
Animal Model Alternatives in Filovirus and Bornavirus Research

 ,
, {kind=link}
{kind=link}
Abstract
1. Filoviruses and Bornaviruses—Two Distinct Families within the Order Mononegavirales
2. Filovirus Animal Models
3. Animal Models in Bornavirus Research
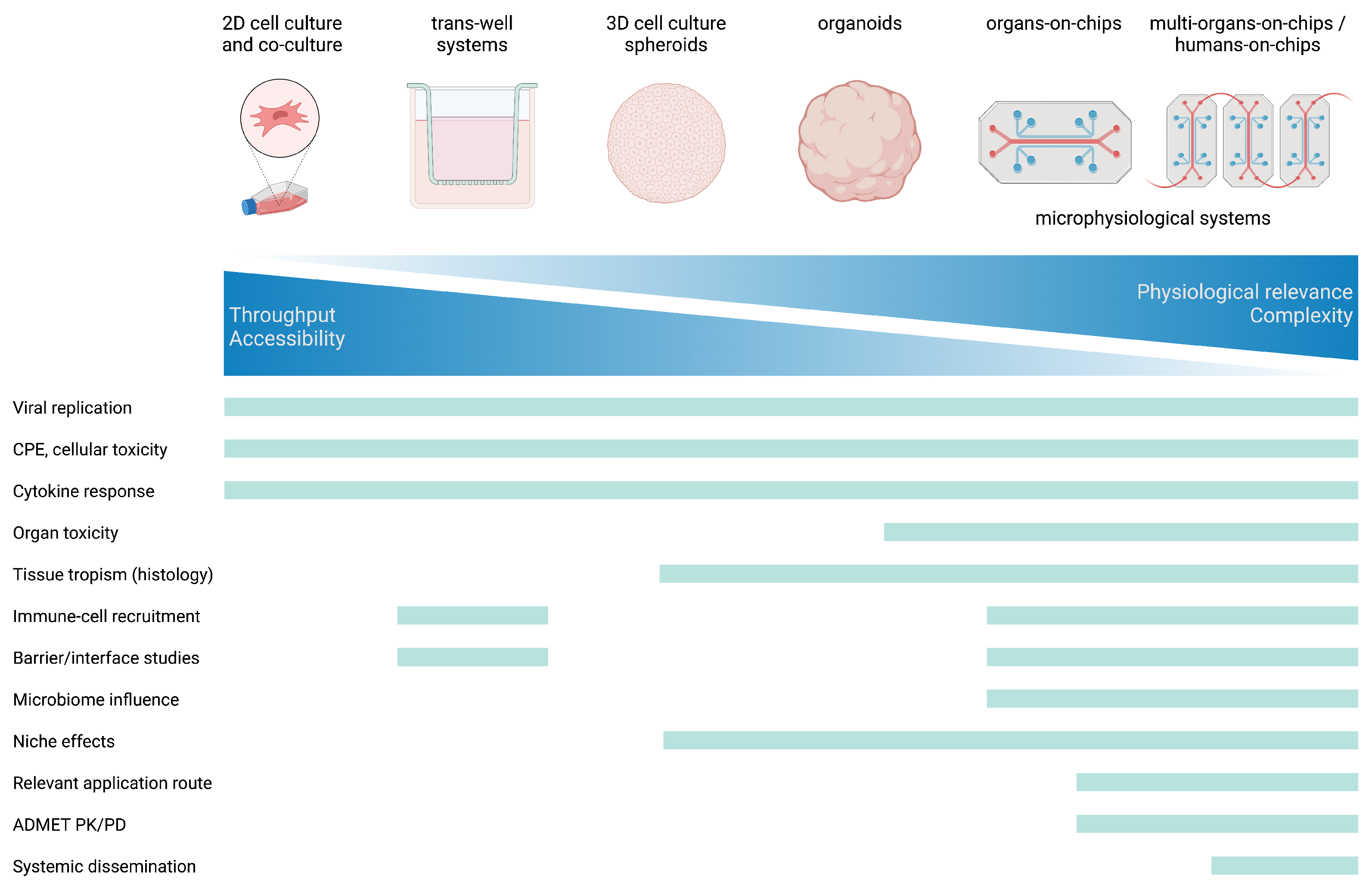
4. Complex In Vitro Models in Viral Research
4.1. 2D In Vitro Models
4.2. Organoids and Other Static 3D Cell Culture Models in Viral Research
Assembloids
4.3. Organs-on-Chips as Microphysiological Systems under Constant Fluid Flow in Viral Research
Multi-Organ-Systems- and Humans-on-a-Chip
5. Caveats for the Usage of Organoids and MPS in Virological Research
6. Organoids and MPS—The Future of Filovirus and Bornavirus Research?
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ADMET | absorption, distribution, metabolism, excretion, toxicity |
| AdSC | adult stem cell |
| ALI | air–liquid interface |
| BBB | blood–brain barrier |
| BDBV | Bundibugyo virus |
| BoDV-1 | Borna disease virus 1 |
| CC-RIX | collaborative cross resource recombinant inbred inter-crossed |
| CFR | case fatality ratio |
| CPE | cytopathic effect |
| CNS | central nervous system |
| DC | dendritic cell |
| DRC | Democratic Republic of the Congo |
| EBOV | Ebola virus |
| ECM | extracellular matrix |
| GCCP | good cell culture practice |
| GPA | guinea-pig-adapted |
| HA | hamster-adapted |
| HBV | hepatitis B virus |
| HCMV | human cytomegalo virus |
| hESC | human embryonic stem cell |
| HSV-1 | herpes simplex virus 1 |
| IFNAR | interferon alpha/beta receptor |
| iPSC | induced pluripotent stem cell |
| OOCs | organs-on-chips |
| MA | mouse-adapted |
| MARV | Marburg virus |
| microCCA | micro cell culture analog |
| MOCs | multi-organs-on-chips |
| MPCC | micropatterned co-culture |
| MPS | microphysiological system |
| NHP | nonhuman primates |
| PD | pharmacodynamics |
| PDMS | poly(dimethylsiloxane) |
| PK | pharmacokinetics |
| R&D | research and development |
| RAVV | Ravn virus |
| RESTV | Reston virus |
| SCID | severe combined immunodeficiency |
| STAT1 | signal transducer and activator of transcription 1 |
| SUDV | Sudan virus |
| TAFV | Taï forest virus |
| TEER | transepithelial/-endothelial electrical resistance |
| VSBV-1 | variegated squirrel bornavirus 1 |
| ZIKV | Zika virus |
References
- Gorbalenya, A.E.; Krupovic, M.; Mushegian, A.; Kropinski, A.M.; Siddell, S.G.; Varsani, A.; Adams, M.J.; Davison, A.J.; Dutilh, B.E.; Harrach, B.; et al. The new scope of virus taxonomy: Partitioning the virosphere into 15 hierarchical ranks. Nat. Microbiol. 2020, 5, 668–674. [Google Scholar]
- Walker, P.J.; Siddell, S.G.; Lefkowitz, E.J.; Mushegian, A.R.; Adriaenssens, E.M.; Alfenas-Zerbini, P.; Davison, A.J.; Dempsey, D.M.; Dutilh, B.E.; García, M.L.; et al. Changes to virus taxonomy and to the International Code of Virus Classification and Nomenclature ratified by the International Committee on Taxonomy of Viruses (2021). Arch. Virol. 2021, 166, 2633–2648. [Google Scholar] [CrossRef] [PubMed]
- ICTV Taxonomy. Available online: https://ictv.global/taxonomy (accessed on 31 August 2022).
- Siegert, R.; Shu, H.L.; Slenczka, W.; Peters, D.; Müller, G. On the etiology of an unknown human infection originating from monkeys. Dtsch. Med. Wochenschr. 1967, 92, 2341–2343. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, H.; Sprecher, A.; Geisbert, T.W. Ebola. N. Engl. J. Med. 2020, 382, 1832–1842. [Google Scholar] [CrossRef] [PubMed]
- Ebola Virus Disease. Available online: https://www.who.int/news-room/fact-sheets/detail/ebola-virus-disease (accessed on 31 August 2022).
- Uganda Declares Ebola Virus Disease outbreak. Available online: https://www.afro.who.int/countries/uganda/news/uganda-declares-ebola-virus-disease-outbreak (accessed on 17 November 2022).
- Ebola Disease Caused by Sudan Ebolavirus—Uganda. Available online: https://www.who.int/emergencies/disease-outbreak-news/item/2022-DON428 (accessed on 29 December 2022).
