
Use of Hu-PBL Mice to Study Pathogenesis of Human-Restricted Viruses

 , , and
, , and
Abstract
1. Introduction
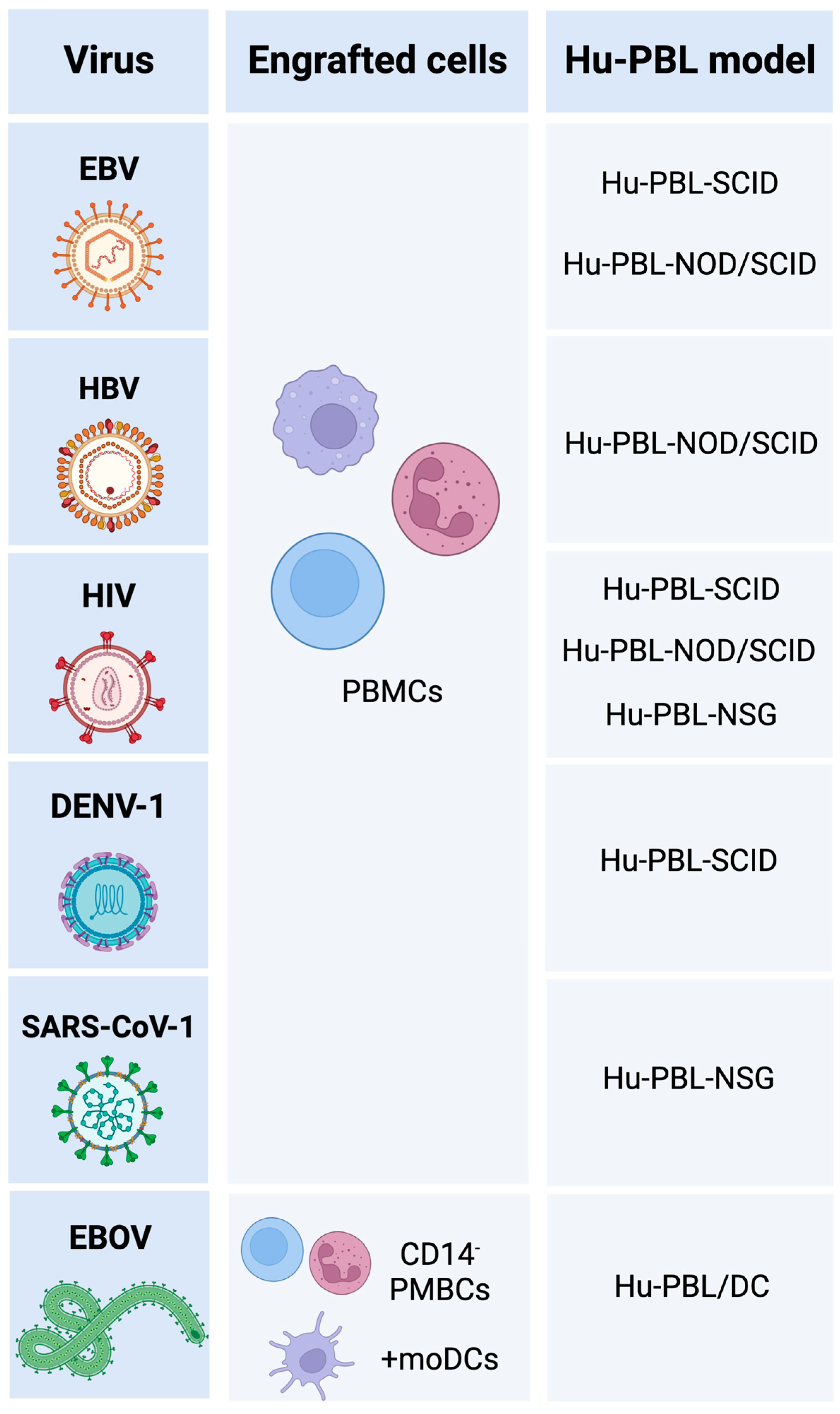
2. Hu-PBL Mouse Models
2.1. Hu-PBL-SCID
2.2. Hu-PBL-NOD/SCID
2.3. Hu-PBL-NSG
2.4. Hu-PBL/DC
3. Research in Virus Immunology Using hu-PBL Mouse Models
3.1. DNA Viruses
3.2. Retroviruses
3.3. Single-Stranded Positive-Sense RNA Viruses
3.4. Single-Stranded Negative-Sense RNA Viruses
4. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shultz, L.D.; Keck, J.; Burzenski, L.; Jangalwe, S.; Vaidya, S.; Greiner, D.L.; Brehm, M.A. Humanized Mouse Models of Immunological Diseases and Precision Medicine. Mamm. Genome 2019, 30, 123–142. [Google Scholar] [CrossRef] [PubMed]
- Stripecke, R.; Münz, C.; Schuringa, J.J.; Bissig, K.; Soper, B.; Meeham, T.; Yao, L.; di Santo, J.P.; Brehm, M.; Rodriguez, E.; et al. Innovations, Challenges, and Minimal Information for Standardization of Humanized Mice. EMBO Mol. Med. 2020, 12, e8662. [Google Scholar] [CrossRef] [PubMed]
- Shultz, L.D.; Ishikawa, F.; Greiner, D.L. Humanized Mice in Translational Biomedical Research. Nat. Rev. Immunol. 2007, 7, 118–130. [Google Scholar] [CrossRef] [PubMed]
- Hirenallur-Shanthappa, D.K.; Ramírez, J.A.; Iritani, B.M. Immunodeficient Mice: The Backbone of Patient-Derived Tumor Xenograft Models. In Patient Derived Tumor Xenograft Models: Promise, Potential and Practice; Elsevier Inc.: Amsterdam, The Netherlands, 2017; pp. 57–73. ISBN 978-0-12-804010-2. [Google Scholar]
- Shultz, L.D.; Brehm, M.A.; Victor Garcia-Martinez, J.; Greiner, D.L. Humanized Mice for Immune System Investigation: Progress, Promise and Challenges. Nat. Rev. Immunol. 2012, 12, 786–798. [Google Scholar] [CrossRef] [PubMed]
- Brehm, M.A.; Wiles, M.V.; Greiner, D.L.; Shultz, L.D. Generation of Improved Humanized Mouse Models for Human Infectious Diseases. J. Immunol. Methods 2014, 410, 3–17. [Google Scholar] [CrossRef]
- Wozniak, D.M.; Lavender, K.J.; Prescott, J.; Spengler, J.R. The Utility of Human Immune System Mice for High-Containment Viral Hemorrhagic Fever Research. Vaccines 2020, 8, 98. [Google Scholar] [CrossRef] [PubMed]
- Mosier, D.E.; Gulizia, R.J.; Baird, S.M.; Wilson, D.B. Transfer of a Functional Human Immune System to Mice with Severe Combined Immunodeficiency. Nature 1988, 335, 256–259. [Google Scholar] [CrossRef]
- Tary-Lehmann, M.; Saxon, A.; Lehmann, P. v The Human Immune System in Hu-PBL-SCID Mice. Immunol. Today 1995, 16, 529–533. [Google Scholar] [CrossRef]
- Bosma, G.C.; Custer, R.P.; Bosma, M.J. A Severe Combined Immunodeficiency Mutation in the Mouse. Nature 1983, 301, 527–530. [Google Scholar] [CrossRef]
- Bosma, M.J.; Carroll, A.M. The SCID Mouse Mutant: Definition, Characterization, and Potential Uses. Annu. Rev. Immunol. 1991, 9, 323–350. [Google Scholar] [CrossRef]
- Duchosal, M.A.; Eming, S.A.; Mcconahey, P.J.; Dixon, F.J. The Hu-PBL-SCID Mouse Model Long-Term Human Serologic Evolution Associated with the Xenogeneic Transfer of Human Peripheral Blood Leukocytes into SCID Mice. Cell Immunol. 1992, 139, 468–477. [Google Scholar] [CrossRef] [PubMed]
- Murphy, W.J.; Bennett, M.; Anver, M.R.; Baseler, M.; Longon, D.L. Human-Mouse Lymphoid Chimeras: Host-vs.-Graft and Graft-vs.-Host Reactions. Eur. J. Immunol. 1992, 22, 1421–1427. [Google Scholar] [CrossRef] [PubMed]
- Martino, G.; Anastasi, J.; Feng, J.; Mc Shan, C.; DeGroot, L.; Quintans, J.; Grimaldi, L.M.E. The Fate of Human Peripheral Blood Lymphocytes after Transplantation into SCID Mice. Eur. J. Immunol. 1993, 23, 1023–1028. [Google Scholar] [CrossRef] [PubMed]
- Hammad, H.; Duez, C.; Fahy, O.; Tsicopoulos, A.; André, C.; Wallaert, B.; Lebecque, S.; Tonnel, A.-B.; Pestel, J. Human Dendritic Cells in the Severe Combined Immunodeficiency Mouse Model: Their Potentiating Role in the Allergic Reaction. Lab. Investig. 2000, 80, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann-Fezer, G.; Gall, C.; Zengerle, U.; Kranz, B.; Thierfelder, S. Immunohistology and Immunocytology of Human T-Cell Chimerism and Graft-Versus-Host Disease in SCID Mice. Blood 1993, 81, 3440–3448. [Google Scholar] [CrossRef]
- Cao, T.; Leroux-Roels, G. Antigen-Specific T Cell Responses in Human Peripheral Blood Leucocyte (Hu-PBL)—Mouse Chimera Conditioned with Radiation and an Antibody Directed against the Mouse IL-2 Receptor Beta-Chain. Clin. Exp. Immunol. 2000, 122, 117–123. [Google Scholar] [CrossRef]
- Tournoy, K.G.; Depraetere, S.; Meuleman, P.; Leroux-Roels, G.; Pauwels, R.A. Murine IL-2 Receptor Beta Chain Blockade Improves Human Leukocyte Engraftment in SCID Mice. Eur. J. Immunol. 1998, 28, 3221–3230. [Google Scholar] [CrossRef]
