
Design and Characterization of Mutated Variants of the Oncotoxic Parvoviral Protein NS1

 , ,
, ,  and
and
Abstract
1. Introduction

2. Materials and Methods
2.1. Materials
2.2. Cloning of NS1 for Cytotoxicity Studies
2.3. Cloning of Plasmids for Proteomics
2.4. Sequencing and Amplification of Plasmid DNA for Transfections
2.5. Cell Culture and Transfection
2.6. MTT Cell Viability Assay
2.7. Sample Preparation for Phospho- and Total Cell Proteomics
2.8. Sample Preparations for (NS1-)GFP-Pulldowns
2.9. Sample Preparations for BioID2
2.10. MS Data Acquisition
2.11. MS Data Analysis
2.12. Statistical Analysis
3. Results
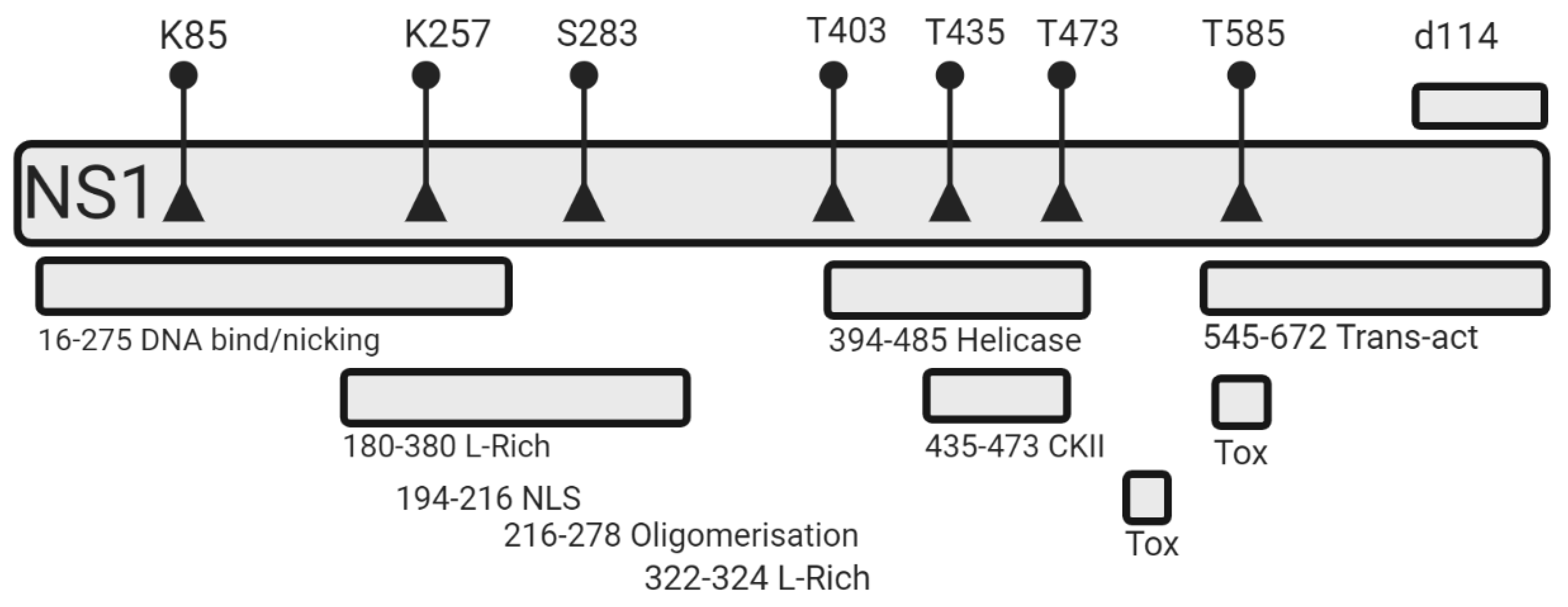
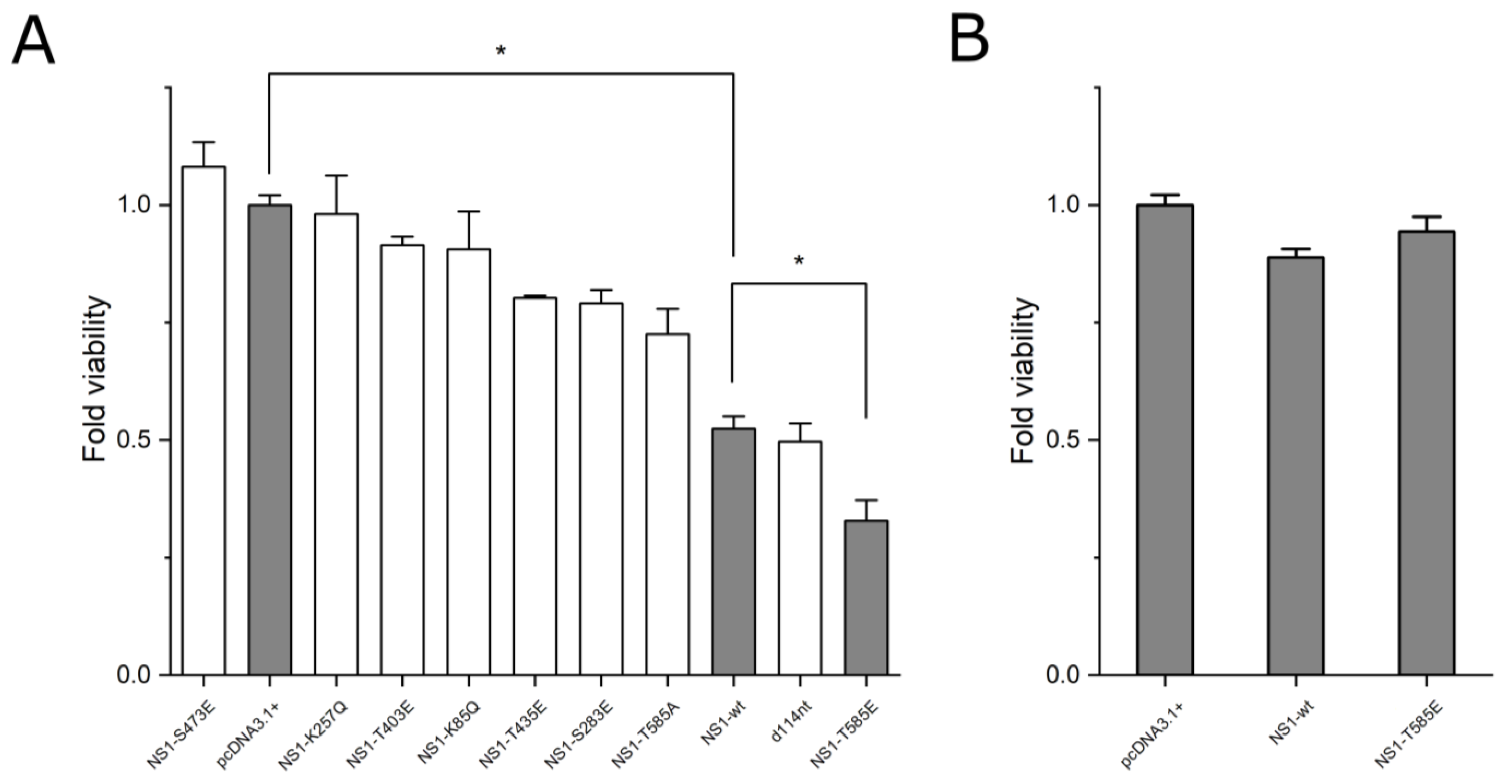
3.1. Effect of NS1 Mutations on Cytotoxicity
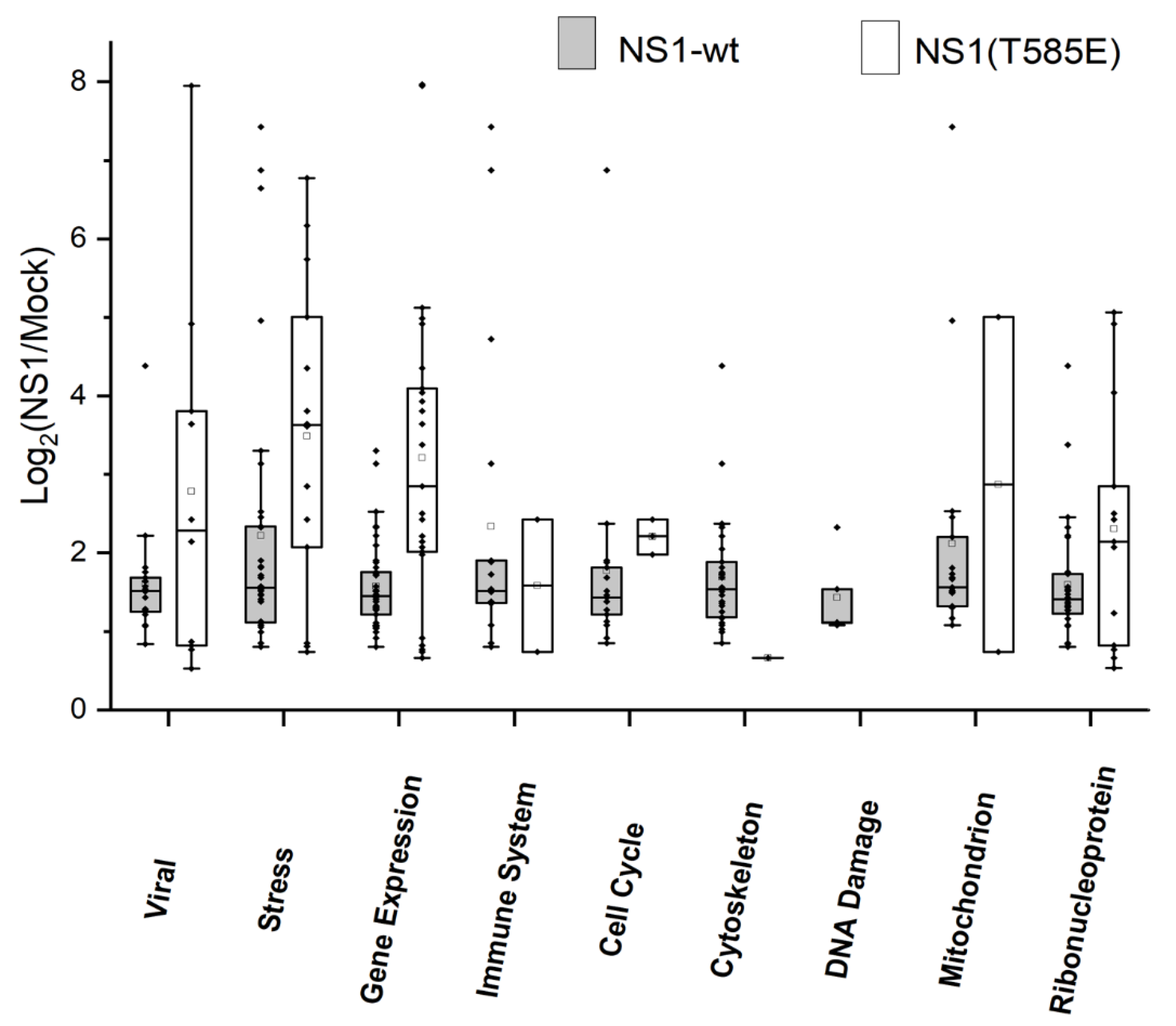
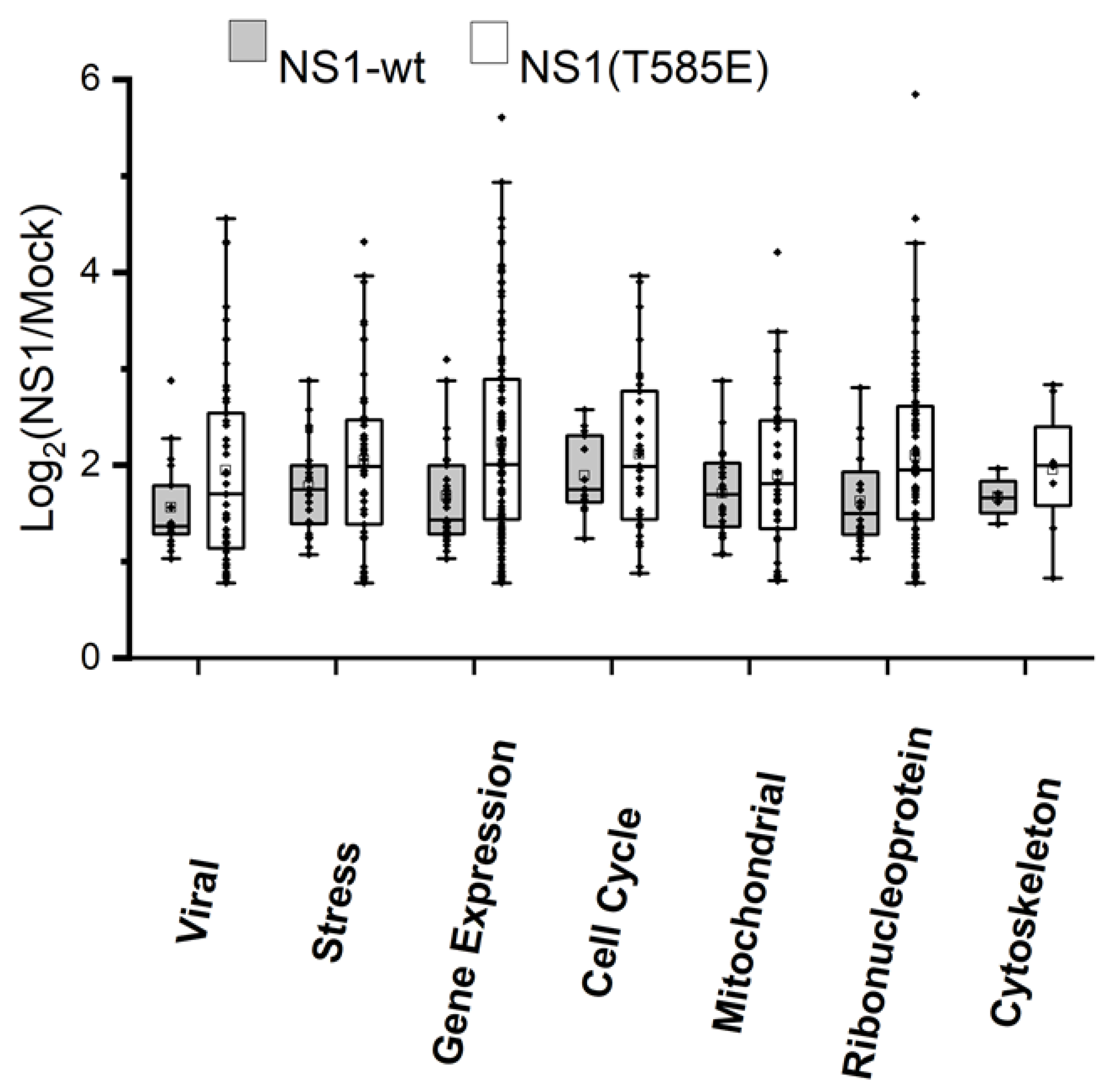
3.2. Proteomics: Effects of NS1-wt and NS1-T585E on the Host-Cell Proteome
3.3. Proteomics: Analysis of Phosphorylation Patterns
3.4. Proteomics: Binding of or Close Proximity to NS1
4. Discussion
4.1. NS1 Mutant Design
4.2. Cell Viability Assays
4.3. Induced Changes in the Host-Cell Proteome afterNS1 Expression
4.4. Impact of NS1 on Phosphorylation Patterns
4.5. Additional NS1 Interaction Partners and Pathways
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lezhnin, Y.N.; Kravchenko, Y.E.; Frolova, E.I.; Chumakov, P.M.; Chumakov, S.P. Oncotoxic Proteins in Cancer Therapy: Mechanisms of Action. Mol. Biol. 2015, 49, 231–243. [Google Scholar] [CrossRef]
- Nüesch, J.P.F.; Lacroix, J.; Marchini, A.; Rommelaere, J. Molecular Pathways: Rodent Parvoviruses—Mechanisms of Oncolysis and Prospects for Clinical Cancer Treatment. Clin. Cancer Res. 2012, 18, 3516–3523. [Google Scholar] [CrossRef]
- Witzigmann, D.; Grossen, P.; Quintavalle, C.; Lanzafame, M.; Schenk, S.H.; Tran, X.-T.; Englinger, B.; Hauswirth, P.; Grünig, D.; van Schoonhoven, S.; et al. Non-Viral Gene Delivery of the Oncotoxic Protein NS1 for Treatment of Hepatocellular Carcinoma. J. Control. Release 2021, 334, 138–152. [Google Scholar] [CrossRef]
- Bretscher, C.; Marchini, A. H-1 Parvovirus as a Cancer-Killing Agent: Past, Present, and Future. Viruses 2019, 11, 562. [Google Scholar] [CrossRef]
- Marchini, A.; Bonifati, S.; Scott, E.M.; Angelova, A.L.; Rommelaere, J. Oncolytic Parvoviruses: From Basic Virology to Clinical Applications. Virol. J. 2015, 12, 6. [Google Scholar] [CrossRef]
- Kaufman, H.L.; Kohlhapp, F.J.; Zloza, A. Oncolytic Viruses: A New Class of Immunotherapy Drugs. Nat. Rev. Drug Discov. 2015, 14, 642–662. [Google Scholar] [CrossRef]
