
Regulation of Stress-Activated Kinases in Response to Tacaribe Virus Infection and Its Implications for Viral Replication


Abstract
1. Introduction
2. Materials and Methods
2.1. Cells
2.2. Virus Infection and Plaque Assay
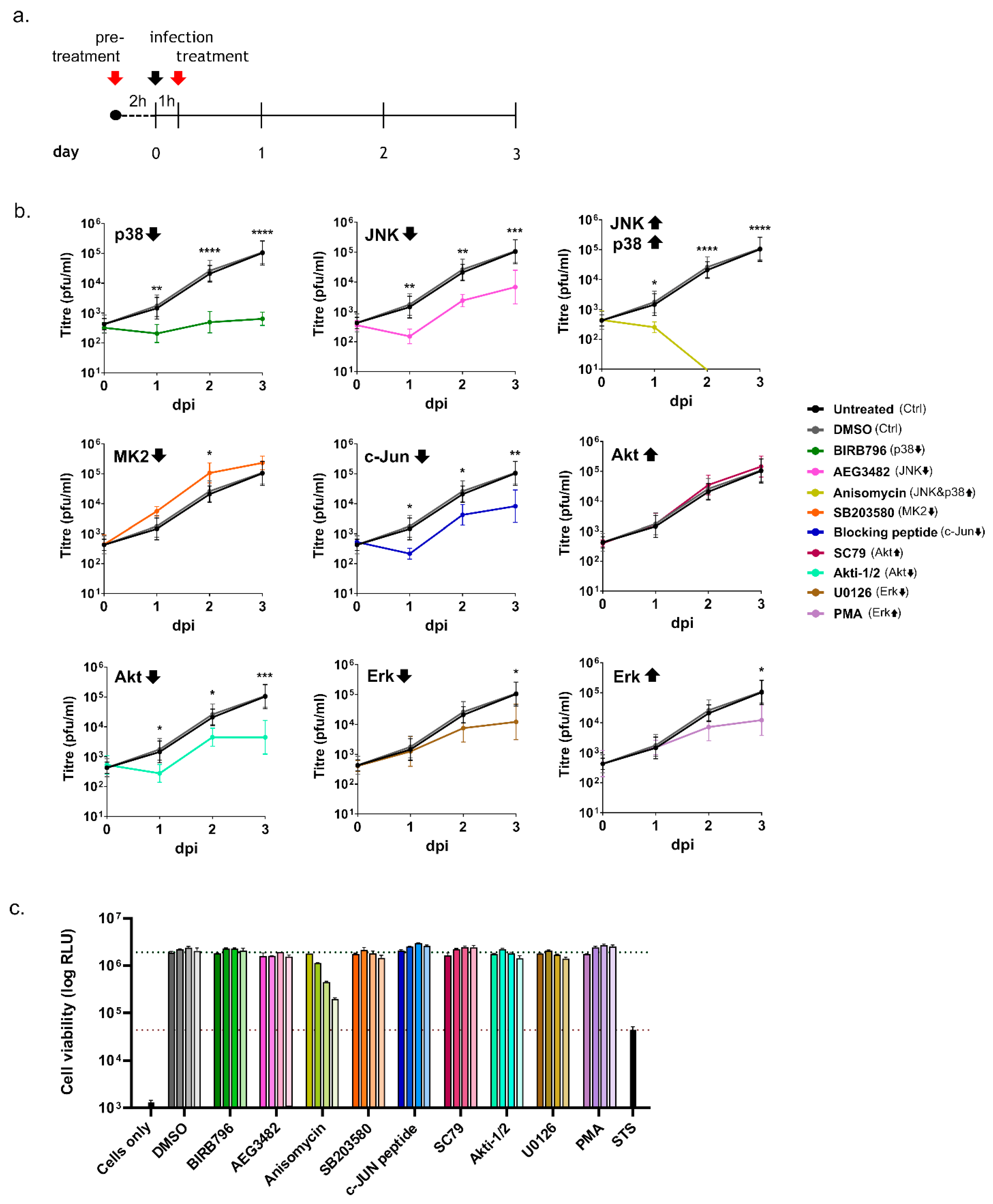
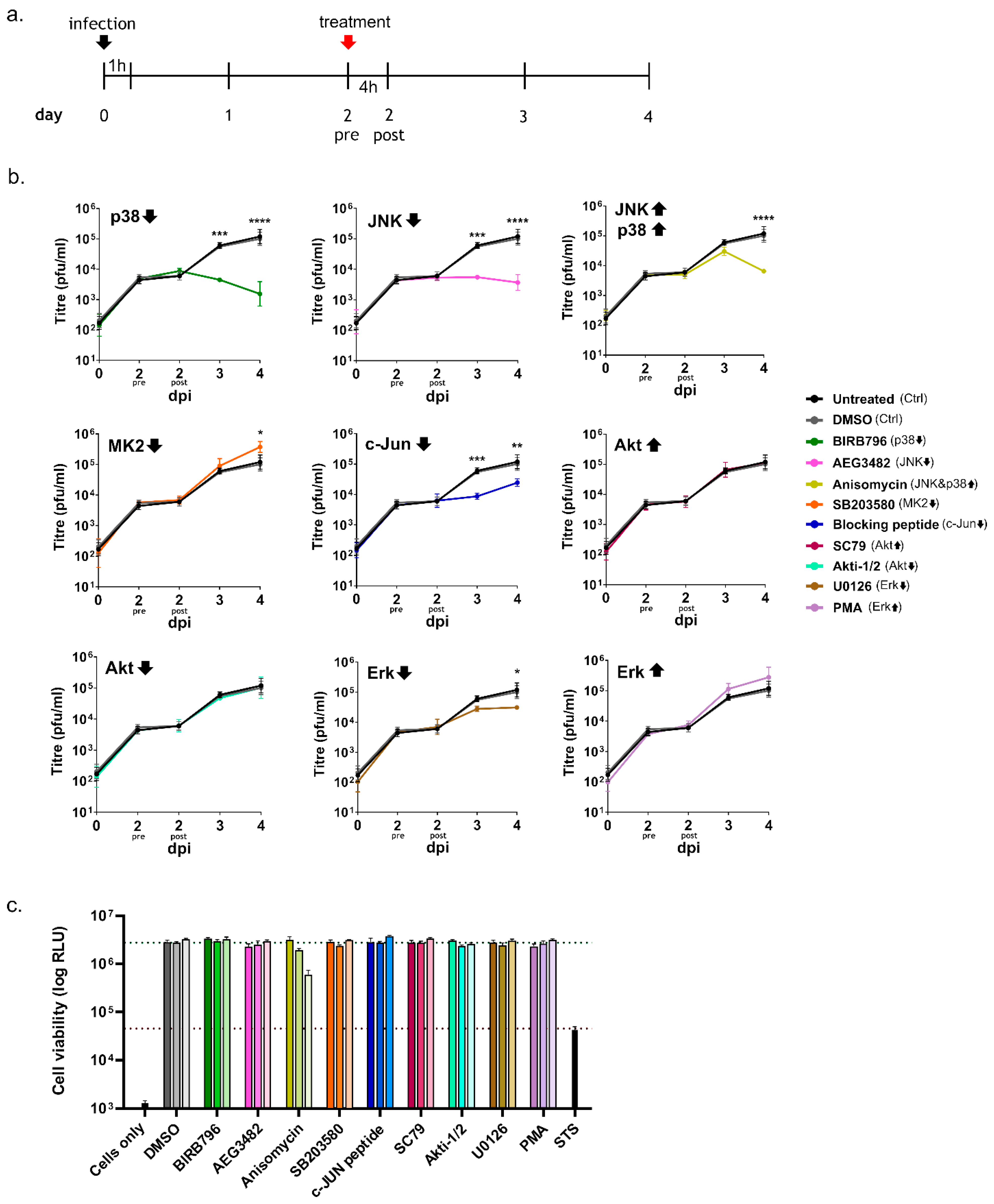
2.3. Chemical Treatments
2.4. Cell Viability Assay
2.5. Western Blot
2.6. Antibodies
2.7. Statistical Analysis
3. Results
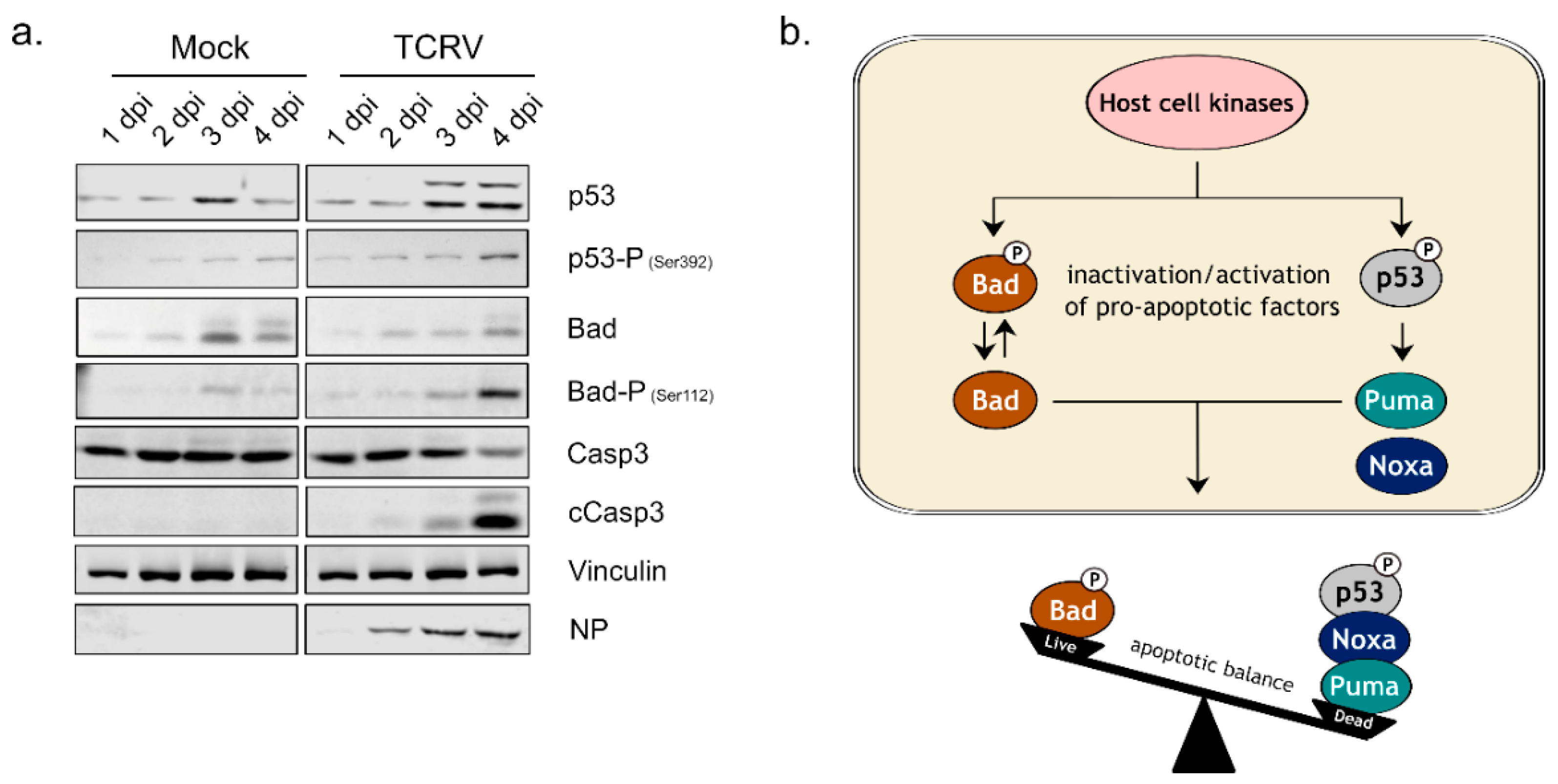
3.1. Activation of Kinase Signaling Pathways by TCRV Infection
3.2. Inhibition of Kinase Activation Significantly Reduces Viral Growth
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Radoshitzky, S.R.; Buchmeier, M.J.; Charrel, R.N.; Clegg, J.C.S.; Gonzalez, J.J.; Günther, S.; Hepojoki, J.; Kuhn, J.H.; Lukashevich, I.S.; Romanowski, V.; et al. ICTV Virus Taxonomy Profile: Arenaviridae. J. Gen. Virol. 2019, 100, 1200–1201. [Google Scholar] [CrossRef] [PubMed]
- Maes, P.; Alkhovsky, S.V.; Bào, Y.; Beer, M.; Birkhead, M.; Briese, T.; Buchmeier, M.J.; Calisher, C.H.; Charrel, R.N.; Choi, I.R.; et al. Taxonomy of the family Arenaviridae and the order Bunyavirales: Update 2018. Arch. Virol. 2018, 163, 2295–2310. [Google Scholar] [CrossRef] [PubMed]
- Childs, J.E.; Peters, C.J. Ecology and Epidemiology of Arenaviruses and Their Hosts. In The Arenaviridae; Salvato, M.S., Ed.; Springer: Boston, MA, USA, 1993; pp. 331–384. [Google Scholar]
- McCormick, J.B.; King, I.J.; Webb, P.A.; Johnson, K.M.; O’Sullivan, R.; Smith, E.S.; Trippel, S.; Tong, T.C. A case-control study of the clinical diagnosis and course of Lassa fever. J. Infect. Dis. 1987, 155, 445–455. [Google Scholar] [CrossRef]
- Carballal, G.; Videla, C.M.; Merani, M.S. Epidemiology of Argentine Hemorrhagic Fever. Eur. J. Epidemiol. 1988, 4, 259–274. [Google Scholar] [CrossRef] [PubMed]
- Charrel, R.N.; de Lamballerie, X. Arenaviruses other than Lassa virus. Antivir. Res. 2003, 57, 89–100. [Google Scholar] [CrossRef]