- Kasai, M.; Yoneda, T.; Habu, S.; Maruyama, Y.; Okumura, K.; Tokunaga, T. In Vivo Effect of Anti-Asialo GM1 Antibody on Natural Killer Activity. Nature 1981, 291, 334–335. [Google Scholar] [CrossRef]
- Habu, S.; Fukui, H.; Shimamura, K.; Kasai, M.; Nagai, Y.; Okumura, K.; Tamaoki, N. In Vivo Effects of Anti-Asialo GM1. I. Reduction of NK Activity and Enhancement of Transplanted Tumor Growth in Nude Mice. J. Immunol. 1981, 127, 34–38. [Google Scholar] [CrossRef]
- Baiocchi, R.A.; Ward, J.S.; Carrodeguas, L.; Eisenbeis, C.F.; Peng, R.; Roychowdhury, S.; Vourganti, S.; Sekula, T.; O’Brien, M.; Moeschberger, M.; et al. GM-CSF and IL-2 Induce Specific Cellular Immunity and Provide Protection against Epstein-Barr Virus Lymphoproliferative Disorder. J. Clin. Investig. 2001, 108, 887–894. [Google Scholar] [CrossRef]
- Murphy, W.J.; Taub, D.D.; Longo, D.L. The HuPBL-SCID Mouse as a Means to Examine Human Immune Function in Vivo. Semin. Immunol. 1996, 8, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Murphy, W.J.; Durum, S.K.; Longo, D.L. Human Growth Hormone Promotes Engraftment of Murine or Human T Cells in Severe Combined Immunodeficient Mice. Immunology 1992, 89, 4481–4485. [Google Scholar] [CrossRef] [PubMed]
- Murphy, W.J.; Durum, S.K.; Anver, M.; Frazier, M.; Longo, D.L. Recombinant Human Growth Hormone Promotes Human Lymphocyte Engraftment in Immunodeficient Mice and Results in an Increased Incidence of Human Epstein Barr Virus-Induced B-Cell Lymphoma. Brain Behav. Immun. 1992, 6, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Zhang, J.; Zhang, C.; Zhang, J.; Liang, S.; Sun, A.; Wang, J.; Tian, Z. Human Prolactin Improves Engraftment and Reconstitution of Human Peripheral Blood Lymphocytes in SCID Mice. Cell. Mol. Immunol. 2004, 129, 129–136. [Google Scholar]
- Bombil, F.; Latinne, D.; Kints, J.-P.; Bazin, H. Human Recombinant Interleukin-4 (HuriL-4) Improves SCID Mouse Reconstitution with Human Peripheral Blood Lymphocytes. Immunobiology 1996, 196, 437–448. [Google Scholar] [CrossRef]
- Coccia, M.A.; Weeks, S.J.; Knott, C.L.; Kuus-Reichel, K. Human IL-6 Enhances Human Lymphocyte Engraftment and Activation but Not Human Antibody Production in SCIDhu PBL Mice. Immunobiology 1997, 198, 396–407. [Google Scholar] [CrossRef]
- Taub, D.D.; Key, M.L.; Longo, D.L.; Murphy, W.J. Chemokine-Induced Human Lymphocyte Infiltration and Engraftment in HuPBL-SCID Mice. Methods Enzymol. 1997, 287, 265–291. [Google Scholar] [CrossRef]
- Brehm, M.A.; Shultz, L.D.; Luban, J.; Greiner, D.L. Overcoming Current Limitations in Humanized Mouse Research. J. Infect. Dis. 2013, 208 (Suppl. S2), S125–S130. [Google Scholar] [CrossRef]
- Avdoshina, D.V.; Kondrashova, A.S.; Belikova, M.G.; Bayurova, E.O. Murine Models of Chronic Viral Infections and Associated Cancers. Mol. Biol. 2022, 56, 649–667. [Google Scholar] [CrossRef]
- Belizário, J.E. Immunodeficient Mouse Models: An Overview. Open Immunol. J. 2009, 2, 79–85. [Google Scholar] [CrossRef]
- Berges, B.K.; Rowan, M.R. The Utility of the New Generation of Humanized Mice to Study HIV-1 Infection: Transmission, Prevention, Pathogenesis, and Treatment. Retrovirology 2011, 8, 65. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Duan, Z.; Zhao, Y. Mouse Models with Human Immunity and Their Application in Biomedical Research. J. Cell. Mol. Med. 2009, 13, 1043–1058. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Hiramatsu, H.; Kobayashi, K.; Suzue, K.; Kawahata, M.; Hioki, K.; Ueyama, Y.; Koyanagi, Y.; Sugamura, K.; Tsuji, K.; et al. NOD/SCID/Γcnull Mouse: An Excellent Recipient Mouse Model for Engraftment of Human Cells. Blood 2002, 100, 3175–3182. [Google Scholar] [CrossRef] [PubMed]
- Sun, A.; Wei, H.; Sun, R.; Xiao, W.; Yang, Y.; Tian, Z. Human Interleukin-15 Improves Engraftment of Human T Cells in NOD-SCID Mice. Clin. Vaccine Immunol. 2006, 13, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Barry, T.S.; Jones, D.M.; Richter, C.B.; Haynes, B.F. Successful Engraftment of Human Postnatal Thymus in Severe Combined Immune Deficient (SCID) Mice: Differential Engraftment of Thymic Components With Irradiation Versus Anti-Asialo GM-1 Immunosuppressive Regimens. J. Exp. Med. 1991, 173, 167–180. [Google Scholar] [CrossRef]
- Wagar, E.J.; Cromwell, M.A.; Shultz, L.D.; Woda, B.A.; Sullivan, J.L.; Hesselton, R.M.; Greiner, D.L. Regulation of Human Cell Engraftment and Development of EBV-Related Lymphoproliferative Disorders in Hu-PBL-Scid Mice. J. Immunol. 2000, 165, 518–527. [Google Scholar] [CrossRef]