- Rommelaere, J.; Geletneky, K.; Angelova, A.L.; Daeffler, L.; Dinsart, C.; Kiprianova, I.; Schlehofer, J.R.; Raykov, Z. Oncolytic Parvoviruses as Cancer Therapeutics. Cytokine Growth Factor Rev. 2010, 21, 185–195. [Google Scholar] [CrossRef]
- Oryx GmbH & Co. KG. Phase I/IIa Study of Intratumoral/Intracerebral or Intravenous/Intracerebral Administration of Parvovirus H-1 (ParvOryx) in Patients with Progressive Primary or Recurrent Glioblastoma Multiforme; Oryx GmbH & Co. KG: Vaterstetten, Germany, 2015. [Google Scholar]
- Hristov, G.; Krämer, M.; Li, J.; El-Andaloussi, N.; Mora, R.; Daeffler, L.; Zentgraf, H.; Rommelaere, J.; Marchini, A. Through Its Nonstructural Protein NS1, Parvovirus H-1 Induces Apoptosis via Accumulation of Reactive Oxygen Species. J. Virol. 2010, 84, 5909–5922. [Google Scholar] [CrossRef]
- Nüesch, J.P.F.; Rommelaere, J. Tumor Suppressing Properties of Rodent Parvovirus NS1 Proteins and Their Derivatives. In Anticancer Genes; Advances in Experimental Medicine and Biology; Grimm, S., Ed.; Springer: London, UK, 2014; pp. 99–124. ISBN 978-1-4471-6458-6. [Google Scholar]
- Kaiser, J. How Safe Is a Popular Gene Therapy Vector? Science 2020, 367, 131. [Google Scholar] [CrossRef]
- Chira, S.; Jackson, C.S.; Oprea, I.; Ozturk, F.; Pepper, M.S.; Diaconu, I.; Braicu, C.; Raduly, L.-Z.; Calin, G.A.; Berindan-Neagoe, I. Progresses towards Safe and Efficient Gene Therapy Vectors. Oncotarget 2015, 6, 30675–30703. [Google Scholar] [CrossRef]
- Daeffler, L.; Hörlein, R.; Rommelaere, J.; Nüesch, J.P.F. Modulation of Minute Virus of Mice Cytotoxic Activities through Site-Directed Mutagenesis within the NS Coding Region. J. Virol. 2003, 77, 12466–12478. [Google Scholar] [CrossRef]
- Corbau, R.; Duverger, V.; Rommelaere, J.; Nüesch, J.P.F. Regulation of MVM NS1 by Protein Kinase C: Impact of Mutagenesis at Consensus Phosphorylation Sites on Replicative Functions and Cytopathic Effects. Virology 2000, 278, 151–167. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Bonifati, S.; Hristov, G.; Marttila, T.; Valmary-Degano, S.; Stanzel, S.; Schnölzer, M.; Mougin, C.; Aprahamian, M.; Grekova, S.P.; et al. Synergistic Combination of Valproic Acid and Oncolytic Parvovirus H-1PV as a Potential Therapy against Cervical and Pancreatic Carcinomas. EMBO Mol. Med. 2013, 5, 1537–1555. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Cole, P.A. Synthetic Approaches to Protein Phosphorylation. Curr. Opin. Chem. Biol. 2015, 28, 115–122. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Zhang, L.; Wang, X.; Huo, L.; Sun, L.; Feng, C.; Jing, X.; Du, D.; Liang, H.; Liu, M.; et al. Systematic Analysis of the Functions of Lysine Acetylation in the Regulation of Tat Activity. PLoS ONE 2013, 8, e67186. [Google Scholar] [CrossRef] [PubMed]