- Charrel, R.N.; de Lamballerie, X. Zoonotic aspects of arenavirus infections. Vet. Microbiol. 2010, 140, 213–220. [Google Scholar] [CrossRef]
- Meyer, B.; Ly, H. Inhibition of Innate Immune Responses Is Key to Pathogenesis by Arenaviruses. J. Virol. 2016, 90, 3810–3818. [Google Scholar] [CrossRef]
- Marta, R.F.; Montero, V.S.; Hack, C.E.; Sturk, A.; Maiztegui, J.I.; Molinas, F.C. Proinflammatory cytokines and elastase-alpha-1-antitrypsin in Argentine hemorrhagic fever. Am. J. Trop. Med. Hyg. 1999, 60, 85–89. [Google Scholar] [CrossRef]
- Baize, S.; Kaplon, J.; Faure, C.; Pannetier, D.; Georges-Courbot, M.C.; Deubel, V. Lassa virus infection of human dendritic cells and macrophages is productive but fails to activate cells. J. Immunol. 2004, 172, 2861–2869. [Google Scholar] [CrossRef]
- Groseth, A.; Hoenen, T.; Weber, M.; Wolff, S.; Herwig, A.; Kaufmann, A.; Becker, S. Tacaribe virus but not junin virus infection induces cytokine release from primary human monocytes and macrophages. PLoS Negl. Trop. Dis. 2011, 5, e1137. [Google Scholar] [CrossRef]
- Negrotto, S.; Mena, H.A.; Ure, A.E.; Jaquenod De Giusti, C.; Bollati-Fogolín, M.; Vermeulen, E.M.; Schattner, M.; Gómez, R.M. Human Plasmacytoid Dendritic Cells Elicited Different Responses after Infection with Pathogenic and Nonpathogenic Junin Virus Strains. J. Virol. 2015, 89, 7409–7413. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Meyer, B.; Groseth, A. Apoptosis during arenavirus infection: Mechanisms and evasion strategies. Microbes Infect. 2018, 20, 65–80. [Google Scholar] [CrossRef] [PubMed]
- Kolokoltsova, O.A.; Grant, A.M.; Huang, C.; Smith, J.K.; Poussard, A.L.; Tian, B.; Brasier, A.R.; Peters, C.J.; Tseng, C.T.; de la Torre, J.C.; et al. RIG-I enhanced interferon independent apoptosis upon Junin virus infection. PLoS ONE 2014, 9, e99610. [Google Scholar] [CrossRef]
