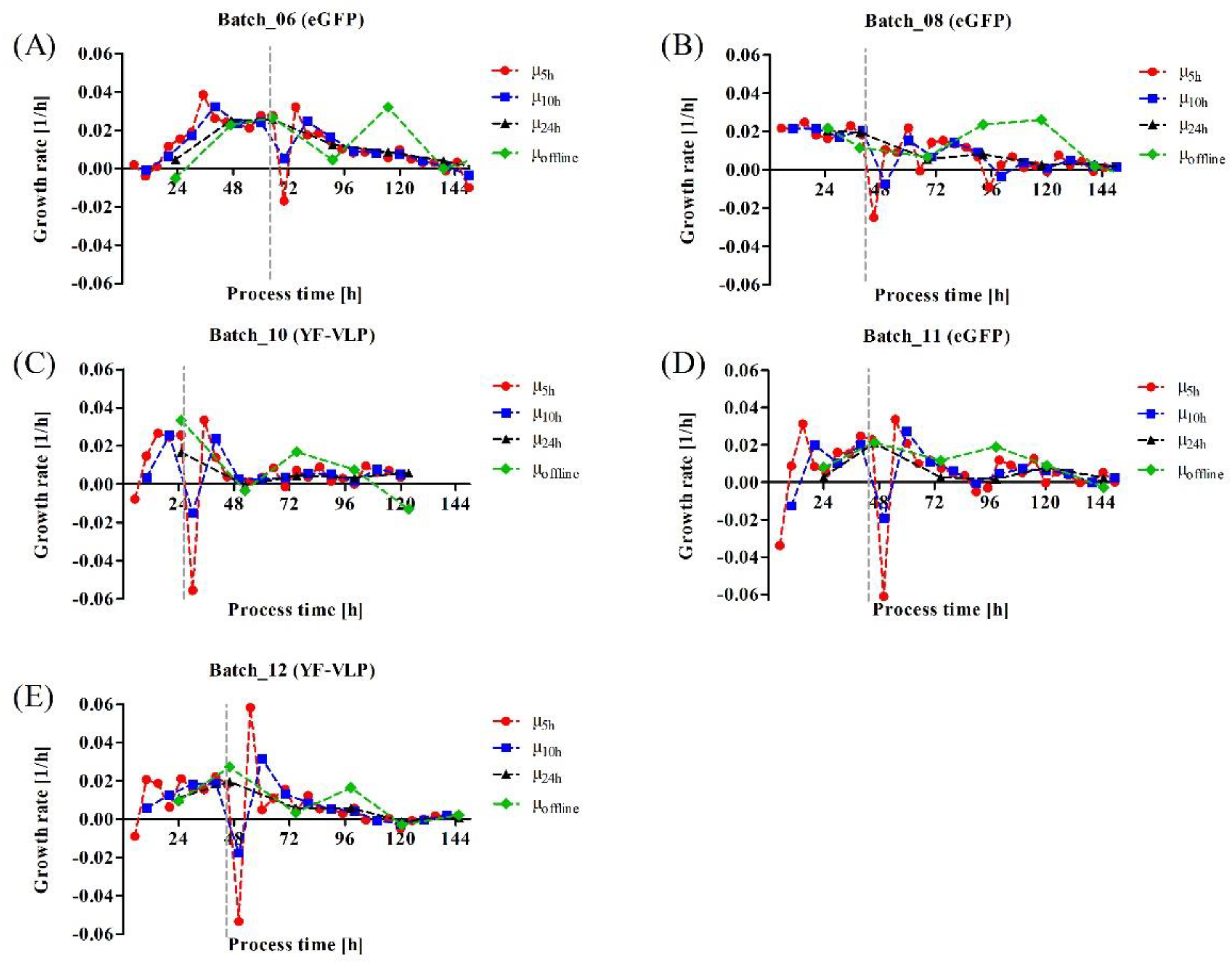
3.3. Monitoring a Transient VLP Production in a Stirred-Tank Bioreactor Using Dielectric Spectroscopy
3.4. Characterization of Produced YF-VLPs
4. Discussion
4.1. Impact of Various Parameters on pDNA Production in E. coli
4.2. Influence of Different Process Parameters on Transient Transfection
4.3. Implementation of Dielectric Spectroscopy into a Transient YF-VLP Production Process
4.4. Characterization of the YF-VLP
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A










References
- Garske, T.; Van Kerkhove, M.D.; Yactayo, S.; Ronveaux, O.; Lewis, R.F.; Staples, J.E.; Perea, W.; Ferguson, N.M.; Yellow Fever Expert Committee. Yellow Fever in Africa: Estimating the burden of disease and impact of mass vaccination from outbreak and serological data. PLoS Med. 2014, 11, e1001638. [Google Scholar] [CrossRef] [PubMed]
- Monath, T.P. Yellow fever vaccine. Expert. Rev. Vaccines 2005, 4, 553–574. [Google Scholar] [CrossRef]
- Barrett, A.D.T.; Higgs, S. Yellow fever: A disease that has yet to be conquered. Annu. Rev. Entomol. 2007, 52, 209–229. [Google Scholar] [CrossRef] [PubMed]
- Frierson, J.G. The Yellow Fever Vaccine: A History. Yale J. Biol. Med. 2020, 83, 77–85. [Google Scholar]
- Grobbelaar, A.A.; Weyer, J.; Moolla, N.; van Vuren, P.J.; Moises, F.; Paweska, J.T. Resurgence of Yellow Fever in Angola, 2015–2016. Emerg. Infect. Dis. 2016, 22, 1854–1855. [Google Scholar] [CrossRef] [PubMed]
- Goldani, L.Z. Yellow fever outbreak in Brazil, 2017. Braz. J. Infect. Dis. 2017, 21, 123–124. [Google Scholar] [CrossRef] [PubMed]
- Noad, R.; Roy, P. Virus-like particles as immunogens. Trends Microbiol. 2013, 11, 438–444. [Google Scholar] [CrossRef] [PubMed]
- Gheysen, D.; Jacobs, E.; Foresta, F.d.; Thiriart, C.; Francotte, M.; Thines, D.; De Wilde, M. Assembly and release of HIV-1 precursor Pr55gag virus-like particles from recombinant baculovirus-infected insect cells. Cell 1989, 59, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Delchambre, M.; Gheysen, D.; Thines, D.; Thiriart, C.; Jacobs, E.; Verdin, E.; Horth, M.; Burny, A.; Bex, F. The GAG precursor of simian immunodeficiency virus assembles into virus-like particles. EMBO J. 1989, 8, 2653–2660. [Google Scholar] [CrossRef]
- Zeltins, A. Construction and characterization of virus-like particles: A review. Mol. Biotechnol. 2013, 53, 92–107. [Google Scholar] [CrossRef]
- Lothert, K.; Dekevic, G.; Loewe, D.; Salzig, D.; Czermak, P.; Wolff, M.W. Upstream and Downstream Processes for Viral Nanoplexes as Vaccines. Methods Mol. Biol. 2021, 2183, 217–248. [Google Scholar] [CrossRef] [PubMed]
- Yamaji, H.; Segawa, M.; Nakamura, M.; Katsuda, T.; Kuwahara, M.; Konishi, E. Production of Japanese encephalitis virus-like particles using the baculovirus-insect cell system. J. Biosci. Bioeng. 2012, 114, 657–662. [Google Scholar] [CrossRef]
- Hirsch, J.; Faber, B.W.; Crowe, J.E.; Verstrepen, B.; Cornelissen, G.E. coli production process yields stable dengue 1 virus-sized particles (VSPs). Vaccine 2020, 38, 3305–3312. [Google Scholar] [CrossRef] [PubMed]
- Ponndorf, D.; Meshcheriakova, Y.; Thuenemann, E.C.; Dobon Alonso, A.; Overman, R.; Holton, N.; Dowall, S.; Kennedy, E.; Stocks, M.; Lomonossoff, G.P.; et al. Plant-made dengue virus-like particles produced by co-expression of structural and non-structural proteins induce a humoral immune response in mice. Plant Biotechnol. J. 2021, 19, 745–756. [Google Scholar] [CrossRef] [PubMed]
- Fischer, D.; Li, Y.; Ahlemeyer, B.; Krieglstein, J.; Kissel, T. In vitro cytotoxicity testing of polycations: Influence of polymerstructure on cell viability an dhemolysis. Biomaterials 2003, 24, 1121–1131. [Google Scholar] [CrossRef]