- Towner, J.S.; Amman, B.R.; Sealy, T.K.; Carroll, S.A.; Comer, J.A.; Kemp, A.; Swanepoel, R.; Paddock, C.D.; Balinandi, S.; Khristova, M.L.; et al. Isolation of genetically diverse Marburg viruses from Egyptian fruit bats. PLoS Pathog. 2009, 5, e1000536. [Google Scholar] [CrossRef]
- Mate, S.E.; Kugelman, J.R.; Nyenswah, T.G.; Ladner, J.T.; Wiley, M.R.; Cordier-Lassalle, T.; Christie, A.; Schroth, G.P.; Gross, S.M.; Davies-Wayne, G.J.; et al. Molecular Evidence of Sexual Transmission of Ebola Virus. N. Engl. J. Med. 2015, 373, 2448–2454. [Google Scholar] [CrossRef]
- Kozak, R.; He, S.; Kroeker, A.; de La Vega, M.A.; Audet, J.; Wong, G.; Urfano, C.; Antonation, K.; Embury-Hyatt, C.; Kobinger, G.P.; et al. Ferrets Infected with Bundibugyo Virus or Ebola Virus Recapitulate Important Aspects of Human Filovirus Disease. J. Virol. 2016, 90, 9209–9223. [Google Scholar] [CrossRef]
- Negredo, A.; Palacios, G.; Vázquez-Morón, S.; González, F.; Dopazo, H.; Molero, F.; Juste, J.; Quetglas, J.; Savji, N.; de la Cruz Martínez, M.; et al. Discovery of an Ebolavirus-Like Filovirus in Europe. PLoS Pathog. 2011, 7, e1002304. [Google Scholar] [CrossRef]
- Brannan, J.M.; Froude, J.W.; Prugar, L.I.; Bakken, R.R.; Zak, S.E.; Daye, S.P.; Wilhelmsen, C.E.; Dye, J.M. Interferon α/β Receptor-Deficient Mice as a Model for Ebola Virus Disease. J. Infect. Dis. 2015, 212 (Suppl. S2), S282–S294. [Google Scholar] [CrossRef]
- Kroeker, A.; He, S.; de La Vega, M.A.; Wong, G.; Embury-Hyatt, C.; Qiu, X. Characterization of Sudan Ebolavirus infection in ferrets. Oncotarget 2017, 8, 46262–46272. [Google Scholar] [CrossRef]
- Miranda, M.E.; Miranda, N.L. Reston ebolavirus in humans and animals in the Philippines: A review. J. Infect. Dis. 2011, 204, S757–S760. [Google Scholar] [CrossRef]
- Le Guenno, B.; Formenty, P.; Wyers, M.; Gounon, P.; Walker, F.; Boesch, C. Isolation and partial characterisation of a new strain of Ebola virus. Lancet 1995, 345, 1271–1274. [Google Scholar] [CrossRef]
- Formenty, P.; Hatz, C.; Le Guenno, B.; Stoll, A.; Rogenmoser, P.; Widmer, A. Human infection due to Ebola virus, subtype Côte d’Ivoire: Clinical and biologic presentation. J. Infect. Dis. 1999, 179 (Suppl. S1), 48–53. [Google Scholar] [CrossRef]
- CDC. Outbreak of Marburg virus hemorrhagic fever–Angola, October 1, 2004-March 29, 2005. MMWR Morb. Mortal. Wkly. Rep. 2005, 54, 308–309. [Google Scholar]
- Bente, D.; Gren, J.; Strong, J.E.; Feldmann, H. Disease modeling for Ebola and Marburg viruses. Dis. Model Mech. 2009, 2, 12–17. [Google Scholar] [CrossRef]
- Nakayama, E.; Saijo, M. Animal models for Ebola and Marburg virus infections. Front Microbiol 2013, 4, 267. [Google Scholar] [CrossRef]
- Siragam, V.; Wong, G.; Qiu, X.G. Animal models for filovirus infections. Zool Res. 2018, 39, 15–24. [Google Scholar] [CrossRef]
- Yamaoka, S.; Banadyga, L.; Bray, M.; Ebihara, H. Small Animal Models for Studying Filovirus Pathogenesis. Curr. Top. Microbiol. Immunol. 2017, 411, 195–227. [Google Scholar]
- Pattyn, S.; van der Groen, G.; Jacob, W.; Piot, P.; Courteille, G. Isolation of Marburg-like virus from a case of haemorrhagic fever in Zaire. Lancet 1977, 1, 573–574. [Google Scholar] [CrossRef]
- Lüdtke, A.; Ruibal, P.; Wozniak, D.M.; Pallasch, E.; Wurr, S.; Bockholt, S.; Gómez-Medina, S.; Qiu, X.; Kobinger, G.P.; Rodríguez, E.; et al. Ebola virus infection kinetics in chimeric mice reveal a key role of T cells as barriers for virus dissemination. Sci. Rep. 2017, 7, 43776. [Google Scholar] [CrossRef]
- Rottstegge, M.; Tipton, T.; Oestereich, L.; Ruibal, P.; Nelson, E.V.; Olal, C.; Port, J.R.; Seibel, J.; Pallasch, E.; Bockholt, S.; et al. Avatar Mice Underscore the Role of the T Cell-Dendritic Cell Crosstalk in Ebola Virus Disease and Reveal Mechanisms of Protection in Survivors. J. Virol. 2022, 96, e0057422. [Google Scholar] [CrossRef] [PubMed]
- McElroy, A.K.; Akondy, R.S.; Davis, C.W.; Ellebedy, A.H.; Mehta, A.K.; Kraft, C.S.; Lyon, G.M.; Ribner, B.S.; Varkey, J.; Sidney, J.; et al. Human Ebola virus infection results in substantial immune activation. Proc. Natl. Acad. Sci. USA 2015, 112, 4719–4724. [Google Scholar] [CrossRef] [PubMed]
- Ruibal, P.; Oestereich, L.; Lüdtke, A.; Becker-Ziaja, B.; Wozniak, D.M.; Kerber, R.; Korva, M.; Cabeza-Cabrerizo, M.; Bore, J.A.; Koundouno, F.R.; et al. Unique human immune signature of Ebola virus disease in Guinea. Nature 2016, 533, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Speranza, E.; Ruibal, P.; Port, J.R.; Feng, F.; Burkhardt, L.; Grundhoff, A.; Günther, S.; Oestereich, L.; Hiscox, J.A.; Connor, J.H.; et al. T-Cell Receptor Diversity and the Control of T-Cell Homeostasis Mark Ebola Virus Disease Survival in Humans. J. Infect. Dis. 2018, 218, S508–S518. [Google Scholar] [CrossRef] [PubMed]
- Korn, K.; Coras, R.; Bobinger, T.; Herzog, S.M.; Lücking, H.; Stöhr, R.; Huttner, H.B.; Hartmann, A.; Ensser, A. Fatal Encephalitis Associated with Borna Disease Virus 1. N. Engl. J. Med. 2018, 379, 1375–1377. [Google Scholar] [CrossRef] [PubMed]
- Schlottau, K.; Forth, L.; Angstwurm, K.; Höper, D.; Zecher, D.; Liesche, F.; Hoffmann, B.; Kegel, V.; Seehofer, D.; Platen, S.; et al. Fatal Encephalitic Borna Disease Virus 1 in Solid-Organ Transplant Recipients. N. Engl. J. Med. 2018, 379, 1377–1379. [Google Scholar] [CrossRef]
- Hoffmann, B.; Tappe, D.; Höper, D.; Herden, C.; Boldt, A.; Mawrin, C.; Niederstraßer, O.; Müller, T.; Jenckel, M.; van der Grinten, E.; et al. A Variegated Squirrel Bornavirus Associated with Fatal Human Encephalitis. N. Engl. J. Med. 2015, 373, 154–162. [Google Scholar] [CrossRef]
- Tappe, D.; Pörtner, K.; Frank, C.; Wilking, H.; Ebinger, A.; Herden, C.; Schulze, C.; Muntau, B.; Eggert, P.; Allartz, P.; et al. Investigation of fatal human Borna disease virus 1 encephalitis outside the previously known area for human cases, Brandenburg, Germany—A case report. BMC Infect. Dis. 2021, 21, 787. [Google Scholar] [CrossRef]
- Niller, H.H.; Angstwurm, K.; Rubbenstroth, D.; Schlottau, K.; Ebinger, A.; Giese, S.; Wunderlich, S.; Banas, B.; Forth, L.F.; Hoffmann, D.; et al. Zoonotic spillover infections with Borna disease virus 1 leading to fatal human encephalitis, 1999–2019: An epidemiological investigation. Lancet Infect. Dis. 2020, 20, 467–477. [Google Scholar] [CrossRef]
- Frank, C.; Wickel, J.; Brämer, D.; Matschke, J.; Ibe, R.; Gazivoda, C.; Günther, A.; Hartmann, C.; Rehn, K.; Cadar, D.; et al. Human Borna disease virus 1 (BoDV-1) encephalitis cases in the north and east of Germany. Emerg. Microbes. Infect. 2022, 11, 6–13. [Google Scholar] [CrossRef]
- Meyer, T.; Tappe, D.; Hasan, D.; Rust, M.; Schulz, J.B.; Schiefer, J.; Tauber, S.C. “Borna disease virus 1” (BoDV-1)-Enzephalitis eines 18-Jährigen außerhalb des bisher bekannten Endemiegebietes. DGNeurologie 2022, 5, 300–304. [Google Scholar] [CrossRef]
- Eisermann, P.; Rubbenstroth, D.; Cadar, D.; Thomé-Bolduan, C.; Eggert, P.; Schlaphof, A.; Leypoldt, F.; Stangel, M.; Fortwängler, T.; Hoffmann, F.; et al. Active Case Finding of Current Bornavirus Infections in Human Encephalitis Cases of Unknown Etiology, Germany, 2018-2020. Emerg. Infect. Dis. 2021, 27, 1371–1379. [Google Scholar] [CrossRef]
- Dürrwald, R.; Kolodziejek, J.; Muluneh, A.; Herzog, S.; Nowotny, N. Epidemiological pattern of classical Borna disease and regional genetic clustering of Borna disease viruses point towards the existence of to-date unknown endemic reservoir host populations. Microbes. Infect. 2006, 8, 917–929. [Google Scholar] [CrossRef]
- Nobach, D.; Justus Liebig University Giessen. Potenzielle Reservoire Bei Neurotropen Bornavirus-Infektionen. Ph.D. Thesis, VVB Laufersweiler Verlag, Giessen, Germany, 2021. [Google Scholar] [CrossRef]
- Puorger, M.E.; Hilbe, M.; Müller, J.P.; Kolodziejek, J.; Nowotny, N.; Zlinszky, K.; Ehrensperger, F. Distribution of Borna disease virus antigen and RNA in tissues of naturally infected bicolored white-toothed shrews, Crocidura leucodon, supporting their role as reservoir host species. Vet. Pathol. 2010, 47, 236–244. [Google Scholar] [CrossRef]