- Weiss, N.; Stroh-Dege, A.; Rommelaere, J.; Dinsart, C.; Salomé, N. An In-Frame Deletion in the NS Protein-Coding Sequence of Parvovirus H-1PV Efficiently Stimulates Export and Infectivity of Progeny Virions. J. Virol. 2012, 86, 7554–7564. [Google Scholar] [CrossRef] [PubMed]
- Geletneky, K.; Hajda, J.; Angelova, A.L.; Leuchs, B.; Capper, D.; Bartsch, A.J.; Neumann, J.-O.; Schöning, T.; Hüsing, J.; Beelte, B.; et al. Oncolytic H-1 Parvovirus Shows Safety and Signs of Immunogenic Activity in a First Phase I/IIa Glioblastoma Trial. Mol. Ther. 2017, 25, 2620–2634. [Google Scholar] [CrossRef]
- Bär, S.; Rommelaere, J.; Nüesch, J.P.F. Vesicular Transport of Progeny Parvovirus Particles through ER and Golgi Regulates Maturation and Cytolysis. PLoS Pathog. 2013, 9, e1003605. [Google Scholar] [CrossRef]
- Daeffler, L.; Nüesch, J.; Rommelaere, J. Zusammensetzung einer Parvovirus VP1-Proteinvariante und eines arvovirus NS1-Proteins zur Induktion von Zytolyse. 2003. [Google Scholar]
- Kerr, J.; Cotmore, S.; Bloom, M.E. Parvoviruses; CRC Press: Boca Raton, FL, USA, 2005; ISBN 978-1-4441-1478-2. Available online: https://patents.google.com/patent/DE60124523T2/de (accessed on 9 January 2023).
- Kiene, K.; Schenk, S.H.; Porta, F.; Ernst, A.; Witzigmann, D.; Grossen, P.; Huwyler, J. PDMS-b-PMOXA Polymersomes for Hepatocyte Targeting and Assessment of Toxicity. Eur. J. Pharm. Biopharm. 2017, 119, 322–332. [Google Scholar] [CrossRef]
- Post, H.; Penning, R.; Fitzpatrick, M.A.; Garrigues, L.B.; Wu, W.; MacGillavry, H.D.; Hoogenraad, C.C.; Heck, A.J.R.; Altelaar, A.F.M. Robust, Sensitive, and Automated Phosphopeptide Enrichment Optimized for Low Sample Amounts Applied to Primary Hippocampal Neurons. J. Proteome Res. 2017, 16, 728–737. [Google Scholar] [CrossRef]
- Ahrné, E.; Glatter, T.; Viganò, C.; von Schubert, C.; Nigg, E.A.; Schmidt, A. Evaluation and Improvement of Quantification Accuracy in Isobaric Mass Tag-Based Protein Quantification Experiments. J. Proteome Res. 2016, 15, 2537–2547. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.I.; Jensen, S.C.; Noble, K.A.; Kc, B.; Roux, K.H.; Motamedchaboki, K.; Roux, K.J. An Improved Smaller Biotin Ligase for BioID Proximity Labeling. Mol. Biol. Cell 2016, 27, 1188–1196. [Google Scholar] [CrossRef]
