

A Repeat Pattern of Founder Events for SARS-CoV-2 Variants in Alaska
 ,
,  ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Retrieving and Analyzing SARS-CoV-2 Sequence Data for Alaska
2.2. SARS-CoV-2 Sequence Data for the Contiguous United States
2.3. Visualizations, Statistical Analyses, and Nextstrain Build
3. Results and Discussion
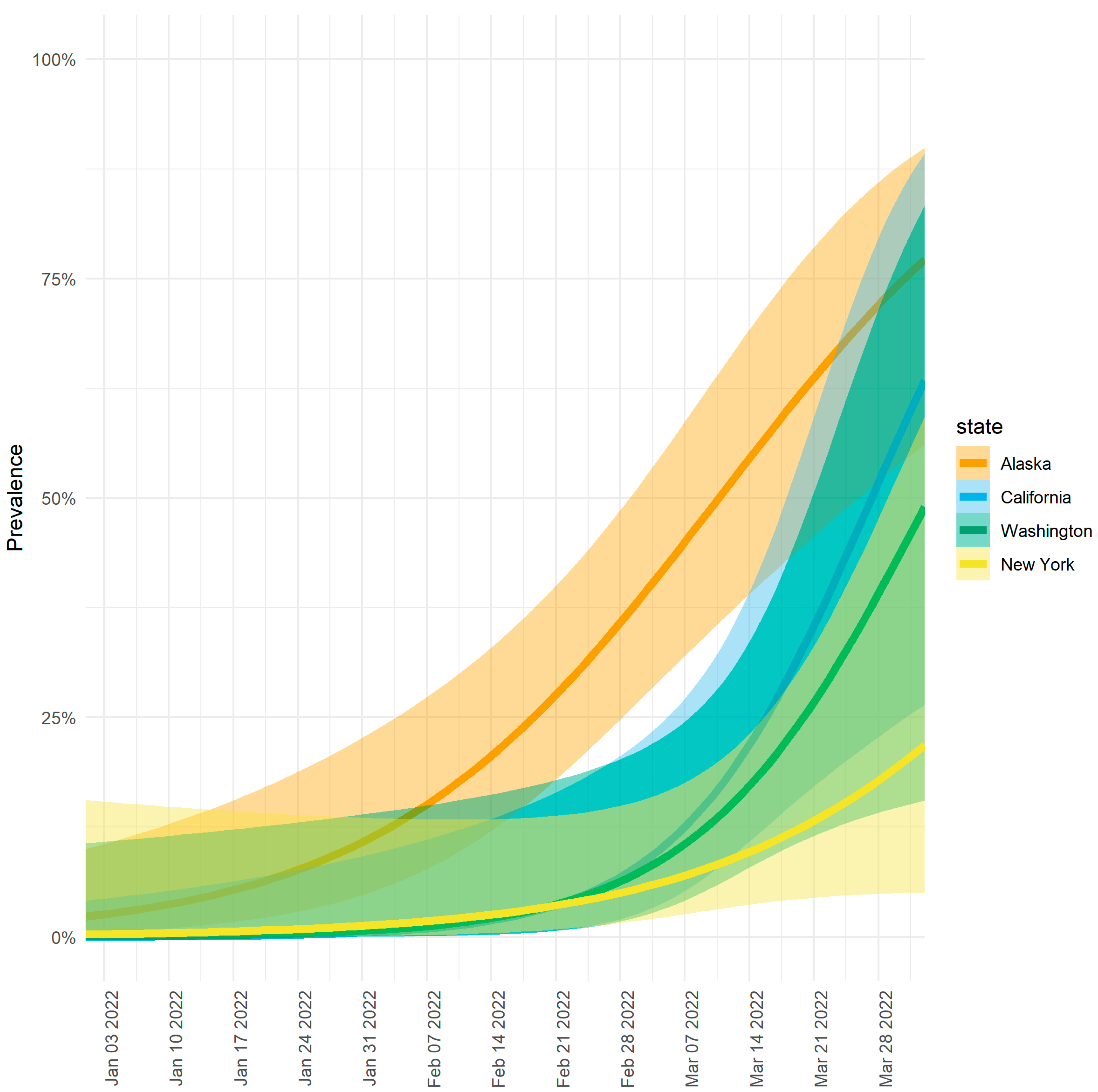
3.1. Higher Prevalence of Omicron Lineage BA.2.3 in Alaska vs. the Contiguous United States
3.2. Modeling Shows Variable Emergence of BA.2.3 across Alaska
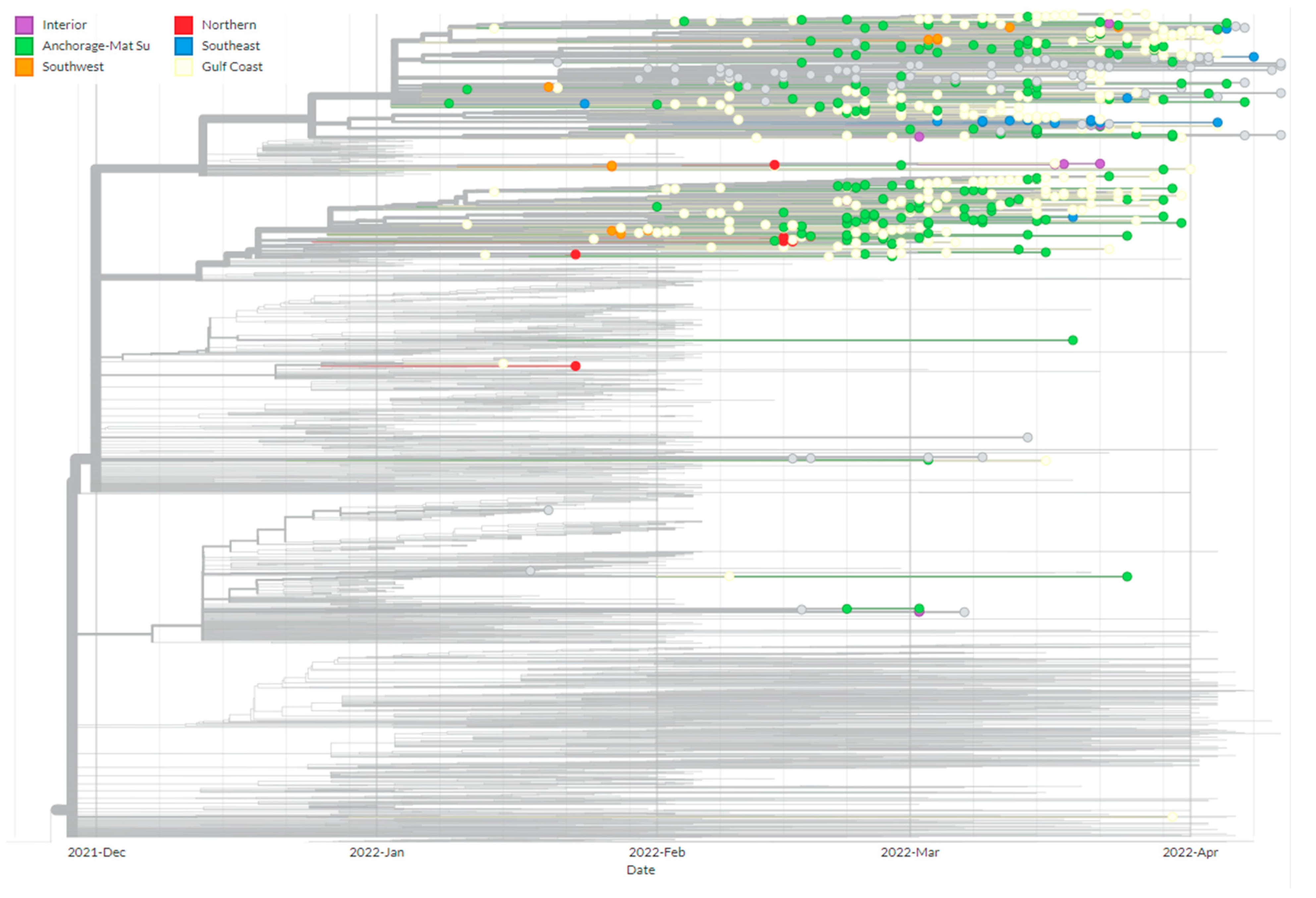
3.3. Phylogenetics of BA.2.3 Provides Evidence of Multiple Introduction Events
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gangavarapu, K.; Latif, A.A.; Mullen, J.L.; Alkuzweny, M.; Hufbauer, E.; Tsueng, G.; Haag, E.; Zeller, M.; Aceves, C.M.; Zaiets, K.; et al. Outbreak.info genomic reports: Scalable and dynamic surveillance of SARS-CoV-2 variants and mutations. medRxiv 2022. [Google Scholar] [CrossRef]
- Alcantara, L.C.J.; Nogueira, E.; Shuab, G.; Tosta, S.; Fristch, H.; Pimentel, V.; Souza-Neto, J.A.; Coutinho, L.L.; Fukumasu, H.; Sampaio, S.C.; et al. SARS-CoV-2 epidemic in Brazil: How the displacement of variants has driven distinct epidemic waves. Virus Res. 2022, 315, 198785. [Google Scholar] [CrossRef]
- Kirby, T. New variant of SARS-CoV-2 in UK causes surge of COVID-19. Lancet Respir. Med. 2021, 9, e20–e21. [Google Scholar] [CrossRef]
- Saha, S.; Tanmoy, A.M.; Tanni, A.A.; Goswami, S.; Sium, S.M.A.; Saha, S.; Islam, S.; Hooda, Y.; Malaker, A.R.; Anik, A.M.; et al. New waves, new variants, old inequity: A continuing COVID-19 crisis. BMJ Glob. Health 2021, 6, e007031. [Google Scholar] [CrossRef]
- Tao, K.; Tzou, P.L.; Nouhin, J.; Gupta, R.K.; de Oliveira, T.; Kosakovsky Pond, S.L.; Fera, D.; Shafer, R.W. The biological and clinical significance of emerging SARS-CoV-2 variants. Nat. Rev. Genet. 2021, 22, 757–773. [Google Scholar] [CrossRef]