- Bourg, M.; Herzog, S.; Encarnação, J.A.; Nobach, D.; Lange-Herbst, H.; Eickmann, M.; Herden, C. Bicolored white-toothed shrews as reservoir for borna disease virus, Bavaria, Germany. Emerg. Infect. Dis. 2013, 19, 2064–2066. [Google Scholar] [CrossRef]
- Hilbe, M.; Herrsche, R.; Kolodziejek, J.; Nowotny, N.; Zlinszky, K.; Ehrensperger, F. Shrews as reservoir hosts of borna disease virus. Emerg. Infect. Dis. 2006, 12, 675–677. [Google Scholar] [CrossRef]
- Matsumoto, Y.; Hayashi, Y.; Omori, H.; Honda, T.; Daito, T.; Horie, M.; Ikuta, K.; Fujino, K.; Nakamura, S.; Schneider, U.; et al. Bornavirus closely associates and segregates with host chromosomes to ensure persistent intranuclear infection. Cell. Host. Microbe. 2012, 11, 492–503. [Google Scholar] [CrossRef]
- Tappe, D.; Frank, C.; Homeier-Bachmann, T.; Wilking, H.; Allendorf, V.; Schlottau, K.; Muñoz-Fontela, C.; Rottstegge, M.; Port, J.R.; Rissland, J.; et al. Analysis of exotic squirrel trade and detection of human infections with variegated squirrel bornavirus 1, Germany, 2005 to 2018. Euro. Surveill. 2019, 24, 1800483. [Google Scholar] [CrossRef]
- Tappe, D.; Schlottau, K.; Cadar, D.; Hoffmann, B.; Balke, L.; Bewig, B.; Hoffmann, D.; Eisermann, P.; Fickenscher, H.; Krumbholz, A.; et al. Occupation-Associated Fatal Limbic Encephalitis Caused by Variegated Squirrel Bornavirus 1, Germany, 2013. Emerg. Infect. Dis. 2018, 24, 978–987. [Google Scholar] [CrossRef]
- Tappe, D.; Frank, C.; Offergeld, R.; Wagner-Wiening, C.; Stark, K.; Rubbenstroth, D.; Giese, S.; Lattwein, E.; Schwemmle, M.; Beer, M.; et al. Low prevalence of Borna disease virus 1 (BoDV-1) IgG antibodies in humans from areas endemic for animal Borna disease of Southern Germany. Sci. Rep. 2019, 9, 20154. [Google Scholar] [CrossRef]
- Bradfute, S.B.; Warfield, K.L.; Bray, M. Mouse models for filovirus infections. Viruses 2012, 4, 1477–1508. [Google Scholar] [CrossRef] [PubMed]
- Shurtleff, A.C.; Bavari, S. Animal models for ebolavirus countermeasures discovery: What defines a useful model? Expert. Opin. Drug Discov. 2015, 10, 685–702. [Google Scholar] [CrossRef] [PubMed]
- Muñoz Fontela, C.; Geisbert, T.W. The gap between animal and human Ebola virus disease. Future Virol. 2017, 12, 61–65. [Google Scholar] [CrossRef]
- Bowen, E.T.; Platt, G.S.; Simpson, D.I.; McArdell, L.B.; Raymond, R.T. Ebola haemorrhagic fever: Experimental infection of monkeys. Trans. R. Soc. Trop. Med. Hyg. 1978, 72, 188–191. [Google Scholar] [CrossRef] [PubMed]
- Haas, R.; Maass, G. Experimental Infection of Monkeys with the Marburg Virus. In Marburg Virus Disease; Martini, G.A., Siegert, R., Eds.; Springer: Berlin/Heidelberg, Germany, 1971; pp. 136–143. [Google Scholar] [CrossRef]
- Hensley, L.E.; Alves, D.A.; Geisbert, J.B.; Fritz, E.A.; Reed, C.; Larsen, T.; Geisbert, T.W. Pathogenesis of Marburg hemorrhagic fever in cynomolgus macaques. J. Infect. Dis. 2011, 204 (Suppl. S3), S1021–S1031. [Google Scholar] [CrossRef]
- Ignatiev, G.M.; Dadaeva, A.A.; Luchko, S.V.; Chepurnov, A.A. Immune and pathophysiological processes in baboons experimentally infected with Ebola virus adapted to guinea pigs. Immunol. Lett. 2000, 71, 131–140. [Google Scholar] [CrossRef]
- Simpson, D. Marburg agent disease: In monkeys. Trans. R. Soc. Trop. Med. Hyg. 1969, 63, 303–309. [Google Scholar] [CrossRef]
- Alves, D.A.; Glynn, A.R.; Steele, K.E.; Lackemeyer, M.G.; Garza, N.L.; Buck, J.G.; Mech, C.; Reed, D.S. Aerosol exposure to the angola strain of marburg virus causes lethal viral hemorrhagic Fever in cynomolgus macaques. Vet. Pathol. 2010, 47, 831–851. [Google Scholar] [CrossRef]
- Johnson, E.D.; Johnson, B.K.; Silverstein, D.; Tukei, P.; Geisbert, T.W.; Sanchez, A.N.; Jahrling, P.B. Characterization of a new Marburg virus isolated from a 1987 fatal case in Kenya. Arch. Virol. Suppl. 1996, 11, 101–114. [Google Scholar]
- Bowen, E.T.; Lloyd, G.; Harris, W.J.; Platt, G.S.; Baskerville, A.; Vella, E.E. Viral haemorrhagic fever in southern Sudan and northern Zaire. Preliminary studies on the aetiological agent. Lancet 1977, 1, 571–573. [Google Scholar] [CrossRef]
- Geisbert, T.W.; Hensley, L.E.; Larsen, T.; Young, H.A.; Reed, D.S.; Geisbert, J.B.; Scott, D.P.; Kagan, E.; Jahrling, P.B.; Davis, K.J. Pathogenesis of Ebola hemorrhagic fever in cynomolgus macaques: Evidence that dendritic cells are early and sustained targets of infection. Am. J. Pathol. 2003, 163, 2347–2370. [Google Scholar] [CrossRef]
- Geisbert, T.W.; Young, H.A.; Jahrling, P.B.; Davis, K.J.; Larsen, T.; Kagan, E.; Hensley, L.E. Pathogenesis of Ebola hemorrhagic fever in primate models: Evidence that hemorrhage is not a direct effect of virus-induced cytolysis of endothelial cells. Am. J. Pathol. 2003, 163, 2371–2382. [Google Scholar] [CrossRef]
- Thi, E.P.; Lee, A.C.; Geisbert, J.B.; Ursic-Bedoya, R.; Agans, K.N.; Robbins, M.; Deer, D.J.; Fenton, K.A.; Kondratowicz, A.S.; MacLachlan, I.; et al. Rescue of non-human primates from advanced Sudan ebolavirus infection with lipid encapsulated siRNA. Nat. Microbiol. 2016, 1, 16142. [Google Scholar] [CrossRef]
- Ellis, D.S.; Bowen, E.T.; Simpson, D.I.; Stamford, S. Ebola virus: A comparison, at ultrastructural level, of the behaviour of the Sudan and Zaire strains in monkeys. Br. J. Exp. Pathol. 1978, 59, 584–593. [Google Scholar]
- Geisbert, T.W.; Daddario-DiCaprio, K.M.; Williams, K.J.; Geisbert, J.B.; Leung, A.; Feldmann, F.; Hensley, L.E.; Feldmann, H.; Jones, S.M. Recombinant vesicular stomatitis virus vector mediates postexposure protection against Sudan Ebola hemorrhagic fever in nonhuman primates. J. Virol. 2008, 82, 5664–5668. [Google Scholar] [CrossRef]
- Mire, C.E.; Geisbert, J.B.; Marzi, A.; Agans, K.N.; Feldmann, H.; Geisbert, T.W. Vesicular stomatitis virus-based vaccines protect nonhuman primates against Bundibugyo ebolavirus. PLoS Negl. Trop. Dis. 2013, 7, e2600. [Google Scholar] [CrossRef]
- Ryabchikova, E.I.; Kolesnikova, L.V.; Luchko, S.V. An analysis of features of pathogenesis in two animal models of Ebola virus infection. J. Infect. Dis. 1999, 179 (Suppl. S1), 199–202. [Google Scholar] [CrossRef]
- Schou, S.; Hansen, A.K. Marburg and Ebola virus infections in laboratory non-human primates: A literature review. Comp. Med. 2000, 50, 108–123. [Google Scholar]
- Baskerville, A.; Bowen, E.T.; Platt, G.S.; McArdell, L.B.; Simpson, D.I. The pathology of experimental Ebola virus infection in monkeys. J. Pathol. 1978, 125, 131–138. [Google Scholar] [CrossRef]
- Ebihara, H.; Rockx, B.; Marzi, A.; Feldmann, F.; Haddock, E.; Brining, D.; LaCasse, R.A.; Gardner, D.; Feldmann, H. Host response dynamics following lethal infection of rhesus macaques with Zaire ebolavirus. J. Infect. Dis. 2011, 204 (Suppl. S3), S991–S999. [Google Scholar] [CrossRef]
- Rollin, P.E.; Williams, R.J.; Bressler, D.S.; Pearson, S.; Cottingham, M.; Pucak, G.; Sanchez, A.; Trappier, S.G.; Peters, R.L.; Greer, P.W.; et al. Ebola (subtype Reston) virus among quarantined nonhuman primates recently imported from the Philippines to the United States. J. Infect. Dis. 1999, 179 (Suppl. S1), S108–S114. [Google Scholar] [CrossRef] [PubMed]
- Jahrling, P.B.; Geisbert, T.W.; Dalgard, D.W.; Johnson, E.D.; Ksiazek, T.G.; Hall, W.C.; Peters, C.J. Preliminary report: Isolation of Ebola virus from monkeys imported to USA. Lancet 1990, 335, 502–505. [Google Scholar] [CrossRef]
- Cross, R.W.; Fenton, K.A.; Geisbert, J.B.; Mire, C.E.; Geisbert, T.W. Modeling the Disease Course of Zaire ebolavirus Infection in the Outbred Guinea Pig. J. Infect. Dis. 2015, 212 (Suppl. S2), S305–S315. [Google Scholar] [CrossRef] [PubMed]
- Marzi, A.; Banadyga, L.; Haddock, E.; Thomas, T.; Shen, K.; Horne, E.J.; Scott, D.P.; Feldmann, H.; Ebihara, H. A hamster model for Marburg virus infection accurately recapitulates Marburg hemorrhagic fever. Sci. Rep. 2016, 6, 39214. [Google Scholar] [CrossRef] [PubMed]
- Cross, R.W.; Mire, C.E.; Borisevich, V.; Geisbert, J.B.; Fenton, K.A.; Geisbert, T.W. The Domestic Ferret (Mustela putorius furo) as a Lethal Infection Model for 3 Species of Ebolavirus. J. Infect. Dis. 2016, 214, 565–569. [Google Scholar] [CrossRef]