- Eden, E.; Navon, R.; Steinfeld, I.; Lipson, D.; Yakhini, Z. GOrilla: A Tool for Discovery and Visualization of Enriched GO Terms in Ranked Gene Lists. BMC Bioinform. 2009, 10, 48. [Google Scholar] [CrossRef] [PubMed]
- Nüesch, J.P.; Christensen, J.; Rommelaere, J. Initiation of Minute Virus of Mice DNA Replication Is Regulated at the Level of Origin Unwinding by Atypical Protein Kinase C Phosphorylation of NS1. J. Virol. 2001, 75, 5730–5739. [Google Scholar] [CrossRef]
- Nüesch, J.P.F.; Lachmann, S.; Corbau, R.; Rommelaere, J. Regulation of Minute Virus of Mice NS1 Replicative Functions by Atypical PKCλ In Vivo. J. Virol. 2003, 77, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Guerra-Castellano, A.; Díaz-Moreno, I.; Velázquez-Campoy, A.; De la Rosa, M.A.; Díaz-Quintana, A. Structural and Functional Characterization of Phosphomimetic Mutants of Cytochrome c at Threonine 28 and Serine 47. Biochim. Biophys. Acta 2016, 1857, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Luwang, J.W.; Natesh, R. Phosphomimetic Mutation Destabilizes the Central Core Domain of Human P53. IUBMB Life 2018, 70, 1023–1031. [Google Scholar] [CrossRef] [PubMed]
- Pecina, P.; Borisenko, G.G.; Belikova, N.A.; Tyurina, Y.Y.; Pecinova, A.; Lee, I.; Samhan-Arias, A.K.; Przyklenk, K.; Kagan, V.E.; Hüttemann, M. Phosphomimetic Substitution of Cytochrome C Tyrosine 48 Decreases Respiration and Binding to Cardiolipin and Abolishes Ability to Trigger Downstream Caspase Activation. Biochemistry 2010, 49, 6705–6714. [Google Scholar] [CrossRef] [PubMed]
- Gorsky, M.K.; Burnouf, S.; Dols, J.; Mandelkow, E.; Partridge, L. Acetylation Mimic of Lysine 280 Exacerbates Human Tau Neurotoxicity in Vivo. Sci. Rep. 2016, 6, 22685. [Google Scholar] [CrossRef]
- Wang, X.; Hayes, J.J. Acetylation Mimics within Individual Core Histone Tail Domains Indicate Distinct Roles in Regulating the Stability of Higher-Order Chromatin Structure. Mol. Cell. Biol. 2008, 28, 227–236. [Google Scholar] [CrossRef]
- Kamieniarz, K.; Schneider, R. Tools to Tackle Protein Acetylation. Chem. Biol. 2009, 16, 1027–1029. [Google Scholar] [CrossRef]
- Hashemi, H.; Condurat, A.-L.; Stroh-Dege, A.; Weiss, N.; Geiss, C.; Pilet, J.; Cornet Bartolomé, C.; Rommelaere, J.; Salomé, N.; Dinsart, C. Mutations in the Non-Structural Protein-Coding Sequence of Protoparvovirus H-1PV Enhance the Fitness of the Virus and Show Key Benefits Regarding the Transduction Efficiency of Derived Vectors. Viruses 2018, 10, 150. [Google Scholar] [CrossRef]