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef]
- Volz, E.; Mishra, S.; Chand, M.; Barrett, J.C.; Johnson, R.; Geidelberg, L.; Hinsley, W.R.; Laydon, D.J.; Dabrera, G.; O’Toole, Á.; et al. Assessing transmissibility of SARS-CoV-2 lineage B.1.1.7 in England. Nature 2021, 593, 266–269. [Google Scholar] [CrossRef]
- Haan, T.J.; Smith, L.K.; DeRonde, S.; House, E.; Zidek, J.; Puhak, D.; Redlinger, M.; Parker, J.; Barnes, B.M.; Burkhead, J.L.; et al. Pattern of SARS-CoV-2 variant B.1.1.519 emergence in Alaska. Sci. Rep. 2022, 12, 20662. [Google Scholar] [CrossRef]
- Rodríguez-Maldonado, A.P.; Vázquez-Pérez, J.A.; Cedro-Tanda, A.; Taboada, B.; Boukadida, C.; Wong-Arámbula, C.; Nuñez-García, T.E.; Cruz-Ortiz, N.; Barrera-Badillo, G.; Hernández-Rivas, L.; et al. Emergence and spread of the potential variant of interest (VOI) B.1.1.519 of SARS-CoV-2 predominantly present in Mexico. Arch. Virol. 2021, 166, 3173–3177. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. Science Brief: Omicron (B.1.1.529) Variant. In CDC COVID-19 Science Briefs; CDC: Atlanta, GA, USA, 2021. [Google Scholar]
- Stegger, M.; Edslev, S.M.; Sieber, R.N.; Cäcilia Ingham, A.; Ng, K.L.; Tang, M.-H.E.; Alexandersen, S.; Fonager, J.; Legarth, R.; Utko, M.; et al. Occurrence and significance of Omicron BA.1 infection followed by BA.2 reinfection. medRxiv 2022. [Google Scholar] [CrossRef]
- Iketani, S.; Liu, L.; Guo, Y.; Liu, L.; Chan, J.F.W.; Huang, Y.; Wang, M.; Luo, Y.; Yu, J.; Chu, H.; et al. Antibody evasion properties of SARS-CoV-2 Omicron sublineages. Nature 2022, 604, 553–556. [Google Scholar] [CrossRef]
- Elbe, S.; Buckland-Merrett, G. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Glob. Chall. 2017, 1, 33–46. [Google Scholar] [CrossRef]
- Shu, Y.; McCauley, J. GISAID: Global initiative on sharing all influenza data – from vision to reality. Eurosurveillance 2017, 22, 30494. [Google Scholar] [CrossRef]