- Wolff, S.; Groseth, A.; Meyer, B.; Jackson, D.; Strecker, T.; Kaufmann, A.; Becker, S. The New World arenavirus Tacaribe virus induces caspase-dependent apoptosis in infected cells. J. Gen. Virol. 2016, 97, 855–866. [Google Scholar] [CrossRef]
- Moreno, H.; Moller, R.; Fedeli, C.; Gerold, G.; Kunz, S. Comparison of the Innate Immune Responses to Pathogenic and Nonpathogenic Clade B New World Arenaviruses. J. Virol. 2019, 93, e00148-19. [Google Scholar] [CrossRef] [PubMed]
- Holzerland, J.; Fénéant, L.; Banadyga, L.; Hölper, J.E.; Knittler, M.R.; Groseth, A. BH3-only Sensors Bad, Noxa and Puma are Key Regulators of Tacaribe Virus-Induced Apoptosis. PLoS Pathog. 2020, 16, e1008948. [Google Scholar] [CrossRef]
- Wolff, S.; Becker, S.; Groseth, A. Cleavage of the Junin virus nucleoprotein serves a decoy function to inhibit the induction of apoptosis during infection. J. Virol. 2013, 87, 224–233. [Google Scholar] [CrossRef]
- Maclaine, N.J.; Hupp, T.R. The regulation of p53 by phosphorylation: A model for how distinct signals integrate into the p53 pathway. Aging 2009, 1, 490–502. [Google Scholar] [CrossRef]
- Ferecatu, I.; Rincheval, V.; Mignotte, B.; Vayssiere, J.L. Tickets for p53 journey among organelles. Front. Biosci. 2009, 14, 4214–4228. [Google Scholar] [CrossRef][Green Version]
- Downward, J. How BAD phosphorylation is good for survival. Nat. Cell Biol. 1999, 1, E33–E35. [Google Scholar] [CrossRef]
- Datta, S.R.; Katsov, A.; Hu, L.; Petros, A.; Fesik, S.W.; Yaffe, M.B.; Greenberg, M.E. 14-3-3 proteins and survival kinases cooperate to inactivate BAD by BH3 domain phosphorylation. Mol. Cell 2000, 6, 41–51. [Google Scholar] [CrossRef]
- Vela, E.M.; Bowick, G.C.; Herzog, N.K.; Aronson, J.F. Genistein treatment of cells inhibits arenavirus infection. Antivir. Res. 2008, 77, 153–156. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, M.E.; Brunetti, J.E.; Wachsman, M.B.; Scolaro, L.A.; Castilla, V. Raf/MEK/ERK pathway activation is required for Junin virus replication. J. Gen. Virol. 2014, 95, 799–805. [Google Scholar] [CrossRef]