- Moghimi, S.M.; Symonds, P.; Murray, J.C.; Hunter, A.C.; Debska, G.; Szewczyk, A. A two-stage poly(ethylenimine)-mediated cytotoxicity: Implications for gene transfer/therapy. Mol. Ther. 2005, 11, 990–995. [Google Scholar] [CrossRef]
- Puckette, M.; Primavera, V.; Martel, E.; Barrera, J.; Hurtle, W.; Clark, B.; Kamicker, B.; Zurita, M.; Brake, D.; Neilan, J. Transiently Transfected Mammalian Cell Cultures: An Adaptable and Effective Platform for Virus-like Particle-Based Vaccines against Foot-and-Mouth Disease Virus. Viruses 2022, 14, 989. [Google Scholar] [CrossRef]
- Cervera, L.; Gutiérrez-Granados, S.; Martínez, M.; Blanco, J.; Gòdia, F.; Segura, M.M. Generation of HIV-1 Gag VLPs by transient transfection of HEK 293 suspension cell cultures using an optimized animal-derived component free medium. J. Biotechnol. 2013, 166, 152–165. [Google Scholar] [CrossRef]
- González-Domínguez, I.; Lorenzo, E.; Bernier, A.; Cervera, L.; Gòdia, F.; Kamen, A. A Four-Step Purification Process for Gag VLPs: From Culture Supernatant to High-Purity Lyophilized Particles. Vaccines 2021, 9, 1154. [Google Scholar] [CrossRef]
- Lavado-García, J.; Jorge, I.; Cervera, L.; Vazquez, J.; Gòdia, F. Multiplexed Quantitative Proteomic Analysis of HEK293 Provides Insights into Molecular Changes Associated with the Cell Density Effect, Transient Transfection, and Virus-Like Particle Production. J. Proteome Res. 2020, 19, 1085–1099. [Google Scholar] [CrossRef]
- Alvim, R.G.F.; Lima, T.M.; Silva, J.L.; Silva, J.L.; de Oliveira, G.A.; Castilho, L.R. Process intensification for the production of yellow fever virus-like particles as potential recombinant vaccine antigen. Biotechnol. Bioeng. 2021, 118, 3581–3592. [Google Scholar] [CrossRef]
- Alvim, R.G.F.; Itabaiana, I.; Castilho, L.R. Zika virus-like particles (VLPs): Stable cell lines and continuous perfusion processes as a new potential vaccine manufacturing platform. Vaccine 2019, 37, 6970–6977. [Google Scholar] [CrossRef] [PubMed]
- Druzinec, D.; Weiss, K.; Elseberg, C.; Salzig, D.; Kraume, M.; Pörtner, R.; Czermak, P. Process analytical technology (PAT) in insect and mammalian cell culture processes: Dielectric spectroscopy and focused beam reflectance measurement (FBRM). Methods Mol. Biol. 2014, 1104, 313–341. [Google Scholar] [CrossRef]
- Markx, G.H.; Davey, C.L. The dielectric properties of biological cells at radiofrequencies: Applications in biotechnology. Enzym. Microb. Technol. 1999, 25, 161–171. [Google Scholar] [CrossRef]
- Negrete, A.; Esteban, G.; Kotin, R.M. Process optimization of large-scale production of recombinant adeno-associated vectors using dielectric spectroscopy. Appl. Microbiol. Biotechnol. 2007, 76, 761–772. [Google Scholar] [CrossRef]
- Flores-Cosío, G.; Herrera-López, E.J.; Arellano-Plaza, M.; Gschaedler-Mathis, A.; Kirchmayr, M.; Amaya-Delgado, L. Application of dielectric spectroscopy to unravel the physiological state of microorganisms: Current state, prospects and limits. Appl. Microbiol. Biotechnol. 2020, 104, 6101–6113. [Google Scholar] [CrossRef] [PubMed]
- Schwan, H.P. Electrical properties of tissue and cell suspensions. Adv. Biol. Med. Phys. 1957, 5, 147–209. [Google Scholar] [CrossRef]
- Cole, K.S.; Cole, R.H. Dispersion and Absorption in Dielectrics, I. Alternating Current Characteristics. J. Chem. Phys. 1041, 9, 341–351. [Google Scholar] [CrossRef]
- Kiviharju, K.; Salonen, K.; Moilanen, U.; Meskanen, E.; Leisola, M.; Eerikäinen, T. On-line biomass measurements in bioreactor cultivations: Comparison study of two on-line probes. J. Ind. Microbiol. Biotechnol. 2007, 34, 561–566. [Google Scholar] [CrossRef]