- Moehler, M.; Blechacz, B.; Weiskopf, N.; Zeidler, M.; Stremmel, W.; Rommelaere, J.; Galle, P.R.; Cornelis, J.J. Effective Infection, Apoptotic Cell Killing and Gene Transfer of Human Hepatoma Cells but Not Primary Hepatocytes by Parvovirus H1 and Derived Vectors. Cancer Gene Ther. 2001, 8, 158–167. [Google Scholar] [CrossRef]
- Hunter, T. Why Nature Chose Phosphate to Modify Proteins. Philos. Trans. R. Soc. B Biol. Sci. 2012, 367, 2513–2516. [Google Scholar] [CrossRef]
- Pérez-Mejías, G.; Velázquez-Cruz, A.; Guerra-Castellano, A.; Baños-Jaime, B.; Díaz-Quintana, A.; González-Arzola, K.; Ángel De la Rosa, M.; Díaz-Moreno, I. Exploring Protein Phosphorylation by Combining Computational Approaches and Biochemical Methods. Comput. Struct. Biotechnol. J. 2020, 18, 1852–1863. [Google Scholar] [CrossRef]
- Huang, W.; Erikson, R.L. Constitutive Activation of Mek1 by Mutation of Serine Phosphorylation Sites. Proc. Natl. Acad. Sci. USA 1994, 91, 8960–8963. [Google Scholar] [CrossRef]
- Lacroix, J.; Leuchs, B.; Li, J.; Hristov, G.; Deubzer, H.E.; Kulozik, A.E.; Rommelaere, J.; Schlehofer, J.R.; Witt, O. Parvovirus H1 Selectively Induces Cytotoxic Effects on Human Neuroblastoma Cells. Int. J. Cancer 2010, 127, 1230–1239. [Google Scholar] [CrossRef]
- Nüesch, J.P.; Tattersall, P. Nuclear Targeting of the Parvoviral Replicator Molecule NS1: Evidence for Self-Association Prior to Nuclear Transport. Virology 1993, 196, 637–651. [Google Scholar] [CrossRef]
- Nüesch, J.P.; Corbau, R.; Tattersall, P.; Rommelaere, J. Biochemical Activities of Minute Virus of Mice Nonstructural Protein NS1 Are Modulated In Vitro by the Phosphorylation State of the Polypeptide. J. Virol. 1998, 72, 8002–8012. [Google Scholar] [CrossRef]
- Nüesch, J.P.F.; Rommelaere, J. NS1 Interaction with CKIIα: Novel Protein Complex Mediating Parvovirus-Induced Cytotoxicity. J. Virol. 2006, 80, 4729–4739. [Google Scholar] [CrossRef]
- Nüesch, J.P.F.; Rommelaere, J. A Viral Adaptor Protein Modulating Casein Kinase II Activity Induces Cytopathic Effects in Permissive Cells. Proc. Natl. Acad. Sci. USA 2007, 104, 12482–12487. [Google Scholar] [CrossRef]
- Gallo, S.; Arcidiacono, M.V.; Tisato, V.; Piva, R.; Penolazzi, L.; Bosi, C.; Feo, C.V.; Gafà, R.; Secchiero, P. Upregulation of the Alternative Splicing Factor NOVA2 in Colorectal Cancer Vasculature. Onco Targets Ther. 2018, 11, 6049–6056. [Google Scholar] [CrossRef]