- Brunetti, J.E.; Foscaldi, S.; Quintana, V.M.; Scolaro, L.A.; Lopez, N.; Castilla, V. Role of the ERK1/2 Signaling Pathway in the Replication of Junin and Tacaribe Viruses. Viruses 2018, 10, 199. [Google Scholar] [CrossRef] [PubMed]
- Rojek, J.M.; Moraz, M.L.; Pythoud, C.; Rothenberger, S.; Van der Goot, F.G.; Campbell, K.P.; Kunz, S. Binding of Lassa virus perturbs extracellular matrix-induced signal transduction via dystroglycan. Cell. Microbiol. 2012, 14, 1122–1134. [Google Scholar] [CrossRef] [PubMed]
- Scheid, M.P.; Schubert, K.M.; Duronio, V. Regulation of bad phosphorylation and association with Bcl-x(L) by the MAPK/Erk kinase. J. Biol. Chem. 1999, 274, 31108–31113. [Google Scholar] [CrossRef]
- Wu, G.S. The functional interactions between the p53 and MAPK signaling pathways. Cancer Biol. Ther. 2004, 3, 156–161. [Google Scholar] [CrossRef]
- Linero, F.N.; Scolaro, L.A. Participation of the phosphatidylinositol 3-kinase/Akt pathway in Junín virus replication In Vitro. Virus Res. 2009, 145, 166–170. [Google Scholar] [CrossRef]
- Linero, F.N.; Fernandez Bell-Fano, P.M.; Cuervo, E.; Castilla, V.; Scolaro, L.A. Inhibition of the PI3K/Akt pathway by Ly294002 does not prevent establishment of persistent Junin virus infection in Vero cells. Arch. Virol. 2015, 160, 469–475. [Google Scholar] [CrossRef]
- Bowick, G.C.; Fennewald, S.M.; Scott, E.P.; Zhang, L.; Elsom, B.L.; Aronson, J.F.; Spratt, H.M.; Luxon, B.A.; Gorenstein, D.G.; Herzog, N.K. Identification of differentially activated cell-signaling networks associated with pichinde virus pathogenesis by using systems kinomics. J. Virol. 2007, 81, 1923–1933. [Google Scholar] [CrossRef]
