
A Genome of Temperate Enterococcus Bacteriophage Placed in a Space of Pooled Viral Dark Matter Sequences
 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Isolation and Description of Enterococcus Phage VEsP-1
2.2. Genome Network Analysis
2.3. Phylogenomic Analysis
2.4. Functional Analysis
3. Results
3.1. Phenotype of Enterococcus Phage VEsP-1
3.2. Taxonomic Analysis
3.3. The Proposed Genus “Vespunovirus”
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sausset, R.; Petit, M.A.; Gaboriau-Routhiau, V.; De Paepe, M. New insights into intestinal phages. Mucosal Immunol. 2020, 13, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Minot, S.; Bryson, A.; Chehoud, C.; Wu, G.D.; Lewis, J.D.; Bushman, F.D. Rapid evolution of the human gut virome. Proc. Natl. Acad. Sci. USA 2013, 110, 12450–12455. [Google Scholar] [CrossRef] [PubMed]
- Davies, E.V.; Winstanley, C.; Fothergill, J.L.; James, C.E. The role of temperate bacteriophages in bacterial infection. FEMS Microbiol. Lett. 2016, 363, fnw015. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.N.; Sheriff, E.K.; Duerkop, B.A.; Chatterjee, A. Let me upgrade you: Impact of mobile genetic elements on enterococcal adaptation and evolution. J. Bacteriol. 2021, 203, e0017721. [Google Scholar] [CrossRef] [PubMed]
- Minot, S.; Sinha, R.; Chen, J.; Li, H.; Keilbaugh, S.A.; Wu, G.D.; Lewis, J.D.; Bushman, F.D. The human gut virome: Inter-individual variation and dynamic response to diet. Genome Res. 2011, 21, 1616–1625. [Google Scholar] [CrossRef]
- Liang, G.; Conrad, M.A.; Kelsen, J.R.; Kessler, L.R.; Breton, J.; Albenberg, L.G.; Marakos, S.; Galgano, A.; Devas, N.; Erlichman, J.; et al. Dynamics of the stool virome in very early- onset inflammatory bowel disease. J. Crohns Colitis 2020, 14, 1600–1610. [Google Scholar] [CrossRef]
- Clooney, A.G.; Sutton, T.D.S.; Shkoporov, A.N.; Holohan, R.K.; Daly, K.M.; O’Regan, O.; Ryan, F.J.; Draper, L.A.; Plevy, S.E.; Ross, R.P.; et al. Whole-virome analysis sheds light on viral dark matter in inflammatory bowel disease. Cell Host Microbe 2019, 26, 764–778.e5. [Google Scholar] [CrossRef]
- Lang, S.; Demir, M.; Martin, A.; Jiang, L.; Zhang, X.; Duan, Y.; Gao, B.; Wisplinghoff, H.; Kasper, P.; Roderburg, C.; et al. Intestinal virome signature associated with severity of nonalcoholic fatty liver disease. Gastroenterology 2020, 159, 1839–1852. [Google Scholar] [CrossRef]
- Jiang, L.; Lang, S.; Duan, Y.; Zhang, X.; Gao, B.; Chopyk, J.; Schwanemann, L.K.; Ventura-Cots, M.; Bataller, R.; Bosques-Padilla, F.; et al. Intestinal virome in patients with alcoholic hepatitis. Hepatology 2020, 72, 2182–2196. [Google Scholar] [CrossRef]
- Nakatsu, G.; Zhou, H.; Wu, W.K.K.; Wong, S.H.; Coker, O.O.; Dai, Z.; Li, X.; Szeto, C.-H.; Sugimura, N.; Lam, T.Y.-T.; et al. Alterations in enteric virome are associated with colorectal cancer and survival outcomes. Gastroenterology 2018, 155, 529–541.e5. [Google Scholar] [CrossRef]
- Aggarwala, V.; Liang, G.; Bushman, F.D. Viral communities of the human gut: Metagenomic analysis of composition and dynamics. Mob. DNA 2017, 8, 12. [Google Scholar] [CrossRef]
- Fitzgerald, C.B.; Shkoporov, A.N.; Upadrasta, A.; Khokhlova, E.V.; Ross, R.P.; Hill, C. Probing the “dark matter” of the human gut phageome: Culture assisted metagenomics enables rapid discovery and host-linking for novel bacteriophages. Front. Cell. Infect. Microbiol. 2021, 11, 616918. [Google Scholar] [CrossRef]
- Shkoporov, A.N.; Hill, C. Bacteriophages of the human gut: The “known unknown” of the microbiome. Cell Host Microbe 2019, 25, 195–209. [Google Scholar] [CrossRef]