- Tang, S.; Zhao, Y.; He, X.; Zhu, J.; Chen, S.; Wen, J.; Deng, Y. Identification of NOVA Family Proteins as Novel β-Catenin RNA-Binding Proteins That Promote Epithelial-Mesenchymal Transition. RNA Biol. 2020, 17, 881–891. [Google Scholar] [CrossRef]
- Shang, S.; Hua, F.; Hu, Z.-W. The Regulation of β-Catenin Activity and Function in Cancer: Therapeutic Opportunities. Oncotarget 2017, 8, 33972–33989. [Google Scholar] [CrossRef]
- Luis, G.; Godfroid, A.; Nishiumi, S.; Cimino, J.; Blacher, S.; Maquoi, E.; Wery, C.; Collignon, A.; Longuespée, R.; Montero-Ruiz, L.; et al. Tumor Resistance to Ferroptosis Driven by Stearoyl-CoA Desaturase-1 (SCD1) in Cancer Cells and Fatty Acid Biding Protein-4 (FABP4) in Tumor Microenvironment Promote Tumor Recurrence. Redox Biol. 2021, 43, 102006. [Google Scholar] [CrossRef]
- Bansal, S.; Berk, M.; Alkhouri, N.; Partrick, D.A.; Fung, J.J.; Feldstein, A. Stearoyl-CoA Desaturase Plays an Important Role in Proliferation and Chemoresistance in Human Hepatocellular Carcinoma. J. Surg. Res. 2014, 186, 29–38. [Google Scholar] [CrossRef]
- Paton, C.M.; Ntambi, J.M. Biochemical and Physiological Function of Stearoyl-CoA Desaturase. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E28–E37. [Google Scholar] [CrossRef]
- Hardy, S.; Langelier, Y.; Prentki, M. Oleate Activates Phosphatidylinositol 3-Kinase and Promotes Proliferation and Reduces Apoptosis of MDA-MB-231 Breast Cancer Cells, Whereas Palmitate Has Opposite Effects. Cancer Res. 2000, 60, 6353–6358. [Google Scholar]
- Chen, L.; Ren, J.; Yang, L.; Li, Y.; Fu, J.; Li, Y.; Tian, Y.; Qiu, F.; Liu, Z.; Qiu, Y. Stearoyl-CoA Desaturase-1 Mediated Cell Apoptosis in Colorectal Cancer by Promoting Ceramide Synthesis. Sci. Rep. 2016, 6, 19665. [Google Scholar] [CrossRef]
- Cziepluch, C.; Kordes, E.; Poirey, R.; Grewenig, A.; Rommelaere, J.; Jauniaux, J.-C. Identification of a Novel Cellular TPR-Containing Protein, SGT, That Interacts with the Nonstructural Protein NS1 of Parvovirus H-1. J. Virol. 1998, 72, 4149–4156. [Google Scholar] [CrossRef]
- Roberts, J.D.; Thapaliya, A.; Martínez-Lumbreras, S.; Krysztofinska, E.M.; Isaacson, R.L. Structural and Functional Insights into Small, Glutamine-Rich, Tetratricopeptide Repeat Protein Alpha. Front. Mol. Biosci. 2015, 2, 71. [Google Scholar] [CrossRef]