- Olsson, L.; Nielsen, J. On-line and in situ monitoring of biomass in submerged cultivations. Trends Biotechnol. 1997, 15, 517–522. [Google Scholar] [CrossRef]
- Sonnleitner, B.; Locher, G.; Fiechter, A. Biomass determination. J. Biotechnol. 1997, 25, 5–22. [Google Scholar] [CrossRef] [PubMed]
- Carvell, J.P.; Dowd, J.E. On-line Measurements and Control of Viable Cell Density in Cell Culture Manufacturing Processes using Radio-frequency Impedance. Cytotechnology 2006, 50, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Konstantinov, K.; Chuppa, S.; Sajan, E.; Tsai, Y.; Yoon, S.; Golini, F. Real-time biomass-concentration monitoring in animal-cell cultures. Trends Biotechnol. 1994, 12, 324–333. [Google Scholar] [CrossRef]
- Brunner, S.; Sauer, T.; Carotta, S.; Cotten, M.; Saltik, M.; Wagner, E. Cell cycle dependence of gene transfer by lipoplex, polyplex and recombinant adenovirus. Gene Ther. 2000, 7, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Kiviharju, K.; Salonen, K.; Moilanen, U.; Eerikäinen, T. Biomass measurement online: The performance of in situ measurements and software sensors. J. Ind. Microbiol. Biotechnol. 2008, 35, 657–665. [Google Scholar] [CrossRef]
- Justice, C.; Brix, A.; Freimark, D.; Kraume, M.; Pfromm, P.; Eichenmueller, B.; Czermak, P. Process control in cell culture technology using dielectric spectroscopy. Biotechnol. Adv. 2011, 29, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Zitzmann, J.; Weidner, T.; Eichner, G.; Salzig, D.; Czermak, P. Dielectric Spectroscopy and Optical Density Measurement for the Online Monitoring and Control of Recombinant Protein Production in Stably Transformed Drosophila melanogaster S2 Cells. Sensors 2018, 18, 900. [Google Scholar] [CrossRef]
- Grein, T.A.; Loewe, D.; Dieken, H.; Salzig, D.; Weidner, T.; Czermak, P. High titer oncolytic measles virus production process by integration of dielectric spectroscopy as online monitoring system. Biotechnol. Bioeng. 2018, 115, 1186–1194. [Google Scholar] [CrossRef]
- Dekevic, G.; Tasto, L.; Czermak, P.; Salzig, D. Statistical experimental designs to optimize the transient transfection of HEK 293T cells and determine a transfer criterion from adherent cells to larger-scale cell suspension cultures. J. Biotechnol. 2022, 346, 23–34. [Google Scholar] [CrossRef]
- Welsh, J.A.; Van Der Pol, E.; Arkesteijn, G.J.A.; Bremer, M.; Brisson, A.; Coumans, F.; Dignat-George, F.; Duggan, E.; Ghiran, I.; Giebel, B.; et al. MIFlowCyt-EV: A framework for standardized reporting of extracellular vesicle flow cytometry experiments. J. Extracell. Vesicles 2020, 9, 1713526. [Google Scholar] [CrossRef]
- Tertel, T.; Bremer, M.; Maire, C.; Lamszus, K.; Peine, S.; Jawad, R.; Andaloussi, S.E.L.; Giebel, B.; Ricklefs, F.L.; Görgens, A. High-Resolution Imaging Flow Cytometry Reveals Impact of Incubation Temperature on Labeling of Extracellular Vesicles with Antibodies. Cytom. A 2020, 97, 602–609. [Google Scholar] [CrossRef] [PubMed]
- Görgens, A.; Bremer, M.; Ferrer-Tur, R.; Murke, F.; Tertel, T.; Horn, P.A.; Thalmann, S.; Welsh, J.A.; Probst, C.; Guerin, C.; et al. Optimisation of imaging flow cytometry for the analysis of single extracellular vesicles by using fluorescence-tagged vesicles as biological reference material. J. Extracell. Vesicles 2019, 8, 1587567. [Google Scholar] [CrossRef] [PubMed]
- Tertel, T.; Görgens, A.; Giebel, B. Analysis of individual extracellular vesicles by imaging flow cytometry. Methods Enzymol. 2020, 645, 55–78. [Google Scholar] [CrossRef]