- Deboutte, W.; Beller, L.; Yinda, C.K.; Maes, P.; de Graaf, D.C.; Matthijnssens, J. Honey-bee-associated prokaryotic viral communities reveal wide viral diversity and a profound metabolic coding potential. Proc. Natl. Acad. Sci. USA 2020, 117, 10511–10519. [Google Scholar] [CrossRef]
- Tkachev, P.V.; Pchelin, I.M.; Azarov, D.V.; Gorshkov, A.N.; Shamova, O.V.; Dmitriev, A.V.; Goncharov, A.E. Two novel lytic bacteriophages infecting Enterococcus spp. are promising candidates for targeted antibacterial therapy. Viruses 2022, 14, 831. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef]
- Overbeek, R.; Olson, R.; Pusch, G.D.; Olsen, G.J.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Parrello, B.; Shukla, M.; et al. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 2014, 42, D206–D214. [Google Scholar] [CrossRef]
- Brettin, T.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Olsen, G.J.; Olson, R.; Overbeek, R.; Parrello, B.; Pusch, G.D.; et al. RASTtk: A modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 2015, 5, 8365. [Google Scholar] [CrossRef]
- Bolduc, B.; Jang, H.B.; Doulcier, G.; You, Z.Q.; Roux, S.; Sullivan, M.B. vConTACT: An iVirus tool to classify double-stranded DNA viruses that infect Archaea and Bacteria. PeerJ. 2017, 5, e3243. [Google Scholar] [CrossRef]
- Buchfink, B.; Reuter, K.; Drost, H.G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods 2021, 18, 366–368. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Göker, M. VICTOR: Genome-based phylogeny and classification of prokaryotic viruses. Bioinformatics 2017, 33, 3396–3404. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Auch, A.F.; Klenk, H.-P.; Göker, M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics 2013, 14, 60. [Google Scholar] [CrossRef]
- Lefort, V.; Desper, R.; Gascuel, O. FastME 2.0: A comprehensive, accurate, and fast distance-based phylogeny inference program. Mol. Biol. Evol. 2015, 32, 2798–2800. [Google Scholar] [CrossRef]
- Göker, M.; García-Blázquez, G.; Voglmayr, H.; Tellería, M.T.; Martín, M.P. Molecular taxonomy of phytopathogenic fungi: A case study in Peronospora. PLoS ONE 2009, 4, e6319. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Hahnke, R.L.; Petersen, J.; Scheuner, C.; Michael, V.; Fiebig, A.; Rohde, C.; Rohde, M.; Fartmann, B.; Goodwin, L.A.; et al. Complete genome sequence of DSM 30083T, the type strain (U5/41T) of Escherichia coli, and a proposal for delineating subspecies in microbial taxonomy. Stand. Genomic Sci. 2014, 9, 2. [Google Scholar] [CrossRef]
- Moraru, C.; Varsani, A.; Kropinski, A.M. VIRIDIC-A Novel Tool to Calculate the Intergenomic Similarities of Prokaryote-Infecting Viruses. Viruses 2020, 12, 1268. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate Maximum-Likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef]
- Gouy, M.; Guindon, S.; Gascuel, O. SeaView version 4: A multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 2010, 27, 221–224. [Google Scholar] [CrossRef]
- Lemoine, F.; Domelevo Entfellner, J.-B.; Wilkinson, E.; Correia, D.; Dávila Felipe, M.; De Oliveira, T.; Gascuel, O. Renewing Felsenstein’s phylogenetic bootstrap in the era of big data. Nature 2018, 556, 452–456. [Google Scholar] [CrossRef]
- Contreras-Moreira, B.; Vinuesa, P. GET_HOMOLOGUES, a versatile software package for scalable and robust microbial pangenome analysis. Appl. Environ. Microbiol. 2013, 79, 7696–7701. [Google Scholar] [CrossRef]
- Lee, D.Y.; Bartels, C.; McNair, K.; Edwards, R.A.; Swairjo, M.A.; Luque, A. Predicting the capsid architecture of phages from metagenomic data. Comput. Struct. Biotechnol. J. 2022, 20, 721–732. [Google Scholar] [CrossRef]
- Seemann, T. ABRicate. Available online: https://github.com/tseemann/abricate (accessed on 22 August 2022).