- Kim, K.C.; Choi, B.S.; Kim, K.C.; Park, K.H.; Lee, H.J.; Cho, Y.K.; Kim, S.I.; Kim, S.S.; Oh, Y.K.; Kim, Y.B. A Simple Mouse Model for the Study of Human Immunodeficiency Virus. AIDS Res. Hum. Retrovir. 2016, 32, 194–202. [Google Scholar] [CrossRef]
- Ishikawa, Y.; Usui, T.; Shiomi, A.; Shimizu, M.; Murakami, K.; Mimori, T. Functional Engraftment of Human Peripheral T and B Cells and Sustained Production of Autoantibodies in NOD/LtSzscid/IL-2Rγ-/- Mice. Eur. J. Immunol. 2014, 44, 3453–3463. [Google Scholar] [CrossRef]
- Holling, T.M.; Schooten, E.; van den Elsen, P.J. Function and Regulation of MHC Class II Molecules in T-Lymphocytes: Of Mice and Men. Hum. Immunol. 2004, 65, 282–290. [Google Scholar] [CrossRef]
- Spranger, S.; Frankenberger, B.; Schendel, D.J. NOD/Scid IL-2Rg Nullmice: A Preclinical Model System to Evaluate Human Dendritic Cell-Based Vaccine Strategies in Vivo. J. Transl. Med. 2012, 10, 30. [Google Scholar] [CrossRef]
- Harui, A.; Kiertscher, S.M.; Roth, M.D. Reconstitution of HuPBL-NSG Mice with Donor-Matched Dendritic Cells Enables Antigen-Specific T-Cell Activation. J. Neuroimmune Pharmacol. 2011, 6, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Rottstegge, M.; Tipton, T.; Oestereich, L.; Ruibal, P.; Nelson, E.V.; Olal, C.; Port, J.R.; Seibel, J.; Pallasch, E.; Bockholt, S.; et al. Avatar Mice Underscore the Role of the T Cell-Dendritic Cell Crosstalk in Ebola Virus Disease and Reveal Mechanisms of Protection in Survivors. J. Virol. 2022, 96, e00574-22. [Google Scholar] [CrossRef] [PubMed]
- Fuzzati-Armentero, M.-T.; Duchosal, M.A. Hu-PBL-SCID Mice: An in Vivo Model of Epstein-Barr Virus-Dependent Lymphoproliferative Disease. Histol. Histopathol. 1998, 13, 68. [Google Scholar] [CrossRef]
- Mosier, D.E. Viral Pathogenesis in Hu-PBL-SCID Mice. Semin. Immunol. 1996, 8, 255–262. [Google Scholar] [CrossRef]
- Baiocchi, R.A.; Caligiuri, M.A. Low-Dose Interleukin 2 Prevents the Development of Epstein-Barr Virus (EBV)-Associated Lymphoproliferative Disease in Scid/Scid Mice Reconstituted i.p. with EBV-Seropositive Human Peripheral Blood Lymphocytes. Proc. Natl. Acad. Sci. USA 1994, 91, 5577–5581. [Google Scholar] [CrossRef] [PubMed]
- Baiocchi, R.A.; Ross, M.E.; Tan, J.C.; Chou, C.-C.; Sullivan, L.; Haldar, S.; Monne, M.; Seiden, M.V.; Narula, S.K.; Sklar, J.; et al. Lymphomagenesis in the SCID-Hu Mouse Involves Abundant Production of Human Interleukin-10. Blood 1995, 85, 1063–1074. [Google Scholar] [CrossRef]
- Burdin, N.; Péronne, C.; Banchereau, J.; Rousset, F. Epstein-Barr Virus Transformation Induces B Lymphocytes to Produce Human Interleukin 10. J. Exp. Med. 1993, 177, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Funakoshi, S.; Longo, D.L.; Beckwith, M.; Conley, D.K.; Tsarfaty, G.; Tsarfaty, L.; Armitage, R.J.; Fanslow, W.C.; Spriggs, M.K.; Murphy, W.J. Inhibition of Human B-Cell Lymphoma Growth by CD40 Stimulation. Blood 1994, 83, 2787–2794. [Google Scholar] [CrossRef]
- Murphy, W.J.; Funakoshi, S.; Beckwith, M.; Rushing, S.E.; Conley, D.K.; Arrnitage, R.J.; Fanslow, W.C.; Rager, H.C.; Taub, D.D.; Ruscetti, F.W.; et al. Antibodies to CD40 Prevent Epstein-Barr Virus-Mediated Human B-Cell Lymphomagenesis in Severe Combined Immune Deficient Mice Given Human Peripheral Blood Lymphocytes. Blood 1995, 86, 1946–1953. [Google Scholar] [CrossRef]
- Rowe, M.; Young, L.S.; Crocker, J.; Stokes, H.; Henderson, S.; Rickinson, A.B. Epstein-Barr Virus (EBV)-Associated Lymphoproliferative Disease in the SCID Mouse Model: Implications for the Pathogenesis of EBVpositive Lymphomas in Man. J. Exp. Med. 1991, 173, 147–158. [Google Scholar] [CrossRef]
- Cao, T.; Lazdina, U.; Desombere, I.; Vanlandschoot, P.; Milich, D.R.; Sällberg, M.; Leroux-Roels, G. Hepatitis B Virus Core Antigen Binds and Activates Naive Human B Cells In Vivo: Studies with a Human PBL-NOD/SCID Mouse Model. J. Virol. 2001, 75, 6359–6366. [Google Scholar] [CrossRef] [PubMed]
- Mosier, D.E.; Gulizia, R.J.; Baird, S.M.; Wilson, D.B.; Spector, D.H.; Spector, S.A. Human Immunodeficiency Virus Infection of Human-PBL-SCID Mice. Science 1991, 251, 791–794. [Google Scholar] [CrossRef] [PubMed]
- Mosier, D.E.; Gulizia, R.J.; MacIsaac, P.D.; Torbett, B.E.; Levy, J.A. Rapid Loss of CD4+ T Cells in Human-PBL-SCID Mice by Noncytopathic HIV Isolates. Science 1993, 260, 689–692. [Google Scholar] [CrossRef] [PubMed]
- Mosier, D.E. Distinct Rates and Patterns of Human CD4 + T-Cell Depletion in Hu-PBL-SCID Mice Infected with Different Isolates of the Human Immunodeficiency Virus. J. Clin. Immunol. 1995, 15, 130S–133S. [Google Scholar] [CrossRef]