- Wang, C.; Börger, V.; Sardari, M.; Murke, F.; Skuljec, J.; Pul, R.; Hagemann, N.; Dzyubenko, E.; Dittrich, R.; Gregorius, J.; et al. Mesenchymal Stromal Cell-Derived Small Extracellular Vesicles Induce Ischemic Neuroprotection by Modulating Leukocytes and Specifically Neutrophils. Stroke 2020, 51, 1825–1834. [Google Scholar] [CrossRef] [PubMed]
- Siebertz, K.; van Bebber, D.; Hochkirchen, T. (Eds.) Statistische Versuchsplanung: Design of Experiments (DoE); Springer: Berlin/Heidelberg, Germany, 2010. [Google Scholar]
- Williams, J.A.; Luke, J.; Langtry, S.; Meskanen, E.; Leisola, M.; Eerikäinen, T. Generic plasmid DNA production platform incorporating low metabolic burden seed-stock and fed-batch fermentation processes. Biotechnol. Bioeng. 2009, 103, 1129–1143. [Google Scholar] [CrossRef] [PubMed]
- Geisse, S.; Henke, M. Large-scale transient transfection of mammalian cells: A newly emerging attractive option for recombinant protein production. J. Struct. Funct. Genom. 2005, 6, 165–170. [Google Scholar] [CrossRef]
- Pear, W. Transient transfection methods for preparation of high-titer retroviral supernatants. Curr. Protoc. Mol. Biol. 2001, 9, mb0911s36. [Google Scholar] [CrossRef]
- Tom, R.; Bisson, L.; Durocher, Y. Transfection of HEK293-EBNA1 Cells in Suspension with Linear PEI for Production of Recombinant Proteins. CSH Protoc. 2008, 3, 1–4. [Google Scholar] [CrossRef][Green Version]
- Droste, M.; Tertel, T.; Jeruschke, S.; Dittrich, R.; Kontopoulou, E.; Walkenfort, B.; Börger, V.; Hoyer, P.F.; Büscher, A.K.; Thakur, B.K.; et al. Single Extracellular Vesicle Analysis Performed by Imaging Flow Cytometry and Nanoparticle Tracking Analysis Evaluate the Accuracy of Urinary Extracellular Vesicle Preparation Techniques Differently. Int. J. Mol. Sci. 2021, 22, 12436. [Google Scholar] [CrossRef]
- Miller, A.; Sitter, R.R. Using the Folded-Over 12-Run Plackett—Burman Design to Consider Interactions. Technometrics 2001, 43, 44–55. [Google Scholar] [CrossRef]
- Yu, X.; Hallett, S.G.; Sheppard, J.; Watson, A.K. Application of the Plackett-Burman experimental design to evaluate nutritional requirements for the production of Colletotrichum coccodes spores. Appl. Microbiol. Biotechnol. 1997, 47, 301–305. [Google Scholar] [CrossRef]
- Stanbury, P.F.; Whitaker, A.; Hall, S.J. Principles of Fermentation Technology, 3rd ed.; Elsevier: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Kepes, A.; Meury, J.; Robin, A.; Jimeno, J. Some Ion Transport Systems in E. coli (Transport of Potassium and of Anionic Sugars). In Biochemistry of Membrane Transport; Semenza, G., Carafoli, E., Eds.; Springer: Berlin/Heidelberg, Germany, 1977; pp. 633–647. [Google Scholar]
- Pinhal, S.; Ropers, D.; Geiselmann, J.; De Jong, H. Acetate Metabolism and the Inhibition of Bacterial Growth by Acetate. J. Bacteriol. 2019, 201, 1–47. [Google Scholar] [CrossRef] [PubMed]
- Luli, G.W.; Strohl, W.R. Comparison of growth, acetate production, and acetate inhibition of Escherichia coli strains in batch and fed-batch fermentations. Appl. Environ. Microbiol. 1990, 56, 1004–1011. [Google Scholar] [CrossRef]
- Ongkudon, C.M.; Pickering, R.; Webster, D.; Danquah, M.K. Cultivation of E. coli carrying a plasmid-based Measles vaccine construct (4.2 kbp pcDNA3F) employing medium optimisation and pH-temperature induction techniques. Microb. Cell Fact. 2011, 10, 16. [Google Scholar] [CrossRef]
- Wang, Z.; Le, G.; Shi, Y.; Węgrzyn, G. Medium design for plasmid DNA production based on stoichiometric model. Process Biochem. 2001, 36, 1085–1093. [Google Scholar] [CrossRef]