- Riabchikova, E.I.; Baranova, S.G.; Tkachev, V.K.; Grazhdantseva, A.A. The morphological changes in Ebola infection in guinea pigs. Vopr. Virusol. 1993, 38, 176–179. [Google Scholar]
- Ryabchikova, E.; Kolesnikova, L.; Smolina, M.; Tkachev, V.; Pereboeva, L.; Baranova, S.; Grazhdantseva, A.; Rassadkin, Y. Ebola virus infection in guinea pigs: Presumable role of granulomatous inflammation in pathogenesis. Arch. Virol. 1996, 141, 909–921. [Google Scholar] [CrossRef]
- Robin, Y.; Brès, P.; Camain, R. Passage of Marburg Virus in Guinea Pigs. In Marburg Virus Disease; Martini, G.A., Siegert, R., Eds.; Springer: Berlin/Heidelberg, Germany, 1971; pp. 117–122. [Google Scholar] [CrossRef]
- Connolly, B.M.; Steele, K.E.; Davis, K.J.; Geisbert, T.W.; Kell, W.M.; Jaax, N.K.; Jahrling, P.B. Pathogenesis of experimental Ebola virus infection in guinea pigs. J. Infect. Dis. 1999, 179 (Suppl. S1), S203–S217. [Google Scholar] [CrossRef]
- Subbotina, E.; Dadaeva, A.; Kachko, A.; Chepurnov, A. Genetic factors of Ebola virus virulence in guinea pigs. Virus Res. 2010, 153, 121–133. [Google Scholar] [CrossRef]
- Wong, G.; He, S.; Wei, H.; Kroeker, A.; Audet, J.; Leung, A.; Cutts, T.; Graham, J.; Kobasa, D.; Embury-Hyatt, C.; et al. Development and Characterization of a Guinea Pig-Adapted Sudan Virus. J. Virol. 2016, 90, 392–399. [Google Scholar] [CrossRef]
- Zlotnik, I. “Marburg Disease” The Pathology of Experimentally Infected Hamsters. In Marburg Virus Disease; Martini, G.A., Siegert, R., Eds.; Springer: Berlin/Heidelberg, Germany, 1971; pp. 129–135. [Google Scholar] [CrossRef]
- Ebihara, H.; Zivcec, M.; Gardner, D.; Falzarano, D.; LaCasse, R.; Rosenke, R.; Long, D.; Haddock, E.; Fischer, E.; Kawaoka, Y.; et al. A Syrian golden hamster model recapitulating ebola hemorrhagic fever. J. Infect. Dis. 2013, 207, 306–318. [Google Scholar] [CrossRef]
- Bray, M. The role of the Type I interferon response in the resistance of mice to filovirus infection. J Gen Virol 2001, 82, 1365–1373. [Google Scholar] [CrossRef]
- Bray, M.; Hatfill, S.; Hensley, L.; Huggins, J.W. Haematological, biochemical and coagulation changes in mice, guinea-pigs and monkeys infected with a mouse-adapted variant of Ebola Zaire virus. J. Comp. Pathol. 2001, 125, 243–253. [Google Scholar] [CrossRef]
- Warfield, K.L.; Alves, D.A.; Bradfute, S.B.; Reed, D.K.; VanTongeren, S.; Kalina, W.V.; Olinger, G.G.; Bavari, S. Development of a model for marburgvirus based on severe-combined immunodeficiency mice. Virol. J. 2007, 4, 108. [Google Scholar] [CrossRef]
- Bray, M.; Davis, K.; Geisbert, T.; Schmaljohn, C.; Huggins, J. A mouse model for evaluation of prophylaxis and therapy of Ebola hemorrhagic fever. J. Infect. Dis. 1998, 178, 651–661. [Google Scholar] [CrossRef]
- Lofts, L.L.; Wells, J.B.; Bavari, S.; Warfield, K.L. Key Genomic Changes Necessary for an In Vivo Lethal Mouse Marburgvirus Variant Selection Process. J. Virol. 2011, 85, 3905–3917. [Google Scholar] [CrossRef]
- Qiu, X.; Wong, G.; Audet, J.; Cutts, T.; Niu, Y.; Booth, S.; Kobinger, G.P. Establishment and characterization of a lethal mouse model for the Angola strain of Marburg virus. J. Virol. 2014, 88, 12703–12714. [Google Scholar] [CrossRef]
- Gupta, M.; Mahanty, S.; Greer, P.; Towner, J.S.; Shieh, W.J.; Zaki, S.R.; Ahmed, R.; Rollin, P.E. Persistent infection with ebola virus under conditions of partial immunity. J. Virol. 2004, 78, 958–967. [Google Scholar] [CrossRef]
- Warfield, K.L.; Bradfute, S.B.; Wells, J.; Lofts, L.; Cooper, M.T.; Alves, D.A.; Reed, D.K.; VanTongeren, S.A.; Mech, C.A.; Bavari, S. Development and characterization of a mouse model for Marburg hemorrhagic fever. J. Virol. 2009, 83, 6404–6415. [Google Scholar] [CrossRef]
- Gibb, T.R.; Bray, M.; Geisbert, T.W.; Steele, K.E.; Kell, W.M.; Davis, K.J.; Jaax, N.K. Pathogenesis of experimental Ebola Zaire virus infection in BALB/c mice. J. Comp. Pathol. 2001, 125, 233–242. [Google Scholar] [CrossRef]
- Lever, M.S.; Piercy, T.J.; Steward, J.A.; Eastaugh, L.; Smither, S.J.; Taylor, C.; Salguero, F.J.; Phillpotts, R.J. Lethality and pathogenesis of airborne infection with filoviruses in A129a/b -/- interferon receptor-deficient mice. J. Med. Microbiol. 2012, 61, 8–15. [Google Scholar] [CrossRef] [PubMed]
- de Wit, E.; Feldmann, H.; Munster, V.J. Tackling Ebola: New insights into prophylactic and therapeutic intervention strategies. Genome. Med. 2011, 3, 5. [Google Scholar] [CrossRef] [PubMed]
- Raymond, J.; Bradfute, S.; Bray, M. Filovirus infection of STAT-1 knockout mice. J. Infect. Dis. 2011, 204 (Suppl. S3), S986–S990. [Google Scholar] [CrossRef] [PubMed]
- Escudero-Pérez, B.; Muñoz-Fontela, C. Role of Type I Interferons on Filovirus Pathogenesis. Vaccines 2019, 7, 22. [Google Scholar] [CrossRef] [PubMed]
- Bird, B.H.; Spengler, J.R.; Chakrabarti, A.K.; Khristova, M.L.; Sealy, T.K.; Coleman-McCray, J.D.; Martin, B.E.; Dodd, K.A.; Goldsmith, C.S.; Sanders, J.; et al. Humanized Mouse Model of Ebola Virus Disease Mimics the Immune Responses in Human Disease. J. Infect. Dis. 2016, 213, 703–711. [Google Scholar] [CrossRef]
- Spengler, J.R.; Prescott, J.; Feldmann, H.; Spiropoulou, C.F. Human immune system mouse models of Ebola virus infection. Curr. Opin. Virol. 2017, 25, 90–96. [Google Scholar] [CrossRef]
- Lüdtke, A.; Oestereich, L.; Ruibal, P.; Wurr, S.; Pallasch, E.; Bockholt, S.; Ip, W.H.; Rieger, T.; Gómez-Medina, S.; Stocking, C.; et al. Ebola virus disease in mice with transplanted human hematopoietic stem cells. J. Virol. 2015, 89, 4700–4704. [Google Scholar] [CrossRef]
- Escudero-Pérez, B.; Ruibal, P.; Rottstegge, M.; Lüdtke, A.; Port, J.R.; Hartmann, K.; Gómez-Medina, S.; Müller-Guhl, J.; Nelson, E.V.; Krasemann, S.; et al. Comparative pathogenesis of Ebola virus and Reston virus infection in humanized mice. JCI Insight 2019, 4, e126070. [Google Scholar] [CrossRef]
- Rasmussen, A.L.; Okumura, A.; Ferris, M.T.; Green, R.; Feldmann, F.; Kelly, S.M.; Scott, D.P.; Safronetz, D.; Haddock, E.; LaCasse, R.; et al. Host genetic diversity enables Ebola hemorrhagic fever pathogenesis and resistance. Science 2014, 346, 987–991. [Google Scholar] [CrossRef]
- Solbrig, M.V. Animal models of CNS viral disease: Examples from borna disease virus models. Interdiscip. Perspect. Infect. Dis. 2010, 2010, 709791. [Google Scholar] [CrossRef]
- Miranda, H.C.; Nunes, S.O.; Calvo, E.S.; Suzart, S.; Itano, E.N.; Watanabe, M.A. Detection of Borna disease virus p24 RNA in peripheral blood cells from Brazilian mood and psychotic disorder patients. J. Affect. Disord. 2006, 90, 43–47. [Google Scholar] [CrossRef]
- Terayama, H.; Nishino, Y.; Kishi, M.; Ikuta, K.; Itoh, M.; Iwahashi, K. Detection of anti-Borna Disease Virus (BDV) antibodies from patients with schizophrenia and mood disorders in Japan. Psychiatry Res. 2003, 120, 201–206. [Google Scholar] [CrossRef]
- Rott, R.; Herzog, S.; Fleischer, B.; Winokur, A.; Amsterdam, J.; Dyson, W.; Koprowski, H. Detection of serum antibodies to Borna disease virus in patients with psychiatric disorders. Science 1985, 228, 755–756. [Google Scholar] [CrossRef]
- Hornig, M.; Briese, T.; Licinio, J.; Khabbaz, R.F.; Altshuler, L.L.; Potkin, S.G.; Schwemmle, M.; Siemetzki, U.; Mintz, J.; Honkavuori, K.; et al. Absence of evidence for bornavirus infection in schizophrenia, bipolar disorder and major depressive disorder. Mol. Psychiatry 2012, 17, 486–493. [Google Scholar] [CrossRef]
- Schlottau, K.; Feldmann, F.; Hanley, P.W.; Lovaglio, J.; Tang-Huau, T.L.; Meade-White, K.; Callison, J.; Williamson, B.N.; Rosenke, R.; Long, D.; et al. Development of a nonhuman primate model for mammalian bornavirus infection. PNAS Nexus 2022, 1, pgac073. [Google Scholar] [CrossRef]
- Stitz, L.; Krey, H.; Ludwig, H. Borna disease in rhesus monkeys as a models for uveo-cerebral symptoms. J. Med. Virol. 1981, 6, 333–340. [Google Scholar] [CrossRef]
- Morales, J.A.; Herzog, S.; Kompter, C.; Frese, K.; Rott, R. Axonal transport of Borna disease virus along olfactory pathways in spontaneously and experimentally infected rats. Med. Microbiol. Immunol. 1988, 177, 51–68. [Google Scholar] [CrossRef]