- Gupta, S.K.; Padmanabhan, B.R.; Diene, S.M.; Lopez-Rojas, R.; Kempf, M.; Landraud, L.; Rolain, J.M. ARG-ANNOT, a new bioinformatic tool to discover antibiotic resistance genes in bacterial genomes. Antimicrob. Agents Chemother. 2014, 58, 212–220. [Google Scholar] [CrossRef]
- Jia, B.; Raphenya, A.R.; Alcock, B.; Waglechner, N.; Guo, P.; Tsang, K.K.; Lago, B.A.; Dave, B.M.; Pereira, S.; Sharma, A.N.; et al. CARD 2017: Expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 2017, 45, D566–D573. [Google Scholar] [CrossRef]
- Doster, E.; Lakin, S.M.; Dean, C.J.; Wolfe, C.; Young, J.G.; Boucher, C.; Belk, K.E.; Noyes, N.R.; Morley, P.S. MEGARes 2.0: A database for classification of antimicrobial drug, biocide and metal resistance determinants in metagenomic sequence data. Nucleic Acids Res. 2020, 48, D561–D569. [Google Scholar] [CrossRef]
- Feldgarden, M.; Brover, V.; Haft, D.H.; Prasad, A.B.; Slotta, D.J.; Tolstoy, I.; Tyson, G.H.; Zhao, S.; Hsu, C.H.; McDermott, P.F. Validating the AMRFinder tool and resistance gene database by using antimicrobial resistance genotype-phenotype correlations in a collection of isolates. Antimicrob. Agents Chemother. 2019, 63, e00483-19. [Google Scholar] [CrossRef]
- Zankari, E.; Hasman, H.; Cosentino, S.; Vestergaard, M.; Rasmussen, S.; Lund, O.; Aarestrup, F.M.; Larsen, M.V. Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 2012, 67, 2640–2644. [Google Scholar] [CrossRef]
- Chen, L.; Zheng, D.; Liu, B.; Yang, J.; Jin, Q. VFDB 2016: Hierarchical and refined dataset for big data analysis—10 years on. Nucleic Acids Res. 2016, 44, D694–D697. [Google Scholar] [CrossRef]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007, 8, 4. [Google Scholar] [CrossRef] [PubMed]
- Zaharieva, N.; Dimitrov, I.; Flower, D.; Doytchinova, I. Immunogenicity prediction by VaxiJen: A ten year overview. J. Proteom. Bioinform. 2017, 10, 11. [Google Scholar] [CrossRef]
- Herrera-Bravo, J.; Farías, J.G.; Contreras, F.P.; Herrera-Belén, L.; Norambuena, J.A.; Beltrán, J.F. VirVACPRED: A web server for prediction of protective viral antigens. Int. J. Pept. Res. Ther. 2022, 28, 35. [Google Scholar] [CrossRef] [PubMed]
- Chen, H. VennDiagram: Generate High-Resolution Venn and Euler Plots. R Package Version 1.7.3. 2022. Available online: https://CRAN.R-project.org/package=VennDiagram (accessed on 1 July 2022).
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021; Available online: https://www.R-project.org/ (accessed on 8 November 2021).
- Tisza, M.J.; Buck, C.B. A catalog of tens of thousands of viruses from human metagenomes reveals hidden associations with chronic diseases. Proc. Natl. Acad. Sci. USA 2021, 118, e2023202118. [Google Scholar] [CrossRef] [PubMed]