- Rizza, P.; Santini, S.M.; Logozzi, M.; Lapenta, C.; Sestili, P.; Gherardi, G.; Lande, R.; Spada, M.; Parlato, S.; Belardelli, F.; et al. T-Cell Dysfunctions in Hu-PBL-SCID Mice Infected with Human Immunodeficiency Virus (HIV) Shortly after Reconstitution: In Vivo Effects of HIV on Highly Activated Human Immune Cells. J. Virol. 1996, 70, 7958–7964. [Google Scholar] [CrossRef] [PubMed]
- Fais, S.; Lapenta, C.; Santini, S.M.; Spada, M.; Parlato, S.; Logozzi, M.; Rizza, P.; Belardelli, F. Human Immunodeficiency Virus Type 1 Strains R5 and X4 Induce Different Pathogenic Effects in Hu-PBL-SCID Mice, Depending on the State of Activation/Differentiation of Human Target Cells at the Time of Primary Infection. J. Virol. 1999, 73, 6453–6459. [Google Scholar] [CrossRef]
- Vieillard, V.; Jouveshomme, S.; Leflour, N.; Jean-Pierre, E.; Debre, P.; de Maeyer, E.; Autran, B. Transfer of Human CD4 T Lymphocytes Producing Beta Interferon in Hu-PBL-SCID Mice Controls Human Immunodeficiency Virus Infection. J. Virol. 1999, 73, 10281–10288. [Google Scholar] [CrossRef]
- Boyle, M.J.; Connors, M.; Flanigan, M.E.; Geiger, S.P.; Ford, H., Jr.; Baseler, M.; Adelsberger, J.; Davey, R.T., Jr.; Lane, H.C. The Human HIV/Peripheral Blood Lymphocyte (PBL)-SCID Mouse. A Modified Human PBL-SCID Model for the Study of HIV Pathogenesis and Therapy. J. Immunol. 1995, 154, 6612–6623. [Google Scholar] [CrossRef]
- van Duyne, R.; Pedati, C.; Guendel, I.; Carpio, L.; Kehn-Hall, K.; Saifuddin, M.; Kashanchi, F. The Utilization of Humanized Mouse Models for the Study of Human Retroviral Infections. Retrovirology 2009, 6, 76. [Google Scholar] [CrossRef]
- Abeynaike, S.; Paust, S. Humanized Mice for the Evaluation of Novel HIV-1 Therapies. Front. Immunol. 2021, 12, 636775. [Google Scholar] [CrossRef]
- Poignard, P.; Sabbe, R.; Picchio, G.R.; Wang, M.; Gulizia, R.J.; Katinger, H.; Parren, P.W.H.I.; Mosier, D.E.; Burton, D.R. Neutralizing Antibodies Have Limited Effects on the Control of Established HIV-1 Infection In Vivo. Immunity 1999, 10, 431–438. [Google Scholar] [CrossRef]
- Gauduin, M.-C.; Allaway, G.P.; Olson, W.C.; Weir, R.; Maddon, P.J.; Koup, R.A. CD4-Immunoglobulin G2 Protects Hu-PBL-SCID Mice against Challenge by Primary Human Immunodeficiency Virus Type 1 Isolates. J. Virol. 1998, 72, 3475–3478. [Google Scholar] [CrossRef] [PubMed]
- Santini, S.M.; Lapenta, C.; Logozzi, M.; Parlato, S.; Spada, M.; di Pucchio, T.; Belardelli, F. Type I Interferon as a Powerful Adjuvant for Monocyte-Derived Dendritic Cell Development and Activity In Vitro and in Hu-PBL-SCID Mice. J. Exp. Med. 2000, 191, 1777–1788. [Google Scholar] [CrossRef] [PubMed]
- Lapenta, C.; Santini, S.M.; Logozzi, M.; Spada, M.; Andreotti, M.; di Pucchio, T.; Parlato, S.; Belardelli, F. Potent Immune Response against HIV-1 and Protection from Virus Challenge in Hu-PBL-SCID Mice Immunized with Inactivated Virus-Pulsed Dendritic Cells Generated in the Presence of IFN-α. J. Exp. Med. 2003, 198, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, A.; Tanaka, R.; Murakami, T.; Takahashi, Y.; Koyanagi, Y.; Nakamura, M.; Ito, M.; Yamamoto, N.; Tanaka, Y. Induction of Protective Immune Responses against R5 Human Immunodeficiency Virus Type 1 (HIV-1) Infection in Hu-PBL-SCID Mice by Intrasplenic Immunization with HIV-1-Pulsed Dendritic Cells: Possible Involvement of a Novel Factor of Human CD4 + T-Cell Origin. J. Virol. 2003, 77, 8719–8728. [Google Scholar] [CrossRef]
- Kodama, A.; Tanaka, R.; Saito, M.; Ansari, A.A.; Tanaka, Y. A Novel and Simple Method for Generation of Human Dendritic Cells from Unfractionated Peripheral Blood Mononuclear Cells within 2 Days: Its Application for Induction of HIV-1-Reactive CD4+ T Cells in the Hu-PBL SCID Mice. Front. Microbiol. 2013, 4, 292. [Google Scholar] [CrossRef]
- Hesselton, R.M.; Greiner, D.L.; Mordes, J.P.; Rajan, T.V.; Sullivan, J.L.; Shultz, L.D. High Levels of Human Peripheral Blood Mononuclear Cell Engraftment and Enhanced Susceptibility to Human Immunodeficiency Virus Type 1 Infection in NOD/LtSz-Scid/Scid Mice. J. Infect. Dis. 1995, 172, 974–982. [Google Scholar] [CrossRef]
- Gorantla, S.; Santos, K.; Meyer, V.; Dewhurst, S.; Bowers, W.J.; Federoff, H.J.; Gendelman, H.E.; Poluektova, L. Human Dendritic Cells Transduced with Herpes Simplex Virus Amplicons Encoding Human Immunodeficiency Virus Type 1 (HIV-1) Gp120 Elicit Adaptive Immune Responses from Human Cells Engrafted into NOD/SCID Mice and Confer Partial Protection against HIV-1 Challenge. J. Virol. 2005, 79, 2124–2132. [Google Scholar] [CrossRef]
- Wu, X.; Liu, L.; Cheung, K.; Wang, H.; Lu, X.; Cheung, A.K.L.; Liu, W.; Huang, X.; Li, Y.; Chen, Z.W.; et al. Brain Invasion by CD4+ T Cells Infected with a Transmitted/Founder HIV-1BJZS7 During Acute Stage in Humanized Mice. J. Neuroimmune Pharmacol. 2016, 11, 572–583. [Google Scholar] [CrossRef]