- Carnes, A.E. Fermentation Design for the Manufacture of Therapeutic Plasmid DNA; BioProcess International: Boston, MI, USA, 2005; pp. 36–44. [Google Scholar]
- Lahijani, R.; Hulley, G.; Soriano, G.; Horn, N.A.; Marquet, M. High-yield production of pBR322-derived plasmids intended for human gene therapy by employing a temperature-controllable point mutation. Hum. Gene Ther. 1996, 7, 1971–1980. [Google Scholar] [CrossRef]
- Wong, E.M.; Muesing, M.A.; Polisky, B. Temperature-sensitive copy number mutants of CoIE1 are located in an untranslated region of the plasmid genome. Proc. Natl. Acad. Sci. USA 1982, 79, 3570–3574. [Google Scholar] [CrossRef]
- Lin-Chao, S.; Chen, W.T.; Wong, T.T. High copy number of the pUC plasmid results from a Rom/Rop-suppressible point mutation in RNA II. Mol. Microbiol. 1992, 6, 3385–3393. [Google Scholar] [CrossRef]
- Carnes, A.E.; Hodgson, C.P.; Williams, J.A. Inducible Escherichia coli fermentation for increased plasmid DNA production. Biotechnol. Appl. Biochem. 2006, 45, 155–166. [Google Scholar] [CrossRef]
- Durland, R.H.; Eastman, E.M. Manufacturing and quality control of plasmid-based gene expression systems. Adv. Drug Deliv. Rev. 1998, 30, 33–48. [Google Scholar] [CrossRef]
- Satyagal, V.N.; Agrawal, P. A generalized model of plasmid replication. Biotechnol. Bioeng. 1989, 33, 1135–1144. [Google Scholar] [CrossRef]
- Seo, J.H.; Bailey, J.E. Effects of recombinant plasmid content on growth properties and cloned gene product formation in Escherichia coli. Biotechnol. Bioeng. 1985, 27, 1668–1674. [Google Scholar] [CrossRef]
- Chen, W.; Graham, C.; Ciccarelli, R.B. Automated fed-batch fermentation with feed-back controls based on dissolved oxygen (DO) and pH for production of DNA vaccines. J. Ind. Microbiol. Biotechnol. 1997, 18, 43–48. [Google Scholar] [CrossRef]
- Kim, B.G.; Shuler, M.L. Analysis of pBR322 replication kinetics and its dependency on growth rate. Biotechnol. Bioeng. 1990, 36, 233–242. [Google Scholar] [CrossRef]
- Reinikainen, P.; Korpela, K.; Nissinen, V.; Olkku, J.; Söderlund, H.; Markkanen, P. Escherichia coli plasmid production in fermenter. Biotechnol. Bioeng. 1989, 33, 386–393. [Google Scholar] [CrossRef] [PubMed]
- Namdev, P.K.; Irwin, N.; Thompson, B.G.; Gray, M.R. Effect of oxygen fluctuations on recombinant Escherichia coli fermentation. Biotechnol. Bioeng. 1993, 41, 666–670. [Google Scholar] [CrossRef] [PubMed]
- Prather, K.J.; Sagar, S.; Murphy, J.; Chartrain, M. Industrial scale production of plasmid DNA for vaccine and gene therapy: Plasmid design, production, and purification. Enzym. Microb. Technol. 2003, 33, 865–883. [Google Scholar] [CrossRef]
- Follmann, M.; Ochrombel, I.; Krämer, R.; Trötschel, C.; Poetsch, A.; Rückert, C.; Hüser, A.; Persicke, M.; Seiferling, D.; Kalinowski, J.; et al. Functional genomics of pH homeostasis in Corynebacterium glutamicum revealed novel links between pH response, oxidative stress, iron homeostasis and methionine synthesis. BMC Genom. 2009, 10, 621. [Google Scholar] [CrossRef] [PubMed]
- Cortés, J.T.; Flores, N.; Bolívar, F.; Lara, A.R.; Ramírez, O.T. Physiological effects of pH gradients on Escherichia coli during plasmid DNA production. Biotechnol. Bioeng. 2016, 113, 598–611. [Google Scholar] [CrossRef]
- O’Mahony, K.; Freitag, R.; Hilbrig, F.; Müller, P.; Schumacher, I. Strategies for high titre plasmid DNA production in Escherichia coli DH5α. Process Biochem. 2007, 42, 1039–1049. [Google Scholar] [CrossRef]
- Derouazi, M.; Girard, P.; van Tilborgh, F.; Iglesias, K.; Muller, N.; Bertschinger, M.; Wurm, F.M. Serum-free large-scale transient transfection of CHO cells. Biotechnol. Bioeng. 2004, 87, 537–545. [Google Scholar] [CrossRef]