- Kilcher, S.; Loessner, M.J.; Klumpp, J. Brochothrix thermosphacta bacteriophages feature heterogeneous and highly mosaic genomes and utilize unique prophage insertion sites. J. Bacteriol. 2010, 192, 5441–5453. [Google Scholar] [CrossRef]
- Brøndsted, L.; Ostergaard, S.; Pedersen, M.; Hammer, K.; Vogensen, F.K. Analysis of the complete DNA sequence of the temperate bacteriophage TP901-1: Evolution, structure, and genome organization of lactococcal bacteriophages. Virology 2001, 283, 93–109. [Google Scholar] [CrossRef]
- Garneau, J.R.; Depardieu, F.; Fortier, L.C.; Bikard, D.; Monot, M. PhageTerm: A tool for fast and accurate determination of phage termini and packaging mechanism using next-generation sequencing data. Sci. Rep. 2017, 7, 8292. [Google Scholar] [CrossRef]
- Sullivan, M.J.; Petty, N.K.; Beatson, S.A. Easyfig: A genome comparison visualizer. Bioinformatics 2011, 27, 1009–1010. [Google Scholar] [CrossRef]
- Denes, T.; Vongkamjan, K.; Ackermann, H.W.; Moreno Switt, A.I.; Wiedmann, M.; den Bakker, H.C. Comparative genomic and morphological analyses of Listeria phages isolated from farm environments. Appl. Environ. Microbiol. 2014, 80, 4616–4625. [Google Scholar] [CrossRef]
- Bose, B.; Auchtung, J.M.; Lee, C.A.; Grossman, A.D. A conserved anti-repressor controls horizontal gene transfer by proteolysis. Mol. Microbiol. 2008, 70, 570–582. [Google Scholar] [CrossRef]
- Alkhalili, R.N.; Wallenius, J.; Canbäck, B. Towards exploring toxin-antitoxin systems in Geobacillus: A screen for type II toxin-antitoxin system families in a thermophilic genus. Int. J. Mol. Sci. 2019, 20, 5869. [Google Scholar] [CrossRef] [PubMed]
- Ortega, A.P.; Villagra, N.A.; Urrutia, I.M.; Valenzuela, L.M.; Talamilla-Espinoza, A.; Hidalgo, A.A.; Rodas, P.I.; Gil, F.; Calderón, I.L.; Paredes-Sabja, D.; et al. Lose to win: marT pseudogenization in Salmonella enterica serovar Typhi contributed to the surV-dependent survival to H2O2, and inside human macrophage-like cells. Infect. Genet. Evol. 2016, 45, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Cowles, K.N.; Goodrich-Blair, H. Expression and activity of a Xenorhabdus nematophila haemolysin required for full virulence towards Manduca sexta insects. Cell Microbiol. 2005, 7, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Da Silva Duarte, V.; Giaretta, S.; Campanaro, S.; Treu, L.; Armani, A.; Tarrah, A.; Oliveira de Paula, S.; Giacomini, A.; Corich, V. A cryptic non-inducible prophage confers phage-immunity on the Streptococcus thermophilus M17PTZA496. Viruses 2018, 11, 7. [Google Scholar] [CrossRef] [PubMed]
- Phadtare, S.; Alsina, J.; Inouye, M. Cold-shock response and cold-shock proteins. Curr. Opin. Microbiol. 1999, 2, 175–180. [Google Scholar] [CrossRef]