- Metcalf Pate, K.A.; Pohlmeyer, C.W.; Walker-Sperling, V.E.; Foote, J.B.; Najarro, K.M.; Cryer, C.G.; Salgado, M.; Gama, L.; Engle, E.L.; Shirk, E.N.; et al. A Murine Viral Outgrowth Assay to Detect Residual HIV Type 1 in Patients with Undetectable Viral Loads. J. Infect. Dis. 2015, 212, 1387–1396. [Google Scholar] [CrossRef]
- Metcalf Pate, K.A.; Blankson, J.N. The Mouse Viral Outgrowth Assay: Avatars for the Detection of HIV-1 Reservoirs. Retrovirology 2017, 14, 52. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Wu, S.; Zhang, Y.; Xia, Y.; Huelsmann, E.J.; Lacek, A.T.; Nabatiyan, A.; Richards, M.H.; Narasipura, S.D.; Lutgen, V.; et al. HIV Infection Accelerates Gastrointestinal Tumor Outgrowth in NSG-HuPBL Mice. AIDS Res. Hum. Retrovir. 2014, 30, 677–684. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Ban, H.S.; Kim, S.S.; Wu, H.; Pearson, T.; Greiner, D.L.; Laouar, A.; Yao, J.; Haridas, V.; Habiro, K.; et al. T Cell-Specific SiRNA Delivery Suppresses HIV-1 Infection in Humanized Mice. Cell 2008, 134, 577–586. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.G.; Bharaj, P.; Abraham, S.; Ma, H.; Yi, G.; Ye, C.; Dang, Y.; Manjunath, N.; Wu, H.; Shankar, P. Multiplexing Seven MiRNA-Based ShRNAs to Suppress HIV Replication. Mol. Ther. 2015, 23, 310–320. [Google Scholar] [CrossRef]
- Bardhi, A.; Wu, Y.; Chen, W.; Li, W.; Zhu, Z.; Zheng, J.H.; Wong, H.; Jeng, E.; Jones, J.; Ochsenbauer, C.; et al. Potent In Vivo NK Cell-Mediated Elimination of HIV-1-Infected Cells Mobilized by a Gp120-Bispecific and Hexavalent Broadly Neutralizing Fusion Protein. J. Virol. 2017, 91, e00937-17. [Google Scholar] [CrossRef]
- Bhargavan, B.; Woollard, S.M.; McMillan, J.E.; Kanmogne, G.D. CCR5 Antagonist Reduces HIV-Induced Amyloidogenesis, Tau Pathology, Neurodegeneration, and Blood-Brain Barrier Alterations in HIV-Infected Hu-PBL-NSG Mice. Mol. Neurodegener. 2021, 16, 78. [Google Scholar] [CrossRef]
- Leibman, R.S.; Richardson, M.W.; Ellebrecht, C.T.; Maldini, C.R.; Glover, J.A.; Secreto, A.J.; Kulikovskaya, I.; Lacey, S.F.; Akkina, S.R.; Yi, Y.; et al. Supraphysiologic Control over HIV-1 Replication Mediated by CD8 T Cells Expressing a Re-Engineered CD4-Based Chimeric Antigen Receptor. PLoS Pathog. 2017, 13, e1006613. [Google Scholar] [CrossRef] [PubMed]
- Anthony-Gonda, K.; Bardhi, A.; Ray, A.; Flerin, N.; Li, M.; Chen, W.; Ochsenbauer, C.; Kappes, J.C.; Krueger, W.; Worden, A.; et al. Multispecific Anti-HIV DuoCAR-T Cells Display Broad in Vitro Antiviral Activity and Potent in Vivo Elimination of HIV-Infected Cells in a Humanized Mouse Model. Sci. Transl. Med. 2019, 11, 5685. [Google Scholar] [CrossRef]
- Seay, K.; Church, C.; Zheng, J.H.; Deneroff, K.; Ochsenbauer, C.; Kappes, J.C.; Liu, B.; Jeng, E.K.; Wong, H.C.; Goldstein, H. In Vivo Activation of Human NK Cells by Treatment with an Interleukin-15 Superagonist Potently Inhibits Acute In Vivo HIV-1 Infection in Humanized Mice. J. Virol. 2015, 89, 6264–6274. [Google Scholar] [CrossRef]
- Abraham, S.; Choi, J.-G.; Ortega, N.M.; Zhang, J.; Shankar, P.; Swamy, N.M. Gene Therapy with Plasmids Encoding IFN-β or IFN-A14 Confers Long-Term Resistance to HIV-1 in Humanized Mice. Oncotarget 2016, 7, 78412–78420. [Google Scholar] [CrossRef]
- Wu, S.J.L.; Hayes, C.G.; Dubois, D.R.; Windheuser, M.G.; Kang, Y.H.; Watts, D.M.; Sieckmann, D.G. Evaluation of the Severe Combined Immunodeficient (SCID) Mouse as an Animal Model for Dengue Viral Infection. Am. J. Trop. Med. Hyg. 1995, 52, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Takemoto, Y.; Okuno, Y.; Hashimoto, S.; Fukunaga, Y.; Tanaka, T.; Kita, Y.; Kuwayama, S.; Muraki, Y.; Kanamaru, N.; et al. Development of Vaccines and Passive Immunotherapy against SARS Coronavirus Using Mouse and SCID-PBL/Hu Mouse Models. In The Nidoviruses: Toward Control of SARS and Other Nidovirus Diseases; Perlman, S., Holmes, K.V., Eds.; Springer: New York, NY, USA, 2006; pp. 561–566. ISBN 978-0-387-33012-9. [Google Scholar]
- Murata, T.; Sugimoto, A.; Inagaki, T.; Yanagi, Y.; Watanabe, T.; Sato, Y.; Kimura, H. Molecular Basis of Epstein–Barr Virus Latency Establishment and Lytic Reactivation. Viruses 2021, 13, 2344. [Google Scholar] [CrossRef] [PubMed]
- Herrscher, C.; Roingeard, P.; Blanchard, E. Hepatitis B Virus Entry into Cells. Cells 2020, 9, 1486. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Li, Y.; Zhang, Z.; Wei, W. Human Immunodeficiency Virus-1 Core: The Trojan Horse in Virus–Host Interaction. Front. Microbiol. 2022, 13, 1002476. [Google Scholar] [CrossRef]