- Yang, S.; Shi, H.; Chu, X.; Zhou, X.; Sun, P. A rapid and efficient polyethylenimine-based transfection method to prepare lentiviral or retroviral vectors: Useful for making iPS cells and transduction of primary cells. Biotechnol. Lett. 2016, 38, 1631–1641. [Google Scholar] [CrossRef]
- Backliwal, G.; Hildinger, M.; Hasija, V.; Wurm, F.M. High-density transfection with HEK-293 cells allows doubling of transient titers and removes need for a priori DNA complex formation with PEI. Biotechnol. Bioeng. 2008, 99, 721–727. [Google Scholar] [CrossRef] [PubMed]
- Schlaeger, E.J.; Christensen, K. Transient gene expression in mammalian cells grown in serum-free suspension culture. Cytotechnology 1999, 30, 71–83. [Google Scholar] [CrossRef]
- Nadeau, I.; Kamen, A. Production of adenovirus vector for gene therapy. Biotechnol. Adv. 2003, 20, 475–489. [Google Scholar] [CrossRef] [PubMed]
- Huynh, H.T.; Tran, T.T.B.; Chan, L.C.L.; Nielsen, L.K.; Reid, S. Effect of the peak cell density of recombinant AcMNPV-infected Hi5 cells on baculovirus yields. Appl. Microbiol. Biotechnol. 2015, 99, 1687–1700. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Dalby, B.; Chen, W.; Kilzer, J.M.; Chiou, H.C. Transient transfection factors for high-level recombinant protein production in suspension cultured mammalian cells. Mol. Biotechnol. 2008, 39, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Fischer, D.; Bieber, T.; Li, Y.; Elsässer, H.P.; Kissel, T. A novel non-viral vector for DNA delivery based on low molecular weight, branched polyethylenimine: Effect of molecular weight on transfection efficiency and cytotoxicity. Pharm. Res. 1999, 16, 1273–1279. [Google Scholar] [CrossRef]
- Ducommun, P.; Kadouri, A.; Stockar, U.; Marison, I.W. On-line determination of animal cell concentration in two industrial high-density culture processes by dielectric spectroscopy. Biotechnol. Bioeng. 2002, 77, 316–323. [Google Scholar] [CrossRef]
- Ducommun, P.; Bolzonella, I.; Rhiel, M.; Pugeaud, P.; Von Stockar, U.; Marison, I.W. On-line determination of animal cell concentration. Biotechnol. Bioeng. 2001, 72, 515–522. [Google Scholar] [CrossRef]
- Choosakoonkriang, S.; Lobo, B.A.; Koe, G.S.; Koe, J.G.; Middaugh, C.R. Biophysical characterization of PEI/DNA complexes. J. Pharm. Sci. 2003, 92, 1710–1722. [Google Scholar] [CrossRef] [PubMed]
- Kircheis, R.; Schüller, S.; Brunner, S.; Ogris, M.; Heider, K.H.; Zauner, W.; Wagner, E. Polycation-based DNA complexes for tumor-targeted gene deliveryin vivo. J. Gene Med. 1999, 1, 111–120. [Google Scholar] [CrossRef]
- Ogris, M.; Brunner, S.; Schüller, S.; Kircheis, R.; Wagner, E. PEGylated DNA/transferrin-PEI complexes: Reduced interaction with blood components, extended circulation in blood and potential for systemic gene delivery. Gene Ther. 1999, 6, 595–605. [Google Scholar] [CrossRef]
- Ansorge, S.; Lanthier, S.; Transfiguracion, J.; Henry, O.; Kamen, A. Monitoring lentiviral vector production kinetics using online permittivity measurements. Biochem. Eng. J. 2011, 54, 16–25. [Google Scholar] [CrossRef]
- Patel, P.; Markx, G.H. Dielectric measurement of cell death. Enzym. Microb. Technol. 2008, 43, 463–470. [Google Scholar] [CrossRef]
- Ron, A.; Fishelson, N.; Croitoriu, N.; Benayahu, D.; Shacham-Diamand, Y. Theoretical examination of aggregation effect on the dielectric characteristics of spherical cellular suspension. Biophys. Chem. 2009, 140, 39–50. [Google Scholar] [CrossRef]