- Kim, D.I.; KC, B.; Zhu, W.; Motamedchaboki, K.; Doye, V.; Roux, K.J. Probing Nuclear Pore Complex Architecture with Proximity-Dependent Biotinylation. Proc. Natl. Acad. Sci. USA 2014, 111, E2453–E2461. [Google Scholar] [CrossRef]
- Op De Beeck, A.; Sobczak-Thepot, J.; Sirma, H.; Bourgain, F.; Brechot, C.; Caillet-Fauquet, P. NS1- and Minute Virus of Mice-Induced Cell Cycle Arrest: Involvement of P53 and P21cip1. J. Virol. 2001, 75, 11071–11078. [Google Scholar] [CrossRef]
- Berchtold, M.W.; Villalobo, A. The Many Faces of Calmodulin in Cell Proliferation, Programmed Cell Death, Autophagy, and Cancer. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2014, 1843, 398–435. [Google Scholar] [CrossRef]
- Sahara, S.; Aoto, M.; Eguchi, Y.; Imamoto, N.; Yoneda, Y.; Tsujimoto, Y. Acinus Is a Caspase-3-Activated Protein Required for Apoptotic Chromatin Condensation. Nature 1999, 401, 168–173. [Google Scholar] [CrossRef]
- Lorain, S.; Quivy, J.-P.; Monier-Gavelle, F.; Scamps, C.; Lécluse, Y.; Almouzni, G.; Lipinski, M. Core Histones and HIRIP3, a Novel Histone-Binding Protein, Directly Interact with WD Repeat Protein HIRA. Mol Cell Biol 1998, 18, 5546–5556. [Google Scholar] [CrossRef]
- Assrir, N.; Filhol, O.; Galisson, F.; Lipinski, M. HIRIP3 Is a Nuclear Phosphoprotein Interacting with and Phosphorylated by the Serine-Threonine Kinase CK2. Biol. Chem. 2007, 388, 391–398. [Google Scholar] [CrossRef]
- Ge, H.; Cheng, N.; Xu, X.; Yang, Z.; Hoffman, R.M.; Zhu, J. AMMECR1 Inhibits Apoptosis and Promotes Cell-Cycle Progression and Proliferation of the A549 Human Lung Cancer Cell Line. Anticancer Res. 2019, 39, 4637–4642. [Google Scholar] [CrossRef]
- Nüesch, J.P.F.; Lachmann, S.; Rommelaere, J. Selective Alterations of the Host Cell Architecture upon Infection with Parvovirus Minute Virus of Mice. Virology 2005, 331, 159–174. [Google Scholar] [CrossRef]
- Christensen, J.; Tattersall, P. Parvovirus Initiator Protein NS1 and RPA Coordinate Replication Fork Progression in a Reconstituted DNA Replication System. J. Virol. 2002, 76, 6518–6531. [Google Scholar] [CrossRef]
- Bashir, T.; Rommelaere, J.; Cziepluch, C. In Vivo Accumulation of Cyclin A and Cellular Replication Factors in Autonomous Parvovirus Minute Virus of Mice-Associated Replication Bodies. J. Virol. 2001, 75, 4394–4398. [Google Scholar] [CrossRef]
- Adeyemi, R.O.; Landry, S.; Davis, M.E.; Weitzman, M.D.; Pintel, D.J. Parvovirus Minute Virus of Mice Induces a DNA Damage Response That Facilitates Viral Replication. PLoS Pathog. 2010, 6, e1001141. [Google Scholar] [CrossRef]