- Yong, Y.K.; Wong, W.F.; Vignesh, R.; Chattopadhyay, I.; Velu, V.; Tan, H.Y.; Zhang, Y.; Larsson, M.; Shankar, E.M. Dengue Infection—Recent Advances in Disease Pathogenesis in the Era of COVID-19. Front. Immunol. 2022, 13, 889196. [Google Scholar] [CrossRef]
- Hui, D.S.C.; Zumla, A. Severe Acute Respiratory Syndrome: Historical, Epidemiologic, and Clinical Features. Infect. Dis. Clin. N. Am. 2019, 33, 869–889. [Google Scholar] [CrossRef]
- McElroy, A.K.; Mühlberger, E.; Muñoz-Fontela, C. Immune Barriers of Ebola Virus Infection. Curr. Opin. Virol. 2018, 28, 152–160. [Google Scholar] [CrossRef]
- Muñoz-Fontela, C.; McElroy, A.K. Ebola Virus Disease in Humans: Pathophysiology and Immunity. In Marburg- and Ebolaviruses: From Ecosystems to Molecules; Mühlberger, E., Hensley, L.L., Towner, J.S., Eds.; Springer: Cham, Switzerland, 2017; Volume 411, pp. 141–169. ISBN 978-3-319-68948-7. [Google Scholar]
- Lüdtke, A.; Ruibal, P.; Wozniak, D.M.; Pallasch, E.; Wurr, S.; Bockholt, S.; Gómez-Medina, S.; Qiu, X.; Kobinger, G.P.; Rodríguez, E.; et al. Ebola Virus Infection Kinetics in Chimeric Mice Reveal a Key Role of T Cells as Barriers for Virus Dissemination. Sci. Rep. 2017, 7, 43776. [Google Scholar] [CrossRef]
- Stephens, P.R.; Sundaram, M.; Ferreira, S.; Gottdenker, N.; Nipa, K.F.; Schatz, A.M.; Schmidt, J.P.; Drake, J.M. Drivers of African Filovirus (Ebola and Marburg) Outbreaks. Vector Borne Zoonotic Dis. 2022, 22, 478–490. [Google Scholar] [CrossRef]
- Kaner, J.; Schaack, S. Understanding Ebola: The 2014 Epidemic. Glob. Health 2016, 12, 53. [Google Scholar] [CrossRef]
- Omilabu, S.; Salu, O.; Oke, B.; James, A. The West African Ebola Virus Disease Epidemic 2014–2015: A Commissioned Review. Niger. Postgrad. Med. J. 2016, 23, 49. [Google Scholar] [CrossRef] [PubMed]
- Port, J.R.; Wozniak, D.M.; Oestereich, L.; Pallasch, E.; Becker-Ziaja, B.; Müller, J.; Rottstegge, M.; Olal, C.; Gómez-Medina, S.; Oyakhliome, J.; et al. Severe Human Lassa Fever Is Characterized by Nonspecific T-Cell Activation and Lymphocyte Homing to Inflamed Tissues. J. Virol. 2020, 94, e01367-20. [Google Scholar] [CrossRef]
- Flatz, L.; Rieger, T.; Merkler, D.; Bergthaler, A.; Regen, T.; Schedensack, M.; Bestmann, L.; Verschoor, A.; Kreutzfeldt, M.; Brück, W.; et al. T Cell-Dependence of Lassa Fever Pathogenesis. PLoS Pathog. 2010, 6, e1000836. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, J.; Reyna, R.A.; Kishimoto-Urata, M.; Urata, S.; Manning, J.T.; Harsell, N.; Cook, R.; Huang, C.; Nikolich-Zugich, J.; Makishima, T.; et al. CD4 T-Cell Depletion Prevents Lassa Fever Associated Hearing Loss in the Mouse Model. PLoS Pathog. 2022, 18, e1010557. [Google Scholar] [CrossRef] [PubMed]
- Abele-Ohl, S.; Leis, M.; Mahmoudian, S.; Weyand, M.; Stamminger, T.; Ensminger, S.M. Rag2-/- γ-Chain-/- Mice as Hosts for Human Vessel Transplantation and Allogeneic Human Leukocyte Reconstitution. Transpl Immunol. 2010, 23, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Abele-Ohl, S.; Leis, M.; Wollin, M.; Mahmoudian, S.; Hoffmann, J.; Müller, R.; Heim, C.; Spriewald, B.M.; Weyand, M.; Stamminger, T.; et al. Human Cytomegalovirus Infection Leads to Elevated Levels of Transplant Arteriosclerosis in a Humanized Mouse Aortic Xenograft Model. Am. J. Transplant. 2012, 12, 1720–1729. [Google Scholar] [CrossRef]
- Steinsvik, T.E.; Gaarder, P.I.; Aabergef, I.S.; L0vik, M.; Steinsvik, T.E. Engraftment and Humoral Immunity in SCID and RAG-2-Deficient Mice Transplanted with Human Peripheral Blood Lymphocytes. Scand. J. Immunol. 1995, 42, 607–616. [Google Scholar] [CrossRef]
- Holguin, L.; Echavarria, L.; Burnett, J.C. Novel Humanized Peripheral Blood Mononuclear Cell Mouse Model with Delayed Onset of Graft-versus-Host Disease for Preclinical HIV Research. J. Virol. 2022, 96, e01394-21. [Google Scholar] [CrossRef]
- McCann, C.D.; van Dorp, C.H.; Danesh, A.; Ward, A.R.; Dilling, T.R.; Mota, T.M.; Zale, E.; Stevenson, E.M.; Patel, S.; Brumme, C.J.; et al. A Participant-Derived Xenograft Model of HIV Enables Long-Term Evaluation of Autologous Immunotherapies. J. Exp. Med. 2021, 218, e20201908. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef]
- Chitlaru, T.; Bar-Haim, E.; Bar-On, L.; Rotem, S.; Cohen, H.; Elia, U.; Gur, D.; Aftalion, M.; Alkalay, R.; Makdasi, E.; et al. Implementation of Adenovirus-Mediated Pulmonary Expression of Human ACE2 in HLA Transgenic Mice Enables Establishment of a COVID-19 Murine Model for Assessment of Immune Responses to SARS-CoV-2 Infection. Pathogens 2021, 10, 940. [Google Scholar] [CrossRef] [PubMed]
- Rathnasinghe, R.; Strohmeier, S.; Amanat, F.; Gillespie, V.L.; Krammer, F.; García-Sastre, A.; Coughlan, L.; Schotsaert, M.; Uccellini, M.B. Comparison of Transgenic and Adenovirus HACE2 Mouse Models for SARS-CoV-2 Infection. Emerg. Microbes Infect. 2020, 9, 2433–2445. [Google Scholar] [CrossRef] [PubMed]