- Narayan, O.; Herzog, S.; Frese, K.; Scheefers, H.; Rott, R. Behavioral disease in rats caused by immunopathological responses to persistent borna virus in the brain. Science 1983, 220, 1401–1403. [Google Scholar] [CrossRef]
- Hirano, N.; Kao, M.; Ludwig, H. Persistent, tolerant or subacute infection in Borna disease virus-infected rats. J. Gen. Virol. 1983, 64, 1521–1530. [Google Scholar] [CrossRef]
- Planz, O.; Bilzer, T.; Sobbe, M.; Stitz, L. Lysis of major histocompatibility complex class I-bearing cells in Borna disease virus-induced degenerative encephalopathy. J. Exp. Med. 1993, 178, 163–174. [Google Scholar] [CrossRef]
- Narayan, O.; Herzog, S.; Frese, K.; Scheefers, H.; Rott, R. Pathogenesis of Borna disease in rats: Immune-mediated viral ophthalmoencephalopathy causing blindness and behavioral abnormalities. J. Infect. Dis. 1983, 148, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Bilzer, T.; Stitz, L. Immune-mediated brain atrophy. CD8+ T cells contribute to tissue destruction during borna disease. J. Immunol. 1994, 153, 818–823. [Google Scholar] [CrossRef] [PubMed]
- Stitz, L.; Planz, O.; Bilzer, T.; Frei, K.; Fontana, A. Transforming growth factor-beta modulates T cell-mediated encephalitis caused by Borna disease virus. Pathogenic importance of CD8+ cells and suppression of antibody formation. J. Immunol. 1991, 147, 3581–3586. [Google Scholar] [CrossRef] [PubMed]
- Stitz, L.; Soeder, D.; Deschl, U.; Frese, K.; Rott, R. Inhibition of immune-mediated meningoencephalitis in persistently Borna disease virus-infected rats by cyclosporine A. J. Immunol. 1989, 143, 4250–4256. [Google Scholar] [CrossRef] [PubMed]
- Sobbe, M.; Bilzer, T.; Gommel, S.; Nöske, K.; Planz, O.; Stitz, L. Induction of degenerative brain lesions after adoptive transfer of brain lymphocytes from Borna disease virus-infected rats: Presence of CD8+ T cells and perforin mRNA. J. Virol. 1997, 71, 2400–2407. [Google Scholar] [CrossRef]
- Rubin, S.A.; Waltrip, R.W.; Bautista, J.R.; Carbone, K.M. Borna disease virus in mice: Host-specific differences in disease expression. J. Virol. 1993, 67, 548–552. [Google Scholar] [CrossRef]
- Kao, M.; Ludwig, H.; Gosztonyi, G. Adaptation of Borna disease virus to the mouse. J. Gen. Virol. 1984, 65, 1845–1849. [Google Scholar] [CrossRef]
- Mayr, A.; Danner, K. In vitro cultivation of Borna virus using brain explants of infected animals. Zentralbl. Veterinarmed. B 1972, 19, 785–800. [Google Scholar] [CrossRef]
- Ackermann, A.; Staeheli, P.; Schneider, U. Adaptation of Borna disease virus to new host species attributed to altered regulation of viral polymerase activity. J. Virol. 2007, 81, 7933–7940. [Google Scholar] [CrossRef]
- Hallensleben, W.; Schwemmle, M.; Hausmann, J.; Stitz, L.; Volk, B.; Pagenstecher, A.; Staeheli, P. Borna disease virus-induced neurological disorder in mice: Infection of neonates results in immunopathology. J. Virol. 1998, 72, 4379–4386. [Google Scholar] [CrossRef]
- Nitzschke, E. Über den Nachweis eines komplementbindenden Antigens und komplementbindender Antikörper bei weissen Ratten, Meerschweinchen und Pferden, die mit dem Virus der Bornaschen Krankheit infiziert waren. Zentralblatt für Veterinärmedizin 1957, 4, 289–296. [Google Scholar] [CrossRef]
- Danner, K.; Mayr, A. Fluorescence serological studies on the appearance of Borna virus antigen in cell cultures from brain explants of infected rabbits. Zentralbl. Veterinarmed. B 1973, 20, 497–508. [Google Scholar] [CrossRef]
- Anzil, A.P.; Blinzinger, K. Electron microscopic studies of rabbit central and peripheral nervous system in experimental Borna disease. Acta Neuropathol. 1972, 22, 305–318. [Google Scholar] [CrossRef]
- Shapshak, P.; Somboonwit, C.; Sinnott, J.T.; Menezes, L.J.; Kangueane, P.; Balaji, S.C.F. Global Virology III: Virology in the 21st Century; Springer: Berlin/Heidelberg, Germany, 2019. [Google Scholar]
- Ingber, D.E. Human organs-on-chips for disease modelling, drug development and personalized medicine. Nat. Rev. Genet. 2022, 23, 467–491. [Google Scholar] [CrossRef]
- Bhatia, S.N.; Ingber, D.E. Microfluidic organs-on-chips. Nat. Biotechnol. 2014, 32, 760–772. [Google Scholar] [CrossRef]
- Victor, I.A.; Andem, A.B.; Archibong, I.A.; Iwok, E.O. Interplay between Cell Proliferation and Cellular Differentiation: A mutually exclusive paradigm. GSJ 2020, 8, 1328–1338. [Google Scholar]
- Pamies, D.; Bal-Price, A.; Chesné, C.; Coecke, S.; Dinnyes, A.; Eskes, C.; Grillari, R.; Gstaunthaler, G.; Hartung, T.; Jennings, P.; et al. Advanced Good Cell Culture Practice for Human Primary, Stem Cell-Derived and Organoid Models as well as Microphysiological Systems. ALTEX 2018, 35, 353–378. [Google Scholar] [CrossRef]
- Brnic, D.; Stevanovic, V.; Cochet, M.; Agier, C.; Richardson, J.; Montero-Menei, C.N.; Milhavet, O.; Eloit, M.; Coulpier, M. Borna disease virus infects human neural progenitor cells and impairs neurogenesis. J. Virol. 2012, 86, 2512–2522. [Google Scholar] [CrossRef][Green Version]
- Scordel, C.; Huttin, A.; Cochet-Bernoin, M.; Szelechowski, M.; Poulet, A.; Richardson, J.; Benchoua, A.; Gonzalez-Dunia, D.; Eloit, M.; Coulpier, M. Borna disease virus phosphoprotein impairs the developmental program controlling neurogenesis and reduces human GABAergic neurogenesis. PLoS Pathog. 2015, 11, e1004859. [Google Scholar] [CrossRef]
- March, S.; Ramanan, V.; Trehan, K.; Ng, S.; Galstian, A.; Gural, N.; Scull, M.A.; Shlomai, A.; Mota, M.M.; Fleming, H.E.; et al. Micropatterned coculture of primary human hepatocytes and supportive cells for the study of hepatotropic pathogens. Nat. Protoc. 2015, 10, 2027–2053. [Google Scholar] [CrossRef]
- Scoon, W.A.; Mancio-Silva, L.; Suder, E.L.; Villacorta-Martin, C.; Lindstrom-Vautrin, J.; Bernbaum, J.G.; Mazur, S.; Johnson, R.F.; Olejnik, J.; Flores, E.Y.; et al. Ebola virus infection induces a delayed type I IFN response in bystander cells and the shutdown of key liver genes in human iPSC-derived hepatocytes. Stem. Cell. Rep. 2022, 17, 2286–2302. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.W.; Khetani, S.R.; Krzyzewski, S.; Duignan, D.B.; Obach, R.S. Assessment of a micropatterned hepatocyte coculture system to generate major human excretory and circulating drug metabolites. Drug Metab. Dispos. 2010, 38, 1900–1905. [Google Scholar] [CrossRef] [PubMed]
- Le, H.; Spearman, P.; Waggoner, S.N.; Singh, K. Ebola virus protein VP40 stimulates IL-12- and IL-18-dependent activation of human natural killer cells. JCI Insight 2022, 7, e158902. [Google Scholar] [CrossRef]
- Lubaki, N.M.; Younan, P.; Santos, R.I.; Meyer, M.; Iampietro, M.; Koup, R.A.; Bukreyev, A. The Ebola Interferon Inhibiting Domains Attenuate and Dysregulate Cell-Mediated Immune Responses. PLoS Pathog. 2016, 12, e1006031. [Google Scholar] [CrossRef] [PubMed]
- Richt, J.A.; Stitz, L. Borna disease virus-infected astrocytes function as antigen-presenting and target cells for virus-specific CD4-bearing lymphocytes. Arch. Virol. 1992, 124, 95–109. [Google Scholar] [CrossRef]
- Hu, W.; Lazar, M.A. Modelling metabolic diseases and drug response using stem cells and organoids. Nat. Rev. Endocrinol. 2022, 18, 744–759. [Google Scholar] [CrossRef]
- Antonucci, J.; Gehrke, L. Cerebral Organoid Models for Neurotropic Viruses. ACS Infect. Dis. 2019, 5, 1976–1979. [Google Scholar] [CrossRef]
- Clevers, H. Modeling Development and Disease with Organoids. Cell 2016, 165, 1586–1597. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Knoblich, J.A. Organogenesis in a dish: Modeling development and disease using organoid technologies. Science 2014, 345, 1247125. [Google Scholar] [CrossRef]
- Kim, J.; Koo, B.K.; Knoblich, J.A. Human organoids: Model systems for human biology and medicine. Nat. Rev. Mol. Cell. Biol. 2020, 21, 571–584. [Google Scholar] [CrossRef]
- Kessler, M.; Hoffmann, K.; Fritsche, K.; Brinkmann, V.; Mollenkopf, H.J.; Thieck, O.; Teixeira da Costa, A.R.; Braicu, E.I.; Sehouli, J.; Mangler, M.; et al. Chronic Chlamydia infection in human organoids increases stemness and promotes age-dependent CpG methylation. Nat. Commun. 2019, 10, 1194. [Google Scholar] [CrossRef]