- Cacaci, M.; Giraud, C.; Leger, L.; Torelli, R.; Martini, C.; Posteraro, B.; Palmieri, V.; Sanguinetti, M.; Bugli, F.; Hartke, A. Expression profiling in a mammalian host reveals the strong induction of genes encoding LysM domain-containing proteins in Enterococcus faecium. Sci. Rep. 2018, 8, 12412. [Google Scholar] [CrossRef]
- Nunoshiba, T.; Demple, B. A cluster of constitutive mutations affecting the C-terminus of the redox-sensitive SoxR transcriptional activator. Nucleic Acids Res. 1994, 22, 2958–2962. [Google Scholar] [CrossRef]
- Rich, J.J.; Kinscherf, T.G.; Kitten, T.; Willis, D.K. Genetic evidence that the gacA gene encodes the cognate response regulator for the lemA sensor in Pseudomonas syringae. J. Bacteriol. 1994, 176, 7468–7475. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; ISBN 978-3-319-24277-4. [Google Scholar]
- Turner, D.; Kropinski, A.M.; Adriaenssens, E.M. A roadmap for genome-based phage taxonomy. Viruses 2021, 13, 506. [Google Scholar] [CrossRef]
- Pope, W.H.; Bowman, C.A.; Russell, D.A.; Jacobs-Sera, D.; Asai, D.J.; Cresawn, S.G.; Jacobs, W.R.; Hendrix, R.W.; Lawrence, J.G.; Hatfull, G.F.; et al. Whole genome comparison of a large collection of mycobacteriophages reveals a continuum of phage genetic diversity. Elife 2015, 4, e06416. [Google Scholar] [CrossRef]
- Dion, M.B.; Oechslin, F.; Moineau, S. Phage diversity, genomics and phylogeny. Nat. Rev. Microbiol. 2020, 18, 125–138. [Google Scholar] [CrossRef]
- Moura de Sousa, J.A.; Pfeifer, E.; Touchon, M.; Rocha, E.P.C. Causes and consequences of bacteriophage diversification via genetic exchanges across lifestyles and bacterial taxa. Mol. Biol. Evol. 2021, 38, 2497–2512. [Google Scholar] [CrossRef]
- Lefkowitz, E.J.; Dempsey, D.M.; Hendrickson, R.C.; Orton, R.J.; Siddell, S.G.; Smith, D.B. Virus taxonomy: The database of the International Committee on Taxonomy of Viruses (ICTV). Nucleic Acids Res. 2018, 46, D708–D717. [Google Scholar] [CrossRef]
- Morgan, G.J.; Pitts, W.B. Evolution without species: The case of mosaic bacteriophages. Brit. J. Phil. Sci. 2008, 59, 745–765. [Google Scholar] [CrossRef]
- Nakonieczna, A.; Rutyna, P.; Fedorowicz, M.; Kwiatek, M.; Mizak, L.; Łobocka, M. Three novel bacteriophages, J5a, F16Ba, and z1a, specific for Bacillus anthracis, define a new clade of historical Wbeta phage relatives. Viruses 2022, 14, 213. [Google Scholar] [CrossRef]
- Lucchini, S.; Desiere, F.; Brüssow, H. The structural gene module in Streptococcus thermophilus bacteriophage phi Sfi11 shows a hierarchy of relatedness to Siphoviridae from a wide range of bacterial hosts. Virology 1998, 246, 63–73. [Google Scholar] [CrossRef]