- Khare, S.; Gurry, C.; Freitas, L.; Schultz, M.B.; Bach, G.; Diallo, A.; Akite, N.; Ho, J.; Lee, R.T.C.; Yeo, W.; et al. GISAID’s Role in Pandemic Response. China CDC Wkly. 2021, 3, 1049–1051. [Google Scholar] [CrossRef]
- O’Toole, Á.; Scher, E.; Underwood, A.; Jackson, B.; Hill, V.; McCrone, J.T.; Colquhoun, R.; Ruis, C.; Abu-Dahab, K.; Taylor, B.; et al. Assignment of epidemiological lineages in an emerging pandemic using the pangolin tool. Virus Evol. 2021, 7, veab064. [Google Scholar] [CrossRef] [PubMed]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef] [PubMed]
- Attwood, S.W.; Hill, S.C.; Aanensen, D.M.; Connor, T.R.; Pybus, O.G. Phylogenetic and phylodynamic approaches to understanding and combating the early SARS-CoV-2 pandemic. Nat. Rev. Genet. 2022, 23, 547–562. [Google Scholar] [CrossRef]
- Rambaut, A.; Posada, D.; Crandall, K.A.; Holmes, E.C. The causes and consequences of HIV evolution. Nat. Rev. Genet. 2004, 5, 52–61. [Google Scholar] [CrossRef]
- Ruan, Y.; Luo, Z.; Tang, X.; Li, G.; Wen, H.; He, X.; Lu, X.; Lu, J.; Wu, C.-I. On the founder effect in COVID-19 outbreaks: How many infected travelers may have started them all? Natl. Sci. Rev. 2021, 8, nwaa246. [Google Scholar] [CrossRef]
- Hodcroft, E.B.; Zuber, M.; Nadeau, S.; Vaughan, T.G.; Crawford, K.H.D.; Althaus, C.L.; Reichmuth, M.L.; Bowen, J.E.; Walls, A.C.; Corti, D.; et al. Spread of a SARS-CoV-2 variant through Europe in the summer of 2020. Nature 2021, 595, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Ozer, E.A.; Simons, L.M.; Adewumi, O.M.; Fowotade, A.A.; Omoruyi, E.C.; Adeniji, J.A.; Olayinka, O.A.; Dean, T.J.; Zayas, J.; Bhimalli, P.P.; et al. Multiple expansions of globally uncommon SARS-CoV-2 lineages in Nigeria. Nat. Commun. 2022, 13, 688. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.W.; Tambyah, P.A.; Hui, D.S.C. Emergence of a new SARS-CoV-2 variant in the UK. J. Infect. 2021, 82, e27–e28. [Google Scholar] [CrossRef] [PubMed]