- Dos Reis, R.S.; Sant, S.; Keeney, H.; Wagner, M.C.E.; Ayyavoo, V. Modeling HIV-1 neuropathogenesis using three-dimensional human brain organoids (hBORGs) with HIV-1 infected microglia. Sci. Rep. 2020, 10, 15209. [Google Scholar] [CrossRef]
- Chen, K.G.; Park, K.; Spence, J.R. Studying SARS-CoV-2 infectivity and therapeutic responses with complex organoids. Nat. Cell. Biol. 2021, 23, 822–833. [Google Scholar] [CrossRef]
- Paşca, S.P. Assembling human brain organoids. Science 2019, 363, 126–127. [Google Scholar] [CrossRef]
- Lin, Y.T.; Seo, J.; Gao, F.; Feldman, H.M.; Wen, H.L.; Penney, J.; Cam, H.P.; Gjoneska, E.; Raja, W.K.; Cheng, J.; et al. APOE4 Causes Widespread Molecular and Cellular Alterations Associated with Alzheimer’s Disease Phenotypes in Human iPSC-Derived Brain Cell Types. Neuron 2018, 98, 1294. [Google Scholar] [CrossRef]
- Gleeson, J.; Wang, L.; Sievert, D.; Clark, A.; Federman, H.; Gastfriend, B.; Shusta, E.V.; Palecek, S.P.; Carlin, A. A Human 3D neural assembloid model for SARS-CoV-2 infection. bioRxiv 2021. [Google Scholar] [CrossRef]
- Birey, F.; Andersen, J.; Makinson, C.D.; Islam, S.; Wei, W.; Huber, N.; Fan, H.C.; Metzler, K.R.C.; Panagiotakos, G.; Thom, N.; et al. Assembly of functionally integrated human forebrain spheroids. Nature 2017, 545, 54–59. [Google Scholar] [CrossRef]
- Cederquist, G.Y.; Asciolla, J.J.; Tchieu, J.; Walsh, R.M.; Cornacchia, D.; Resh, M.D.; Studer, L. Specification of positional identity in forebrain organoids. Nat. Biotechnol. 2019, 37, 436–444. [Google Scholar] [CrossRef]
- Watanabe, M.; Buth, J.E.; Vishlaghi, N.; de la Torre-Ubieta, L.; Taxidis, J.; Khakh, B.S.; Coppola, G.; Pearson, C.A.; Yamauchi, K.; Gong, D.; et al. Self-Organized Cerebral Organoids with Human-Specific Features Predict Effective Drugs to Combat Zika Virus Infection. Cell. Rep. 2017, 21, 517–532. [Google Scholar] [CrossRef]
- Zhou, J.; Li, C.; Sachs, N.; Chiu, M.C.; Wong, B.H.; Chu, H.; Poon, V.K.; Wang, D.; Zhao, X.; Wen, L.; et al. Differentiated human airway organoids to assess infectivity of emerging influenza virus. Proc. Natl. Acad. Sci. USA 2018, 115, 6822–6827. [Google Scholar] [CrossRef]
- Zhou, J.; Li, C.; Liu, X.; Chiu, M.C.; Zhao, X.; Wang, D.; Wei, Y.; Lee, A.; Zhang, A.J.; Chu, H.; et al. Infection of bat and human intestinal organoids by SARS-CoV-2. Nat. Med. 2020, 26, 1077–1083. [Google Scholar] [CrossRef] [PubMed]
- Ayala-Nunez, N.V.; Follain, G.; Delalande, F.; Hirschler, A.; Partiot, E.; Hale, G.L.; Bollweg, B.C.; Roels, J.; Chazal, M.; Bakoa, F.; et al. Zika virus enhances monocyte adhesion and transmigration favoring viral dissemination to neural cells. Nat. Commun. 2019, 10, 4430. [Google Scholar] [CrossRef] [PubMed]
- Salahudeen, A.A.; Choi, S.S.; Rustagi, A.; Zhu, J.; van Unen, V.; de la O, S.M.; Flynn, R.A.; Margalef-Català, M.; Santos, A.J.M.; Ju, J.; et al. Progenitor identification and SARS-CoV-2 infection in human distal lung organoids. Nature 2020, 588, 670–675. [Google Scholar] [CrossRef] [PubMed]
- Giobbe, G.G.; Bonfante, F.; Jones, B.C.; Gagliano, O.; Luni, C.; Zambaiti, E.; Perin, S.; Laterza, C.; Busslinger, G.; Stuart, H.; et al. SARS-CoV-2 infection and replication in human gastric organoids. Nat. Commun. 2021, 12, 6610. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Duan, X.; Yang, L.; Nilsson-Payant, B.E.; Wang, P.; Duan, F.; Tang, X.; Yaron, T.M.; Zhang, T.; Uhl, S.; et al. Identification of SARS-CoV-2 inhibitors using lung and colonic organoids. Nature 2021, 589, 270–275. [Google Scholar] [CrossRef]
- Sano, E.; Suzuki, T.; Hashimoto, R.; Itoh, Y.; Sakamoto, A.; Sakai, Y.; Saito, A.; Okuzaki, D.; Motooka, D.; Muramoto, Y.; et al. Cell response analysis in SARS-CoV-2 infected bronchial organoids. Commun. Biol. 2022, 5, 516. [Google Scholar] [CrossRef]
- Chiu, M.C.; Li, C.; Liu, X.; Yu, Y.; Huang, J.; Wan, Z.; Xiao, D.; Chu, H.; Cai, J.P.; Zhou, B.; et al. A bipotential organoid model of respiratory epithelium recapitulates high infectivity of SARS-CoV-2 Omicron variant. Cell. Discov. 2022, 8, 57. [Google Scholar] [CrossRef]
- Dickson, I. Organoids demonstrate gut infection by SARS-CoV-2. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 383. [Google Scholar] [CrossRef]
- Krenn, V.; Bosone, C.; Burkard, T.R.; Spanier, J.; Kalinke, U.; Calistri, A.; Salata, C.; Rilo Christoff, R.; Pestana Garcez, P.; Mirazimi, A.; et al. Organoid modeling of Zika and herpes simplex virus 1 infections reveals virus-specific responses leading to microcephaly. Cell Stem Cell 2021, 28, 1362–1379. [Google Scholar] [CrossRef]
- Zhang, B.; He, Y.; Xu, Y.; Mo, F.; Mi, T.; Shen, Q.S.; Li, C.; Li, Y.; Liu, J.; Wu, Y.; et al. Differential antiviral immunity to Japanese encephalitis virus in developing cortical organoids. Cell. Death. Dis. 2018, 9, 719. [Google Scholar] [CrossRef]
- Li, V.S.W. Modelling intestinal inflammation and infection using ’mini-gut’ organoids. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 89–90. [Google Scholar] [CrossRef]
- Qian, X.; Nguyen, H.N.; Jacob, F.; Song, H.; Ming, G.L. Using brain organoids to understand Zika virus-induced microcephaly. Development 2017, 144, 952–957. [Google Scholar] [CrossRef]
- Hoffmann, K.; Obermayer, B.; Hönzke, K.; Fatykhova, D.; Demir, Z.; Löwa, A.; Alves, L.G.T.; Wyler, E.; Lopez-Rodriguez, E.; Mieth, M.; et al. Human alveolar progenitors generate dual lineage bronchioalveolar organoids. Commun. Biol. 2022, 5, 875. [Google Scholar] [CrossRef]
- Koster, S.; Gurumurthy, R.K.; Kumar, N.; Prakash, P.G.; Dhanraj, J.; Bayer, S.; Berger, H.; Kurian, S.M.; Drabkina, M.; Mollenkopf, H.J.; et al. Modelling Chlamydia and HPV co-infection in patient-derived ectocervix organoids reveals distinct cellular reprogramming. Nat. Commun. 2022, 13, 1030. [Google Scholar] [CrossRef]
- Purwada, A.; Singh, A. Immuno-engineered organoids for regulating the kinetics of B-cell development and antibody production. Nat. Protoc. 2017, 12, 168–182. [Google Scholar] [CrossRef]
- Huh, D.; Hamilton, G.A.; Ingber, D.E. From 3D cell culture to organs-on-chips. Trends Cell. Biol. 2011, 21, 745–754. [Google Scholar] [CrossRef]
- Derda, R.; Tang, S.K.; Laromaine, A.; Mosadegh, B.; Hong, E.; Mwangi, M.; Mammoto, A.; Ingber, D.E.; Whitesides, G.M. Multizone paper platform for 3D cell cultures. PLoS ONE 2011, 6, e18940. [Google Scholar] [CrossRef]
- Huh, D.D. A human breathing lung-on-a-chip. Ann. Am. Thorac. Soc. 2015, 12 (Suppl. S1), S42–S44. [Google Scholar] [CrossRef]
- Lee, S.E.; Choi, H.; Shin, N.; Kong, D.; Kim, N.G.; Kim, H.Y.; Kim, M.J.; Choi, S.W.; Kim, Y.B.; Kang, K.S. Zika virus infection accelerates Alzheimer’s disease phenotypes in brain organoids. Cell. Death. Discov. 2022, 8, 153. [Google Scholar] [CrossRef]
- Duffy, D.C.; McDonald, J.C.; Schueller, O.J.; Whitesides, G.M. Rapid Prototyping of Microfluidic Systems in Poly(dimethylsiloxane). Anal. Chem. 1998, 70, 4974–4984. [Google Scholar] [CrossRef]
- Chiu, D.T.; Jeon, N.L.; Huang, S.; Kane, R.S.; Wargo, C.J.; Choi, I.S.; Ingber, D.E.; Whitesides, G.M. Patterned deposition of cells and proteins onto surfaces by using three-dimensional microfluidic systems. Proc. Natl. Acad. Sci. USA 2000, 97, 2408–2413. [Google Scholar] [CrossRef] [PubMed]
- Folch, A.; Toner, M. Cellular micropatterns on biocompatible materials. Biotechnol. Prog. 1998, 14, 388–392. [Google Scholar] [CrossRef] [PubMed]
- Folch, A.; Ayon, A.; Hurtado, O.; Schmidt, M.A.; Toner, M. Molding of deep polydimethylsiloxane microstructures for microfluidics and biological applications. J. Biomech. Eng. 1999, 121, 28–34. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Takayama, S.; Ostuni, E.; LeDuc, P.; Naruse, K.; Ingber, D.E.; Whitesides, G.M. Subcellular positioning of small molecules. Nature 2001, 411, 1016. [Google Scholar] [CrossRef] [PubMed]
- Li Jeon, N.; Baskaran, H.; Dertinger, S.K.; Whitesides, G.M.; Van de Water, L.; Toner, M. Neutrophil chemotaxis in linear and complex gradients of interleukin-8 formed in a microfabricated device. Nat. Biotechnol. 2002, 20, 826–830. [Google Scholar] [CrossRef]
- Selimović, S.; Sim, W.Y.; Kim, S.B.; Jang, Y.H.; Lee, W.G.; Khabiry, M.; Bae, H.; Jambovane, S.; Hong, J.W.; Khademhosseini, A. Generating nonlinear concentration gradients in microfluidic devices for cell studies. Anal. Chem. 2011, 83, 2020–2028. [Google Scholar] [CrossRef]