- Wong, L.Y.R.; Li, K.; Sun, J.; Zhuang, Z.; Zhao, J.; McCray, P.B.; Perlman, S. Sensitization of Non-Permissive Laboratory Mice to SARS-CoV-2 with a Replication-Deficient Adenovirus Expressing Human ACE2. STAR Protoc. 2020, 1, 100169. [Google Scholar] [CrossRef]
- Oladunni, F.S.; Park, J.G.; Pino, P.A.; Gonzalez, O.; Akhter, A.; Allué-Guardia, A.; Olmo-Fontánez, A.; Gautam, S.; Garcia-Vilanova, A.; Ye, C.; et al. Lethality of SARS-CoV-2 Infection in K18 Human Angiotensin-Converting Enzyme 2 Transgenic Mice. Nat. Commun. 2020, 11, 6122. [Google Scholar] [CrossRef] [PubMed]
- McCray, P.B.; Pewe, L.; Wohlford-Lenane, C.; Hickey, M.; Manzel, L.; Shi, L.; Netland, J.; Jia, H.P.; Halabi, C.; Sigmund, C.D.; et al. Lethal Infection of K18- HACE2 Mice Infected with Severe Acute Respiratory Syndrome Coronavirus. J. Virol. 2007, 81, 813–821. [Google Scholar] [CrossRef]
- Zheng, J.; Wong, L.Y.R.; Li, K.; Verma, A.K.; Ortiz, M.E.; Wohlford-Lenane, C.; Leidinger, M.R.; Knudson, C.M.; Meyerholz, D.K.; McCray, P.B.; et al. COVID-19 Treatments and Pathogenesis Including Anosmia in K18-HACE2 Mice. Nature 2021, 589, 603–607. [Google Scholar] [CrossRef]
- Glazkova, D.V.; Bogoslovskaya, E.V.; Urusov, F.A.; Kartashova, N.P.; Glubokova, E.A.; Gracheva, A.V.; Faizuloev, E.B.; Trunova, G.V.; Khokhlova, V.A.; Bezborodova, O.A.; et al. Generation of SARS-CoV-2 Mouse Model by Transient Expression of the Human ACE2 Gene Mediated by Intranasal Administration of AAV-HACE2. Mol. Biol. 2022, 56, 705–712. [Google Scholar] [CrossRef]
- Strain NOD.Cg-Prkdcscid Il2rgtm1Wjl Ace2tm1(ACE2)Dwnt/J. Available online: https://www.jax.org/strain/035002 (accessed on 5 January 2023).
- Strain NOD.Cg-Gt(ROSA)26Sorem27(KRT18-ACE2)Mvw Prkdcscid Il2rgtm1Wjl/MvwJ. Available online: https://www.jax.org/strain/035959 (accessed on 5 January 2023).
- Strain NOD.Cg-Tg(K18-ACE2)2Prlmn Prkdcscid Il2rgtm1Wjl/J. Available online: https://www.jax.org/strain/034901 (accessed on 5 January 2023).
- Browning, J.; Horner, J.W.; Pettoello-Mantovani, M.; Raker, C.; Yurasov, S.; DePinho, R.A.; Goldstein, H. Mice Transgenic for Human CD4 and CCR5 Are Susceptible to HIV Infection. Proc. Natl. Acad. Sci. USA 1997, 94, 14637–14641. [Google Scholar] [CrossRef]
- Strain C57BL/6J-Tg(SLC10A1)39Mvw/J. Available online: https://www.jax.org/strain/030535 (accessed on 21 December 2022).
- Doyle, J.D.; Barbeau, D.J.; Cartwright, H.N.; McElroy, A.K. Immune Correlates of Protection Following Rift Valley Fever Virus Vaccination. NPJ. Vaccines 2022, 7, 129. [Google Scholar] [CrossRef]
- Liao, S.-H.; Chang, W.-J.; Hsu, C.-Y.; Ming-Fang Yen, A.; Lin, T.-Y.; Li-Sheng Chen, S.; Hsiu-Hsi Chen, T. Evaluating Correlates of Protection for Mix-Match Vaccine against COVID-19 VOCs with Potential of Evading Immunity. Vaccine 2022, 40, 6864–6872. [Google Scholar] [CrossRef]
- Atti, A.; Insalata, F.; Carr, E.J.; Otter, A.D.; Castillo-Olivares, J.; Wu, M.; Harvey, R.; Howell, M.; Chan, A.; Lyall, J.; et al. Antibody Correlates of Protection from SARS-CoV-2 Reinfection Prior to Vaccination: A Nested Case-Control within the SIREN Study. J. Infect. 2022, 85, 545–556. [Google Scholar] [CrossRef]
- Reynolds, C.J.; Pade, C.; Gibbons, J.M.; Otter, A.D.; Lin, K.M.; Sandoval, D.M.; Pieper, F.P.; Butler, D.K.; Liu, S.; Joy, G.; et al. Immune Boosting by B.1.1.529 (Omicron) Depends on Previous SARS-CoV-2 Exposure. Science 2022, 377, eabq1841. [Google Scholar] [CrossRef]
- Chi, W.Y.; Li, Y.D.; Huang, H.C.; Chan, T.E.H.; Chow, S.Y.; Su, J.H.; Ferrall, L.; Hung, C.F.; Wu, T.C. COVID-19 Vaccine Update: Vaccine Effectiveness, SARS-CoV-2 Variants, Boosters, Adverse Effects, and Immune Correlates of Protection. J. Biomed. Sci. 2022, 29, 82. [Google Scholar] [CrossRef]
- Vivaldi, G.; Jolliffe, D.A.; Faustini, S.; Shields, A.M.; Holt, H.; Perdek, N.; Talaei, M.; Tydeman, F.; Chambers, E.S.; Cai, W.; et al. Correlation Between Postvaccination Anti-Spike Antibody Titers and Protection Against Breakthrough Severe Acute Respiratory Syndrome Coronavirus 2 Infection: A Population-Based Longitudinal Study. J. Infect Dis. 2022, 226, 1903–1908. [Google Scholar] [CrossRef]
- Özbay Kurt, F.G.; Lepper, A.; Gerhards, C.; Roemer, M.; Lasser, S.; Arkhypov, I.; Bitsch, R.; Bugert, P.; Altevogt, P.; Gouttefangeas, C.; et al. Booster Dose of MRNA Vaccine Augments Waning T Cell and Antibody Responses against SARS-CoV-2. Front. Immunol. 2022, 13, 1012526. [Google Scholar] [CrossRef]