- McDonnell, B.; Mahony, J.; Hanemaaijer, L.; Neve, H.; Noben, J.P.; Lugli, G.A.; Ventura, M.; Kouwen, T.R.; van Sinderen, D. Global survey and genome exploration of bacteriophages infecting the lactic acid bacterium Streptococcus thermophilus. Front. Microbiol. 2017, 8, 1754. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
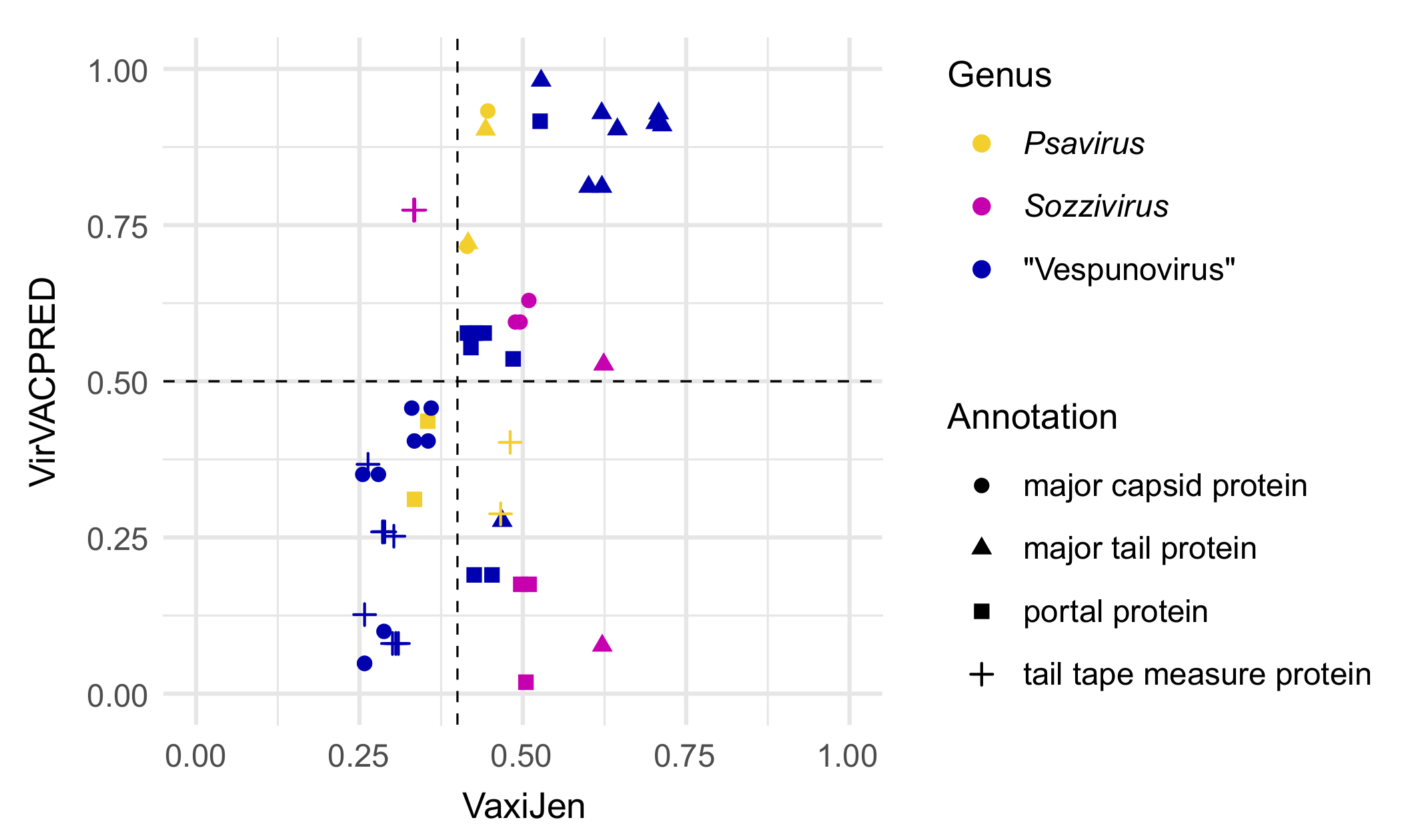{kind=link}
| Phage Name | Sample Location | Sample Type | GenBank Accession | Length, bp | ORFs | tRNAs | GC Content, % | Terminase Large Subunit | Top Bacterial BLAST Hit; Query Cover/Identity, % |
|---|---|---|---|---|---|---|---|---|---|
| cts681 | ND * | Human feces | BK058556 | 40,006 | 63 | 2 | 35.4 | XtmB | Enterococcus faecalis; 71/99 |
| ct63w4 | Estonia | — “ — | BK017522 | 38,654 | 59 | 2 | 35.4 | XtmB | E. faecalis; 72/96 |
| ctinV5 | Denmark | — “ — | BK025106 | 41,201 | 66 | 3 | 35.0 | XtmB | E. faecalis; 69/95 |
| VEsP-1 | Vietnam | River water | MZ333456 | 39,221 | 63 | 1 | 35.1 | XtmB | E. faecalis; 48/95 |
| SEsuP-1 | Russia | Human feces | MZ333458 | 39,319 | 51 | 2 | 33.5 | XtmB | E. faecalis; 96/100 |
| cthD61 | USA | — “ — | BK033083 | 39,498 | 54 | 1 | 34.1 | XtmB | E. faecalis; 99/100 |
| ct40X5 | Russia | — “ — | BK020573 | 35,370 | 52 | 2 | 33.9 | COG5362 | E. faecalis; 100/100 |
| ctyrd11 | Ethiopia | — “ — | BK030209 | 38,401 | 52 | 0 | 33.8 | DEXDc | E. hirae; 83/98 |
| ctCNj1 | USA | — “ — | BK016516 | 42,320 | 66 | 1 | 37.9 | COG5362 ** | E. avium; 23/100 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pchelin, I.M.; Tkachev, P.V.; Azarov, D.V.; Gorshkov, A.N.; Drachko, D.O.; Zlatogursky, V.V.; Dmitriev, A.V.; Goncharov, A.E. A Genome of Temperate Enterococcus Bacteriophage Placed in a Space of Pooled Viral Dark Matter Sequences. Viruses 2023, 15, 216. https://doi.org/10.3390/v15010216
Pchelin IM, Tkachev PV, Azarov DV, Gorshkov AN, Drachko DO, Zlatogursky VV, Dmitriev AV, Goncharov AE. A Genome of Temperate Enterococcus Bacteriophage Placed in a Space of Pooled Viral Dark Matter Sequences. Viruses. 2023; 15(1):216. https://doi.org/10.3390/v15010216
Chicago/Turabian StylePchelin, Ivan M., Pavel V. Tkachev, Daniil V. Azarov, Andrey N. Gorshkov, Daria O. Drachko, Vasily V. Zlatogursky, Alexander V. Dmitriev, and Artemiy E. Goncharov. 2023. "A Genome of Temperate Enterococcus Bacteriophage Placed in a Space of Pooled Viral Dark Matter Sequences" Viruses 15, no. 1: 216. https://doi.org/10.3390/v15010216
APA StylePchelin, I. M., Tkachev, P. V., Azarov, D. V., Gorshkov, A. N., Drachko, D. O., Zlatogursky, V. V., Dmitriev, A. V., & Goncharov, A. E. (2023). A Genome of Temperate Enterococcus Bacteriophage Placed in a Space of Pooled Viral Dark Matter Sequences. Viruses, 15(1), 216. https://doi.org/10.3390/v15010216