- Harris, C.E.; Boden, R.A.; Astell, C.R. A Novel Heterogeneous Nuclear Ribonucleoprotein-Like Protein Interacts with NS1 of the Minute Virus of Mice. J. Virol. 1999, 73, 72–80. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Plasmid | PCR Template | Primers | Expressed Protein |
|---|---|---|---|
| pcDNA3.1+ | original plasmid (Invitrogen) | - | no protein expression |
| pcDNA3.1—NS1 | Witzigmann et al. [3] | - | NS1-wt |
| pcDNA3.1—NS1 (K85Q) | pcDNA3.1—NS1 | p1, p7; p6, p2 | NS1-K85Q |
| pcDNA3.1—NS1 (K257Q) | pcDNA3.1—NS1 | p1, p9; p8, p2 | NS1-K257Q |
| pcDNA3.1—NS1 (S283E) | pcDNA3.1—NS1 | p1, p11; p10, p2 | NS1-S283E |
| pcDNA3.1—NS1 (T435E) | pcDNA3.1—NS1 | p1, p13; p12, p2 | NS1-T435E |
| pcDNA3.1—NS1 (S473E) | pcDNA3.1—NS1 | p1, 15; p14, p2 | NS1-S473E |
| pcDNA3.1—NS1 (T585A) | pcDNA3.1—NS1 | p1, 17; p16, p2 | NS1-T585A |
| pcDNA3.1—NS1 (T585E) | pcDNA3.1—NS1 | p1, p19; p18, p2 | NS1-T585E |
| pcDNA3.1—NS1 (d114) | pcDNA3.1—NS1 | p1, p20; p21, p2 | NS1-d114 |
| pTag-GFP-N | original plasmid (Evrogen) | - | GFP |
| pTag-NS1-GFP | Witzigmann et al. [3] | - | NS1-wt-GFP |
| pTag-NS1(T585E)-GFP | pcDNA3.1—NS1 (T585E) | p1, p3 | NS1-T585E-GFP |
| MCS-BioID2-HA | original plasmid (Addgene) | - | no protein expression |
| pBioID2 | MCS-BioID2-HA | p22, p23 | BioID2 |
| pNS1-BioID2 | pcDNA3.1—NS1 | p4, p5 | NS1-wt-BioID2 |
| pNS1(T585E)-BioID2 | (T585E) | p4, p5 | NS1-T585E-BioID2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hauswirth, P.; Graber, P.; Buczak, K.; Mancuso, R.V.; Schenk, S.H.; Nüesch, J.P.F.; Huwyler, J. Design and Characterization of Mutated Variants of the Oncotoxic Parvoviral Protein NS1. Viruses 2023, 15, 209. https://doi.org/10.3390/v15010209
Hauswirth P, Graber P, Buczak K, Mancuso RV, Schenk SH, Nüesch JPF, Huwyler J. Design and Characterization of Mutated Variants of the Oncotoxic Parvoviral Protein NS1. Viruses. 2023; 15(1):209. https://doi.org/10.3390/v15010209
Chicago/Turabian StyleHauswirth, Patrick, Philipp Graber, Katarzyna Buczak, Riccardo Vincenzo Mancuso, Susanne Heidi Schenk, Jürg P. F. Nüesch, and Jörg Huwyler. 2023. "Design and Characterization of Mutated Variants of the Oncotoxic Parvoviral Protein NS1" Viruses 15, no. 1: 209. https://doi.org/10.3390/v15010209
APA StyleHauswirth, P., Graber, P., Buczak, K., Mancuso, R. V., Schenk, S. H., Nüesch, J. P. F., & Huwyler, J. (2023). Design and Characterization of Mutated Variants of the Oncotoxic Parvoviral Protein NS1. Viruses, 15(1), 209. https://doi.org/10.3390/v15010209