- Brunetti, J.E.; Quintana, V.M.; Scolaro, L.A.; Castilla, V. Inhibitors of the p38 cell signaling pathway as antiviral compounds against Junín virus. Arch. Virol. 2022, 167, 935–940. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, C.J.; Mudhasani, R.R.; Altamura, L.A.; Campbell, C.E.; Tran, J.P.; Beitzel, B.F.; Narayanan, A.; de la Fuente, C.L.; Kehn-Hall, K.; Smith, J.M.; et al. Junin Virus Activates p38 MAPK and HSP27 upon Entry. Front. Cell. Infect. Microbiol. 2022, 12, 798978. [Google Scholar] [CrossRef]
- Oda, E.; Ohki, R.; Murasawa, H.; Nemoto, J.; Shibue, T.; Yamashita, T.; Tokino, T.; Taniguchi, T.; Tanaka, N. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 2000, 288, 1053–1058. [Google Scholar] [CrossRef] [PubMed]
- Nakano, K.; Vousden, K.H. PUMA, a novel proapoptotic gene, is induced by p53. Mol. Cell 2001, 7, 683–694. [Google Scholar] [CrossRef]
- Cargnello, M.; Roux, P.P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef]
- Herring, S.; Oda, J.M.; Wagoner, J.; Kirchmeier, D.; O’Connor, A.; Nelson, E.A.; Huang, Q.; Liang, Y.; DeWald, L.E.; Johansen, L.M.; et al. Inhibition of Arenaviruses by Combinations of Orally Available Approved Drugs. Antimicrob. Agents Chemother. 2021, 65, e01146-20. [Google Scholar] [CrossRef]
- Enria, D.A.; Briggiler, A.M.; Sanchez, Z. Treatment of Argentine Hemorrhagic Fever. Antivir. Res. 2008, 78, 132–139. [Google Scholar] [CrossRef]
- Enria, D.A.; Maiztegui, J.I. Antiviral Treatment of Argentine Hemorrhagic Fever. Antivir. Res. 1994, 23, 23–31. [Google Scholar] [CrossRef]
- Maiztegui, J.I.; McKee, K.T., Jr.; Barrera Oro, J.G.; Harrison, L.H.; Gibbs, P.H.; Feuillade, M.R.; Enria, D.A.; Briggiler, A.M.; Levis, S.C.; Ambrosio, A.M.; et al. Protective efficacy of a live attenuated vaccine against Argentine hemorrhagic fever. AHF Study Group. J. Infect. Dis. 1998, 177, 277–283. [Google Scholar] [CrossRef]
- Briese, T.; Paweska, J.T.; McMullan, L.K.; Hutchison, S.K.; Street, C.; Palacios, G.; Khristova, M.L.; Weyer, J.; Swanepoel, R.; Egholm, M.; et al. Genetic detection and characterization of Lujo virus, a new hemorrhagic fever-associated arenavirus from southern Africa. PLoS Pathog. 2009, 5, e1000455. [Google Scholar] [CrossRef]