- Grubaugh, N.D.; Hanage, W.P.; Rasmussen, A.L. Making Sense of Mutation: What D614G Means for the COVID-19 Pandemic Remains Unclear. Cell 2020, 182, 794–795. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. COVID Data Tracker. Available online: https://covid.cdc.gov/covid-data-tracker (accessed on 11 May 2022).
- Alaska Department of Labor and Workforce Development. 2020 Population Estimates by Borough, Census Area, and Economic Region. 2021. Available online: https://live.laborstats.alaska.gov/pop/index.cfm (accessed on 5 November 2021).






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haan, T.J.; Smith, L.K.; DeRonde, S.; House, E.; Zidek, J.; Puhak, D.; Mullen, L.; Redlinger, M.; Parker, J.; Barnes, B.M.; et al. A Repeat Pattern of Founder Events for SARS-CoV-2 Variants in Alaska. Viruses 2023, 15, 222. https://doi.org/10.3390/v15010222
Haan TJ, Smith LK, DeRonde S, House E, Zidek J, Puhak D, Mullen L, Redlinger M, Parker J, Barnes BM, et al. A Repeat Pattern of Founder Events for SARS-CoV-2 Variants in Alaska. Viruses. 2023; 15(1):222. https://doi.org/10.3390/v15010222
Chicago/Turabian StyleHaan, Tracie J., Lisa K. Smith, Stephanie DeRonde, Elva House, Jacob Zidek, Diana Puhak, Logan Mullen, Matthew Redlinger, Jayme Parker, Brian M. Barnes, and et al. 2023. "A Repeat Pattern of Founder Events for SARS-CoV-2 Variants in Alaska" Viruses 15, no. 1: 222. https://doi.org/10.3390/v15010222
APA StyleHaan, T. J., Smith, L. K., DeRonde, S., House, E., Zidek, J., Puhak, D., Mullen, L., Redlinger, M., Parker, J., Barnes, B. M., Burkhead, J. L., Knall, C., Bortz, E., Chen, J., & Drown, D. M. (2023). A Repeat Pattern of Founder Events for SARS-CoV-2 Variants in Alaska. Viruses, 15(1), 222. https://doi.org/10.3390/v15010222