- Ansorge, S.; Esteban, G.; Schmid, G. Multifrequency permittivity measurements enable on-line monitoring of changes in intracellular conductivity due to nutrient limitations during batch cultivations of CHO cells. Biotechnol. Prog. 2010, 26, 272–283. [Google Scholar] [CrossRef] [PubMed]
- Ansorge, S.; Esteban, G.; Schmid, G. On-line monitoring of responses to nutrient feed additions by multi-frequency permittivity measurements in fed-batch cultivations of CHO cells. Cytotechnology 2010, 62, 121–132. [Google Scholar] [CrossRef]
- Liste-Calleja, L.; Lecina, M.; Lopez-Repullo, J.; Albiol, J.; Solà, C.; Cairó, J.J. Lactate and glucose concomitant consumption as a self-regulated pH detoxification mechanism in HEK293 cell cultures. Appl. Microbiol. Biotechnol. 2015, 99, 9951–9960. [Google Scholar] [CrossRef]
- Petiot, E.; El-Wajgali, A.; Esteban, G.; Gény, C.; Pinton, H.; Marc, A. Real-time monitoring of adherent Vero cell density and apoptosis in bioreactor processes. Cytotechnology 2012, 64, 429–441. [Google Scholar] [CrossRef]
- Ma, F.; Zhang, A.; Chang, D.; Velev, O.D.; Wiltberger, K.; Kshirsagar, R. Real-time monitoring and control of CHO cell apoptosis by in situ multifrequency scanning dielectric spectroscopy. Proc. Biochem. 2019, 80, 138–145. [Google Scholar] [CrossRef]
- González-Correa, C.A.; Colina-Gallo, E.; Miranda-Mercado, D.A. The alpha parameter of the Cole-Cole model as an indicator of fibromyalgia. J. Phys. Conf. Ser. 2019, 1272, 12003. [Google Scholar] [CrossRef]
- Boigard, H.; Alimova, A.; Martin, G.R.; Katz, A.; Gottlieb, P.; Galarza, J.M. Zika virus-like particle (VLP) based vaccine. PLoS Negl. Trop. Dis. 2017, 11, e0005608. [Google Scholar] [CrossRef]
- Kümmerer, B.M.; Rice, C.M. Mutations in the yellow fever virus nonstructural protein NS2A selectively block production of infectious particles. J. Virol. 2002, 76, 4773–4784. [Google Scholar] [CrossRef] [PubMed]
- Bonaldo, M.C.; Sequeira, P.C.; Galler, R. The yellow fever 17D virus as a platform for new live attenuated vaccines. Hum. Vaccines Immunother. 2014, 10, 1256–1265. [Google Scholar] [CrossRef]
- Beeck, A.; Molenkamp, R.; Caron, M.; Ben Younes, A.; Bredenbeek, P.; Dubuisson, J. Role of the transmembrane domains of prM and E proteins in the formation of yellow fever virus envelope. J. Virol. 2003, 77, 813–820. [Google Scholar] [CrossRef]
- Nooraei, S.; Bahrulolum, H.; Hoseini, Z.S.; Katalani, C.; Hajizade, A.; Easton, A.J.; Ahmadian, G. Virus-like particles: Preparation, immunogenicity and their roles as nanovaccines and drug nanocarriers. J. Nanobiotechnol. 2021, 19, 59. [Google Scholar] [CrossRef] [PubMed]
- Yan, Q.; Wu, L.; Chen, L.; Qin, Y.; Pan, Z.; Chen, M. Vesicular stomatitis virus-based vaccines expressing EV71 virus-like particles elicit strong immune responses and protect newborn mice from lethal challenges. Vaccine 2016, 34, 4196–4204. [Google Scholar] [CrossRef]
- Steppert, P.; Burgstaller, D.; Klausberger, M.; Berger, E.; Pereira Aguilar, P.; Schneider, T.A.; Kramberger, P.; Tover, A.; Nöbauer, K.; Razzazi-Fazeli, E.; et al. Purification of HIV-1 gag virus-like particles and separation of other extracellular particles. J. Chromatogr. A 2016, 1455, 93–101. [Google Scholar] [CrossRef]
- Lin, S.Y.; Chiu, H.Y.; Chiang, B.L.; Hu, Y.C. Development of EV71 virus-like particle purification processes. Vaccine 2015, 33, 5966–5973. [Google Scholar] [CrossRef]
- Negrete, A.; Pai, A.; Shiloach, J. Use of hollow fiber tangential flow filtration for the recovery and concentration of HIV virus-like particles produced in insect cells. J. Virol. Methods 2014, 195, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Staubach, S.; Bauer, F.N.; Tertel, T.; Börger, V.; Stambouli, O.; Salzig, D.; Giebel, B. Scaled preparation of extracellular vesicles from conditioned media. Adv. Drug Deliv. Rev. 2021, 177, 113940. [Google Scholar] [CrossRef] [PubMed]