- Delgado, S.; Erickson, B.R.; Agudo, R.; Blair, P.J.; Vallejo, E.; Albariño, C.G.; Vargas, J.; Comer, J.A.; Rollin, P.E.; Ksiazek, T.G.; et al. Chapare virus, a newly discovered arenavirus isolated from a fatal hemorrhagic fever case in Bolivia. PLoS Pathog. 2008, 4, e1000047. [Google Scholar] [CrossRef] [PubMed]
- Raghuvanshi, R.; Bharate, S.B. Recent Developments in the Use of Kinase Inhibitors for Management of Viral Infections. J. Med. Chem. 2022, 65, 893–921. [Google Scholar] [CrossRef] [PubMed]
- Bhullar, K.S.; Lagarón, N.O.; McGowan, E.M.; Parmar, I.; Jha, A.; Hubbard, B.P.; Rupasinghe, H.P.V. Kinase-targeted cancer therapies: Progress, challenges and future directions. Mol. Cancer 2018, 17, 48. [Google Scholar] [CrossRef] [PubMed]
- Pillaiyar, T.; Laufer, S. Kinases as Potential Therapeutic Targets for Anti-coronaviral Therapy. J. Med. Chem. 2022, 65, 955–982. [Google Scholar] [CrossRef]
- Manning, J.T.; Seregin, A.V.; Yun, N.E.; Koma, T.; Huang, C.; Barral, J.; de la Torre, J.C.; Paessler, S. Absence of an N-Linked Glycosylation Motif in the Glycoprotein of the Live-Attenuated Argentine Hemorrhagic Fever Vaccine, Candid #1, Results in Its Improper Processing, and Reduced Surface Expression. Front. Cell. Infect. Microbiol. 2017, 7, 20. [Google Scholar] [CrossRef][Green Version]
- Manning, J.T.; Yun, N.E.; Seregin, A.V.; Koma, T.; Sattler, R.; Ezeomah, C.; Huang, C.; de la Torre, J.C.; Paessler, S. The glycoprotein of the live-attenuated Junin virus vaccine strain induces ER stress and forms aggregates prior to degradation in the lysosome. J. Virol. 2020, 94, e01693-19. [Google Scholar] [CrossRef]
- Tabas, I.; Ron, D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 2011, 13, 184–190. [Google Scholar] [CrossRef]
- Grollman, A.P. Inhibitors of protein biosynthesis. II. Mode of action of anisomycin. J. Biol. Chem. 1967, 242, 3226–3233. [Google Scholar] [CrossRef]