- Song, J.W.; Gu, W.; Futai, N.; Warner, K.A.; Nor, J.E.; Takayama, S. Computer-controlled microcirculatory support system for endothelial cell culture and shearing. Anal. Chem. 2005, 77, 3993–3999. [Google Scholar] [CrossRef]
- Agarwal, A.; Goss, J.A.; Cho, A.; McCain, M.L.; Parker, K.K. Microfluidic heart on a chip for higher throughput pharmacological studies. Lab. Chip. 2013, 13, 3599–3608. [Google Scholar] [CrossRef]
- Mondadori, C.; Palombella, S.; Salehi, S.; Talò, G.; Visone, R.; Rasponi, M.; Redaelli, A.; Sansone, V.; Moretti, M.; Lopa, S. Recapitulating monocyte extravasation to the synovium in an organotypic microfluidic model of the articular joint. Biofabrication 2021, 13. [Google Scholar] [CrossRef]
- Kerns, S.J.; Belgur, C.; Petropolis, D.; Kanellias, M.; Barrile, R.; Sam, J.; Weinzierl, T.; Fauti, T.; Freimoser-Grundschober, A.; Eckmann, J.; et al. Human immunocompetent Organ-on-Chip platforms allow safety profiling of tumor-targeted T-cell bispecific antibodies. Elife 2021, 10, e67106. [Google Scholar] [CrossRef]
- El-Ali, J.; Sorger, P.K.; Jensen, K.F. Cells on chips. Nature 2006, 442, 403–411. [Google Scholar] [CrossRef]
- Meyvantsson, I.; Beebe, D.J. Cell culture models in microfluidic systems. Annu. Rev. Anal. Chem. (Palo Alto Calif) 2008, 1, 423–449. [Google Scholar] [CrossRef]
- Singhvi, R.; Kumar, A.; Lopez, G.P.; Stephanopoulos, G.N.; Wang, D.I.; Whitesides, G.M.; Ingber, D.E. Engineering cell shape and function. Science 1994, 264, 696–698. [Google Scholar] [CrossRef]
- Lee, P.J.; Hung, P.J.; Lee, L.P. An artificial liver sinusoid with a microfluidic endothelial-like barrier for primary hepatocyte culture. Biotechnol. Bioeng. 2007, 97, 1340–1346. [Google Scholar] [CrossRef]
- Kim, J.; Lee, K.T.; Lee, J.S.; Shin, J.; Cui, B.; Yang, K.; Choi, Y.S.; Choi, N.; Lee, S.H.; Lee, J.H.; et al. Fungal brain infection modelled in a human-neurovascular-unit-on-a-chip with a functional blood–brain barrier. Nat. Biomed. Eng. 2021, 5, 830–846. [Google Scholar] [CrossRef]
- Maschmeyer, I.; Lorenz, A.K.; Schimek, K.; Hasenberg, T.; Ramme, A.P.; Hübner, J.; Lindner, M.; Drewell, C.; Bauer, S.; Thomas, A.; et al. A four-organ-chip for interconnected long-term co-culture of human intestine, liver, skin and kidney equivalents. Lab. Chip. 2015, 15, 2688–2699. [Google Scholar] [CrossRef]
- Chou, D.B.; Frismantas, V.; Milton, Y.; David, R.; Pop-Damkov, P.; Ferguson, D.; MacDonald, A.; Vargel Bölükbaşı, O.; Joyce, C.E.; Moreira Teixeira, L.S.; et al. On-chip recapitulation of clinical bone marrow toxicities and patient-specific pathophysiology. Nat. Biomed. Eng. 2020, 4, 394–406. [Google Scholar] [CrossRef]
- Ramsden, D.; Belair, D.G.; Agarwal, S.; Andersson, P.; Humphreys, S.; Dalmas, D.A.; Stahl, S.H.; Maclauchlin, C.; Cichocki, J.A. Leveraging microphysiological systems to address challenges encountered during development of oligonucleotide therapeutics. ALTEX 2022, 39, 273–296. [Google Scholar] [CrossRef]
- Viravaidya, K.; Shuler, M.L. Incorporation of 3T3-L1 cells to mimic bioaccumulation in a microscale cell culture analog device for toxicity studies. Biotechnol. Prog. 2004, 20, 590–597. [Google Scholar] [CrossRef]
- Sin, A.; Chin, K.C.; Jamil, M.F.; Kostov, Y.; Rao, G.; Shuler, M.L. The design and fabrication of three-chamber microscale cell culture analog devices with integrated dissolved oxygen sensors. Biotechnol. Prog. 2004, 20, 338–345. [Google Scholar] [CrossRef]
- Mahler, G.J.; Esch, M.B.; Glahn, R.P.; Shuler, M.L. Characterization of a gastrointestinal tract microscale cell culture analog used to predict drug toxicity. Biotechnol. Bioeng. 2009, 104, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Sung, J.H.; Shuler, M.L. A micro cell culture analog (microCCA) with 3-D hydrogel culture of multiple cell lines to assess metabolism-dependent cytotoxicity of anti-cancer drugs. Lab. Chip. 2009, 9, 1385–1394. [Google Scholar] [CrossRef] [PubMed]
- Miller, P.G.; Shuler, M.L. Design and demonstration of a pumpless 14 compartment microphysiological system. Biotechnol. Bioeng. 2016, 113, 2213–2227. [Google Scholar] [CrossRef] [PubMed]
- Esch, M.B.; King, T.L.; Shuler, M.L. The role of body-on-a-chip devices in drug and toxicity studies. Annu. Rev. Biomed. Eng. 2011, 13, 55–72. [Google Scholar] [CrossRef] [PubMed]
- Pires de Mello, C.P.; Carmona-Moran, C.; McAleer, C.W.; Perez, J.; Coln, E.A.; Long, C.J.; Oleaga, C.; Riu, A.; Note, R.; Teissier, S.; et al. Microphysiological heart-liver body-on-a-chip system with a skin mimic for evaluating topical drug delivery. Lab. Chip. 2020, 20, 749–759. [Google Scholar] [CrossRef]
- Chen, W.L.K.; Edington, C.; Suter, E.; Yu, J.; Velazquez, J.J.; Velazquez, J.G.; Shockley, M.; Large, E.M.; Venkataramanan, R.; Hughes, D.J.; et al. Integrated gut/liver microphysiological systems elucidates inflammatory inter-tissue crosstalk. Biotechnol. Bioeng. 2017, 114, 2648–2659. [Google Scholar] [CrossRef]
- Maoz, B.M.; Herland, A.; FitzGerald, E.A.; Grevesse, T.; Vidoudez, C.; Pacheco, A.R.; Sheehy, S.P.; Park, T.E.; Dauth, S.; Mannix, R.; et al. A linked organ-on-chip model of the human neurovascular unit reveals the metabolic coupling of endothelial and neuronal cells. Nat. Biotechnol. 2018, 36, 865–874. [Google Scholar] [CrossRef]
- Li, Z.A.; Tuan, R.S. Towards establishing human body-on-a-chip systems. Stem. Cell. Res. Ther. 2022, 13, 431. [Google Scholar] [CrossRef]
- Herland, A.; Maoz, B.M.; Das, D.; Somayaji, M.R.; Prantil-Baun, R.; Novak, R.; Cronce, M.; Huffstater, T.; Jeanty, S.S.F.; Ingram, M.; et al. Quantitative prediction of human pharmacokinetic responses to drugs via fluidically coupled vascularized organ chips. Nat. Biomed. Eng. 2020, 4, 421–436. [Google Scholar] [CrossRef]
- Sasserath, T.; Rumsey, J.W.; McAleer, C.W.; Bridges, L.R.; Long, C.J.; Elbrecht, D.; Schuler, F.; Roth, A.; Bertinetti-LaPatki, C.; Shuler, M.L.; et al. Differential Monocyte Actuation in a Three-Organ Functional Innate Immune System-on-a-Chip. Adv. Sci. (Weinh) 2020, 7, 2000323. [Google Scholar] [CrossRef]
- Ronaldson-Bouchard, K.; Teles, D.; Yeager, K.; Tavakol, D.N.; Zhao, Y.; Chramiec, A.; Tagore, S.; Summers, M.; Stylianos, S.; Tamargo, M.; et al. A multi-organ chip with matured tissue niches linked by vascular flow. Nat. Biomed. Eng. 2022, 6, 351–371. [Google Scholar] [CrossRef]
- Benam, K.H.; Villenave, R.; Lucchesi, C.; Varone, A.; Hubeau, C.; Lee, H.H.; Alves, S.E.; Salmon, M.; Ferrante, T.C.; Weaver, J.C.; et al. Small airway-on-a-chip enables analysis of human lung inflammation and drug responses. Nat. Methods 2016, 13, 151–157. [Google Scholar] [CrossRef]
- Zhang, M.; Wang, P.; Luo, R.; Wang, Y.; Li, Z.; Guo, Y.; Yao, Y.; Li, M.; Tao, T.; Chen, W.; et al. Biomimetic Human Disease Model of SARS-CoV-2-Induced Lung Injury and Immune Responses on Organ Chip System. Adv. Sci. (Weinh) 2021, 8, 2002928. [Google Scholar] [CrossRef]
- Bai, H.; Si, L.; Jiang, A.; Belgur, C.; Zhai, Y.; Plebani, R.; Oh, C.Y.; Rodas, M.; Patil, A.; Nurani, A.; et al. Mechanical control of innate immune responses against viral infection revealed in a human lung alveolus chip. Nat. Commun. 2022, 13, 1928. [Google Scholar] [CrossRef]
- Si, L.; Bai, H.; Rodas, M.; Cao, W.; Oh, C.Y.; Jiang, A.; Moller, R.; Hoagland, D.; Oishi, K.; Horiuchi, S.; et al. A human-airway-on-a-chip for the rapid identification of candidate antiviral therapeutics and prophylactics. Nat. Biomed. Eng. 2021, 5, 815–829. [Google Scholar] [CrossRef]
- Ortega-Prieto, A.M.; Skelton, J.K.; Wai, S.N.; Large, E.; Lussignol, M.; Vizcay-Barrena, G.; Hughes, D.; Fleck, R.A.; Thursz, M.; Catanese, M.T.; et al. 3D microfluidic liver cultures as a physiological preclinical tool for hepatitis B virus infection. Nat. Commun. 2018, 9, 682. [Google Scholar] [CrossRef]
- Johnson, B.N.; Lancaster, K.Z.; Hogue, I.B.; Meng, F.; Kong, Y.L.; Enquist, L.W.; McAlpine, M.C. Correction: 3D printed nervous system on a chip. Lab. Chip. 2016, 16, 1946. [Google Scholar] [CrossRef]