- Garg, H.; Mehmetoglu-Gurbuz, T.; Joshi, A. Virus Like Particles (VLP) as multivalent vaccine candidate against Chikungunya, Japanese Encephalitis, Yellow Fever and Zika Virus. Sci. Rep. 2020, 10, 4017. [Google Scholar] [CrossRef] [PubMed]
- Krol, E.; Brzuska, G.; Szewczyk, B. Production and Biomedical Application of Flavivirus-like Particles. Trends Biotechnol. 2019, 37, 1202–1216. [Google Scholar] [CrossRef] [PubMed]
- Hausig-Punke, F.; Dekevic, G.; Sobotta, F.H.; Solomun, J.I.; Richter, F.; Salzig, D.; Traeger, A.; Brendel, J.C. Efficient Transfection via an Unexpected Mechanism by Near Neutral Polypiperazines with Tailored Response to Endosomal pH. Macromol. Biosci. 2023, 23, e2200517. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Standard | 8 | 6 | 5 | 1 | 7 | 3 | 4 | 12 | 11 | 9 | 10 | 2 | Center Point | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Run | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 |
| LB | 1 | −1 | −1 | 1 | 1 | 1 | −1 | −1 | 1 | 1 | −1 | −1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Meat-peptone | 1 | −1 | −1 | 1 | −1 | −1 | 1 | −1 | −1 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Casein-peptone | −1 | −1 | 1 | −1 | −1 | 1 | −1 | −1 | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Yeast extract | −1 | 1 | −1 | 1 | −1 | 1 | 1 | −1 | 1 | −1 | 1 | −1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Glucose | −1 | −1 | 1 | 1 | 1 | −1 | 1 | −1 | 1 | −1 | −1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Glycerol | 1 | 1 | 1 | 1 | −1 | 1 | −1 | −1 | −1 | −1 | −1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Phosphate * | −1 | 1 | −1 | −1 | 1 | 1 | 1 | −1 | −1 | 1 | −1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| NaCl | 1 | −1 | 1 | −1 | 1 | 1 | 1 | −1 | −1 | −1 | 1 | −1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| MgSO4 | 1 | 1 | 1 | −1 | −1 | −1 | 1 | −1 | 1 | 1 | −1 | −1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Dummy 1 | −1 | 1 | 1 | 1 | 1 | −1 | −1 | −1 | −1 | 1 | 1 | −1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Dummy 2 | 1 | 1 | −1 | −1 | 1 | −1 | −1 | −1 | 1 | −1 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dekevic, G.; Tertel, T.; Tasto, L.; Schmidt, D.; Giebel, B.; Czermak, P.; Salzig, D. A Bioreactor-Based Yellow Fever Virus-like Particle Production Process with Integrated Process Analytical Technology Based on Transient Transfection. Viruses 2023, 15, 2013. https://doi.org/10.3390/v15102013
Dekevic G, Tertel T, Tasto L, Schmidt D, Giebel B, Czermak P, Salzig D. A Bioreactor-Based Yellow Fever Virus-like Particle Production Process with Integrated Process Analytical Technology Based on Transient Transfection. Viruses. 2023; 15(10):2013. https://doi.org/10.3390/v15102013
Chicago/Turabian StyleDekevic, Gregor, Tobias Tertel, Lars Tasto, Deborah Schmidt, Bernd Giebel, Peter Czermak, and Denise Salzig. 2023. "A Bioreactor-Based Yellow Fever Virus-like Particle Production Process with Integrated Process Analytical Technology Based on Transient Transfection" Viruses 15, no. 10: 2013. https://doi.org/10.3390/v15102013
APA StyleDekevic, G., Tertel, T., Tasto, L., Schmidt, D., Giebel, B., Czermak, P., & Salzig, D. (2023). A Bioreactor-Based Yellow Fever Virus-like Particle Production Process with Integrated Process Analytical Technology Based on Transient Transfection. Viruses, 15(10), 2013. https://doi.org/10.3390/v15102013