- Soni, S.; Anand, P.; Padwad, Y.S. MAPKAPK2: The master regulator of RNA-binding proteins modulates transcript stability and tumor progression. J. Exp. Clin. Cancer Res. 2019, 38, 121. [Google Scholar] [CrossRef]
- Chang, L.; Karin, M. Mammalian MAP kinase signalling cascades. Nature 2001, 410, 37–40. [Google Scholar] [CrossRef]
- Huang, G.; Shi, L.Z.; Chi, H. Regulation of JNK and p38 MAPK in the immune system: Signal integration, propagation and termination. Cytokine 2009, 48, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Waetzig, V.; Czeloth, K.; Hidding, U.; Mielke, K.; Kanzow, M.; Brecht, S.; Goetz, M.; Lucius, R.; Herdegen, T.; Hanisch, U.K. c-Jun N-terminal kinases (JNKs) mediate pro-inflammatory actions of microglia. Glia 2005, 50, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Pirkmajer, S.; Chibalin, A.V. Serum starvation: Caveat emptor. Am. J. Physiol. Cell Physiol. 2011, 301, C272–C279. [Google Scholar] [CrossRef] [PubMed]
- Rashid, M.U.; Coombs, K.M. Serum-reduced media impacts on cell viability and protein expression in human lung epithelial cells. J. Cell. Physiol. 2019, 234, 7718–7724. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, M.L.; Oldenburg, J.; Reignier, T.; Holt, N.; Hamilton, G.A.; Martin, V.K.; Cannon, P.M. New world clade B arenaviruses can use transferrin receptor 1 (TfR1)-dependent and -independent entry pathways, and glycoproteins from human pathogenic strains are associated with the use of TfR1. J. Virol. 2008, 82, 938–948. [Google Scholar] [CrossRef]
- Radoshitzky, S.R.; Abraham, J.; Spiropoulou, C.F.; Kuhn, J.H.; Nguyen, D.; Li, W.; Nagel, J.; Schmidt, P.J.; Nunberg, J.H.; Andrews, N.C.; et al. Transferrin receptor 1 is a cellular receptor for New World haemorrhagic fever arenaviruses. Nature 2007, 446, 92–96. [Google Scholar] [CrossRef]
- Harada, H.; Becknell, B.; Wilm, M.; Mann, M.; Huang, L.J.; Taylor, S.S.; Scott, J.D.; Korsmeyer, S.J. Phosphorylation and inactivation of BAD by mitochondria-anchored protein kinase A. Mol. Cell 1999, 3, 413–422. [Google Scholar] [CrossRef]