- Villenave, R.; Wales, S.Q.; Hamkins-Indik, T.; Papafragkou, E.; Weaver, J.C.; Ferrante, T.C.; Bahinski, A.; Elkins, C.A.; Kulka, M.; Ingber, D.E. Human Gut-On-A-Chip Supports Polarized Infection of Coxsackie B1 Virus In Vitro. PLoS ONE 2017, 12, e0169412. [Google Scholar] [CrossRef]
- Bein, A.; Kim, S.; Goyal, G.; Cao, W.; Fadel, C.; Naziripour, A.; Sharma, S.; Swenor, B.; LoGrande, N.; Nurani, A.; et al. Enteric Coronavirus Infection and Treatment Modeled With an Immunocompetent Human Intestine-On-A-Chip. Front. Pharmacol. 2021, 12, 718484. [Google Scholar] [CrossRef]
- Guo, Y.; Luo, R.; Wang, Y.; Deng, P.; Song, T.; Zhang, M.; Wang, P.; Zhang, X.; Cui, K.; Tao, T.; et al. SARS-CoV-2 induced intestinal responses with a biomimetic human gut-on-chip. Sci. Bull. 2021, 66, 783–793. [Google Scholar] [CrossRef]
- Wang, J.; Wang, C.; Xu, N.; Liu, Z.F.; Pang, D.W.; Zhang, Z.L. A virus-induced kidney disease model based on organ-on-a-chip: Pathogenesis exploration of virus-related renal dysfunctions. Biomaterials 2019, 219, 119367. [Google Scholar] [CrossRef] [PubMed]
- Gard, A.L.; Luu, R.J.; Miller, C.R.; Maloney, R.; Cain, B.P.; Marr, E.E.; Burns, D.M.; Gaibler, R.; Mulhern, T.J.; Wong, C.A.; et al. High-throughput human primary cell-based airway model for evaluating influenza, coronavirus, or other respiratory viruses. Sci. Rep. 2021, 11, 14961. [Google Scholar] [CrossRef] [PubMed]
- Junaid, A.; Tang, H.; van Reeuwijk, A.; Abouleila, Y.; Wuelfroth, P.; van Duinen, V.; Stam, W.; van Zonneveld, A.J.; Hankemeier, T.; Mashaghi, A. Ebola Hemorrhagic Shock Syndrome-on-a-Chip. iScience 2020, 23, 100765. [Google Scholar] [CrossRef] [PubMed]
- Cable, J.; Lutolf, M.P.; Fu, J.; Park, S.E.; Apostolou, A.; Chen, S.; Song, C.J.; Spence, J.R.; Liberali, P.; Lancaster, M.; et al. Organoids as tools for fundamental discovery and translation-A Keystone Symposia report. Ann. N. Y. Acad. Sci. 2022, 1518, 196–208. [Google Scholar] [CrossRef] [PubMed]
- Cecen, B.; Karavasili, C.; Nazir, M.; Bhusal, A.; Dogan, E.; Shahriyari, F.; Tamburaci, S.; Buyukoz, M.; Kozaci, L.D.; Miri, A.K. Multi-Organs-on-Chips for Testing Small-Molecule Drugs: Challenges and Perspectives. Pharmaceutics 2021, 13, 1657. [Google Scholar] [CrossRef]
- Leung, C.M.; de Haan, P.; Ronaldson-Bouchard, K.; Kim, G.-A.; Ko, J.; Rho, H.S.; Chen, Z.; Habibovic, O.; Jeon, N.L.; Takayama, S.; et al. A guide to the organ-on-a-chip. Nat. Rev. Methods Prim. 2022, 2, 33. [Google Scholar] [CrossRef]
- Greek, R.; Menache, A. Systematic reviews of animal models: Methodology versus epistemology. Int. J. Med. Sci. 2013, 10, 206–221. [Google Scholar] [CrossRef]
- Bailey, J.; Thew, M.; Balls, M. An analysis of the use of animal models in predicting human toxicology and drug safety. Altern. Lab. Anim. 2014, 42, 181–199. [Google Scholar] [CrossRef]
- Seok, J.; Warren, H.S.; Cuenca, A.G.; Mindrinos, M.N.; Baker, H.V.; Xu, W.; Richards, D.R.; McDonald-Smith, G.P.; Gao, H.; Hennessy, L.; et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc. Natl. Acad. Sci. USA 2013, 110, 3507–3512. [Google Scholar] [CrossRef]
- Pain, B.; Baquerre, C.; Coulpier, M. Cerebral organoids and their potential for studies of brain diseases in domestic animals. Vet. Res. 2021, 52, 65. [Google Scholar] [CrossRef]
- Dunning, J.; Kennedy, S.B.; Antierens, A.; Whitehead, J.; Ciglenecki, I.; Carson, G.; Kanapathipillai, R.; Castle, L.; Howell-Jones, R.; Pardinaz-Solis, R.; et al. Experimental Treatment of Ebola Virus Disease with Brincidofovir. PLoS ONE 2016, 11, e0162199. [Google Scholar] [CrossRef]
- Notaras, M.; Lodhi, A.; Dündar, F.; Collier, P.; Sayles, N.M.; Tilgner, H.; Greening, D.; Colak, D. Schizophrenia is defined by cell-specific neuropathology and multiple neurodevelopmental mechanisms in patient-derived cerebral organoids. Mol. Psychiatry 2022, 27, 1416–1434. [Google Scholar] [CrossRef]
- Esteban, M.A.; Wang, T.; Qin, B.; Yang, J.; Qin, D.; Cai, J.; Li, W.; Weng, Z.; Chen, J.; Ni, S.; et al. Vitamin C enhances the generation of mouse and human induced pluripotent stem cells. Cell Stem Cell 2010, 6, 71–79. [Google Scholar] [CrossRef]
- Lorenzo, I.M.; Fleischer, A.; Bachiller, D. Generation of mouse and human induced pluripotent stem cells (iPSC) from primary somatic cells. Stem. Cell. Rev. Rep. 2013, 9, 435–450. [Google Scholar] [CrossRef]
- Stauske, M.; Rodriguez Polo, I.; Haas, W.; Knorr, D.Y.; Borchert, T.; Streckfuss-Bömeke, K.; Dressel, R.; Bartels, I.; Tiburcy, M.; Zimmermann, W.H.; et al. Non-Human Primate iPSC Generation, Cultivation, and Cardiac Differentiation under Chemically Defined Conditions. Cells 2020, 9, 1349. [Google Scholar] [CrossRef]
- D’Souza, S.S.; Kumar, A.; Maufort, J.; Weinfurter, J.T.; Raymond, M.; Strelchenko, N.S.; Perrin, E.; Coonen, J.; Mejia, A.; Simmons, H.A.; et al. Assessment of safety and immunogenicity of MHC homozygous iPSC-derived CD34+ hematopoietic progenitors in an NHP model. Blood Adv. 2022, 6, 5267–5278. [Google Scholar] [CrossRef]
- Rodriguez-Polo, I.; Mißbach, S.; Petkov, S.; Mattern, F.; Maierhofer, A.; Grządzielewska, I.; Tereshchenko, Y.; Urrutia-Cabrera, D.; Haaf, T.; Dressel, R.; et al. A piggyBac-based platform for genome editing and clonal rhesus macaque iPSC line derivation. Sci. Rep. 2021, 11, 15439. [Google Scholar] [CrossRef]
- Petkov, S.; Glage, S.; Nowak-Imialek, M.; Niemann, H. Long-Term Culture of Porcine Induced Pluripotent Stem-Like Cells Under Feeder-Free Conditions in the Presence of Histone Deacetylase Inhibitors. Stem. Cells Dev. 2016, 25, 386–394. [Google Scholar] [CrossRef]
- Ezashi, T.; Yuan, Y.; Roberts, R.M. Pluripotent Stem Cells from Domesticated Mammals. Annu. Rev. Anim. Biosci. 2016, 4, 223–253. [Google Scholar] [CrossRef]
- Gao, X.; Ruan, D.; Liu, P. Reprogramming Porcine Fibroblast to EPSCs. Methods Mol. Biol. 2021, 2239, 199–211. [Google Scholar]
- Nagy, K.; Sung, H.K.; Zhang, P.; Laflamme, S.; Vincent, P.; Agha-Mohammadi, S.; Woltjen, K.; Monetti, C.; Michael, I.P.; Smith, L.C.; et al. Induced pluripotent stem cell lines derived from equine fibroblasts. Stem. Cell. Rev. Rep. 2011, 7, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Breton, A.; Sharma, R.; Diaz, A.C.; Parham, A.G.; Graham, A.; Neil, C.; Whitelaw, C.B.; Milne, E.; Donadeu, F.X. Derivation and characterization of induced pluripotent stem cells from equine fibroblasts. Stem. Cells Dev. 2013, 22, 611–621. [Google Scholar] [CrossRef] [PubMed]
- Sumer, H.; Liu, J.; Malaver-Ortega, L.F.; Lim, M.L.; Khodadadi, K.; Verma, P.J. NANOG is a key factor for induction of pluripotency in bovine adult fibroblasts. J. Anim. Sci. 2011, 89, 2708–2716. [Google Scholar] [CrossRef] [PubMed]
- Bogliotti, Y.S.; Wu, J.; Vilarino, M.; Okamura, D.; Soto, D.A.; Zhong, C.; Sakurai, M.; Sampaio, R.V.; Suzuki, K.; Izpisua Belmonte, J.C.; et al. Efficient derivation of stable primed pluripotent embryonic stem cells from bovine blastocysts. Proc. Natl. Acad. Sci. USA 2018, 115, 2090–2095. [Google Scholar] [CrossRef]
- Liu, J.; Balehosur, D.; Murray, B.; Kelly, J.M.; Sumer, H.; Verma, P.J. Generation and characterization of reprogrammed sheep induced pluripotent stem cells. Theriogenology 2012, 77, 338–346. [Google Scholar] [CrossRef]
- Kimura, H.; Sakai, Y.; Fujii, T. Organ/body-on-a-chip based on microfluidic technology for drug discovery. Drug Metab. Pharmacokinet. 2018, 33, 43–48. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Widerspick, L.; Steffen, J.F.; Tappe, D.; Muñoz-Fontela, C. Animal Model Alternatives in Filovirus and Bornavirus Research. Viruses 2023, 15, 158. https://doi.org/10.3390/v15010158
Widerspick L, Steffen JF, Tappe D, Muñoz-Fontela C. Animal Model Alternatives in Filovirus and Bornavirus Research. Viruses. 2023; 15(1):158. https://doi.org/10.3390/v15010158
Chicago/Turabian StyleWiderspick, Lina, Johanna Friederike Steffen, Dennis Tappe, and César Muñoz-Fontela. 2023. "Animal Model Alternatives in Filovirus and Bornavirus Research" Viruses 15, no. 1: 158. https://doi.org/10.3390/v15010158
APA StyleWiderspick, L., Steffen, J. F., Tappe, D., & Muñoz-Fontela, C. (2023). Animal Model Alternatives in Filovirus and Bornavirus Research. Viruses, 15(1), 158. https://doi.org/10.3390/v15010158