- Tan, Y.; Ruan, H.; Demeter, M.R.; Comb, M.J. p90(RSK) blocks bad-mediated cell death via a protein kinase C-dependent pathway. J. Biol. Chem. 1999, 274, 34859–34867. [Google Scholar] [CrossRef]
- Urata, S.; Ngo, N.; de la Torre, J.C. The PI3K/Akt pathway contributes to arenavirus budding. J. Virol. 2012, 86, 4578–4585. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Chemical Name | Supplier | Final Conc. |
|---|---|---|---|
| Akt ↓ | Akti-1/2 | Tocris # 5773 | 20 µM |
| Akt ↑ | SC79 | Tocris # 4635 | 30 µM |
| Erk↓ | U0126 | Tocris # 1144 | 30 µM |
| Erk ↑ | phorbol 12-myristate 13-acetate (PMA) | Sigma # P8139 | 162 nM |
| JNK ↓ | AEG3482 | Tocris # 2651 | 40 µM |
| p38 ↓ | BIRB796 | Tocris # 5989 | 30 µM |
| JNK/p38 ↑ | Anisomycin | Tocris # 1290 | 1 µM |
| MK2 ↓ | SB203580 | Tocris # 1202 | 30 µM |
| c-Jun ↓ | c-JUN blocking peptide | Tocris # 1989 | 50 µM |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Holzerland, J.; Fénéant, L.; Groseth, A. Regulation of Stress-Activated Kinases in Response to Tacaribe Virus Infection and Its Implications for Viral Replication. Viruses 2022, 14, 2018. https://doi.org/10.3390/v14092018
Holzerland J, Fénéant L, Groseth A. Regulation of Stress-Activated Kinases in Response to Tacaribe Virus Infection and Its Implications for Viral Replication. Viruses. 2022; 14(9):2018. https://doi.org/10.3390/v14092018
Chicago/Turabian StyleHolzerland, Julia, Lucie Fénéant, and Allison Groseth. 2022. "Regulation of Stress-Activated Kinases in Response to Tacaribe Virus Infection and Its Implications for Viral Replication" Viruses 14, no. 9: 2018. https://doi.org/10.3390/v14092018
APA StyleHolzerland, J., Fénéant, L., & Groseth, A. (2022). Regulation of Stress-Activated Kinases in Response to Tacaribe Virus Infection and Its Implications for Viral Replication. Viruses, 14(9), 2018. https://doi.org/10.3390/v14092018