- Altarawneh, H.N.; Chemaitelly, H.; Hasan, M.R.; Ayoub, H.H.; Qassim, S.; AlMukdad, S.; Coyle, P.; Yassine, H.M.; Al-Khatib, H.A.; Benslimane, F.M.; et al. Protection against the Omicron Variant from Previous SARS-CoV-2 Infection. N. Engl. J. Med. 2022, 386, 1288–1290. [Google Scholar] [CrossRef]
- Corbett, K.S.; Nason, M.C.; Flach, B.; Gagne, M.; O’Connell, S.; Johnston, T.S.; Shah, S.N.; Edara, V.V.; Floyd, K.; Lai, L.; et al. Immune Correlates of Protection by MRNA-1273 Vaccine against SARS-CoV-2 in Nonhuman Primates. Science 2021, 373, eabj0299. [Google Scholar] [CrossRef]
- Koch, T.; Mellinghoff, S.C.; Shamsrizi, P.; Addo, M.M.; Dahlke, C. Correlates of Vaccine-Induced Protection against Sars-Cov-2. Vaccines 2021, 9, 238. [Google Scholar] [CrossRef]
- McMahan, K.; Yu, J.; Mercado, N.B.; Loos, C.; Tostanoski, L.H.; Chandrashekar, A.; Liu, J.; Peter, L.; Atyeo, C.; Zhu, A.; et al. Correlates of Protection against SARS-CoV-2 in Rhesus Macaques. Nature 2021, 590, 630–634. [Google Scholar] [CrossRef]
- Ma, J.; Boudewijns, R.; Sanchez-Felipe, L.; Mishra, N.; Vercruysse, T.; Buh Kum, D.; Thibaut, H.J.; Neyts, J.; Dallmeier, K. Comparing Immunogenicity and Protective Efficacy of the Yellow Fever 17D Vaccine in Mice. Emerg. Microbes Infect. 2021, 10, 2279–2290. [Google Scholar] [CrossRef]
- Wu, F.; Qin, M.; Wang, H.; Sun, X. Nanovaccines to Combat Virus-Related Diseases. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2022, 2, e1857. [Google Scholar] [CrossRef]
- Ghattas, M.; Dwivedi, G.; Lavertu, M.; Alameh, M.G. Vaccine Technologies and Platforms for Infectious Diseases: Current Progress, Challenges, and Opportunities. Vaccines 2021, 9, 1490. [Google Scholar] [CrossRef]
- Han, J.; Sun, J.; Zhang, G.; Chen, H. Dcs-Based Therapies: Potential Strategies in Severe Sars-Cov-2 Infection. Int. J. Med. Sci. 2021, 18, 406–418. [Google Scholar] [CrossRef]
- Patham, B.; Simmons, G.L.; Subramanya, S. Advances in Dendritic Cell-Based Vaccines for HIV. Curr. Med. Chem. 2011, 18, 3987–3994. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, Y.; Yao, Z.; Moorman, J.P.; Jia, Z. Dendritic Cell-Based Immunity and Vaccination against Hepatitis C Virus Infection. Immunology 2012, 136, 385–396. [Google Scholar] [CrossRef]
- Norton, T.D.; Tada, T.; Leibowitz, R.; van der Heide, V.; Homann, D.; Landau, N.R. Lentiviral-Vector-Based Dendritic Cell Vaccine Synergizes with Checkpoint Blockade to Clear Chronic Viral Infection. Mol. Ther. 2020, 28, 1795–1805. [Google Scholar] [CrossRef]
- Norton, T.D.; Zhen, A.; Tada, T.; Kim, J.; Kitchen, S.; Landau, N.R. Lentiviral Vector-Based Dendritic Cell Vaccine Suppresses HIV Replication in Humanized Mice. Mol. Ther. 2019, 27, 960–973. [Google Scholar] [CrossRef]
- Tada, T.; Norton, T.D.; Leibowitz, R.; Landau, N.R. Directly Injected Lentiviral Vector-Based T Cell Vaccine Protects Mice against Acute and Chronic Viral Infection. JCI Insight 2022, 7, e161598. [Google Scholar] [CrossRef]
- Fehér, C.; Pastor-Lbáñez, R.; Leal, L.; Plana, M.; Arnedo, M.; van den Ham, H.J.; Andeweg, A.C.; Gruters, R.A.; Díez-Fuertes, F.; Alcamí, J.; et al. Association of Transcriptomic Signatures of Inflammatory Response with Viral Control after Dendritic Cell-Based Therapeutic Vaccination in Hiv-1 Infected Individuals. Vaccines 2021, 9, 799. [Google Scholar] [CrossRef]
- Sadat Larijani, M.; Ramezani, A.; Mashhadi Abolghasem Shirazi, M.; Bolhassani, A.; Pouriayevali, M.H.; Shahbazi, S.; Sadat, S.M. Evaluation of Transduced Dendritic Cells Expressing HIV-1 P24-Nef Antigens in HIV-Specific Cytotoxic T Cells Induction as a Therapeutic Candidate Vaccine. Virus Res. 2021, 298, 198403. [Google Scholar] [CrossRef]
- Espinar-Buitrago, M.; Muñoz-Fernández, M.A. New Approaches to Dendritic Cell-Based Therapeutic Vaccines Against HIV-1 Infection. Front. Immunol. 2022, 12, 719664. [Google Scholar] [CrossRef] [PubMed]
- Allard, S.D.; de Keersmaecker, B.; de Goede, A.L.; Verschuren, E.J.; Koetsveld, J.; Reedijk, M.L.; Wylock, C.; de Bel, A.V.; Vandeloo, J.; Pistoor, F.; et al. A Phase I/IIa Immunotherapy Trial of HIV-1-Infected Patients with Tat, Rev. and Nef Expressing Dendritic Cells Followed by Treatment Interruption. Clin. Immunol. 2012, 142, 252–268. [Google Scholar] [CrossRef] [PubMed]
- Zabaleta, A.; D’Avola, D.; Echeverria, I.; Llopiz, D.; Silva, L.; Villanueva, L.; Riezu-Boj, J.I.; Larrea, E.; Pereboev, A.; Lasarte, J.J.; et al. Clinical Testing of a Dendritic Cell Targeted Therapeutic Vaccine in Patients with Chronic Hepatitis C Virus Infection. Mol. Ther. Methods Clin. Dev. 2015, 2, 15006. [Google Scholar] [CrossRef]
- García, F.; Climent, N.; Guardo, A.C.; Gil, C.; León, A.; Autran, B.; Lifson, J.D.; Martínez-Picado, J.; Dalmau, J.; Clotet, B.; et al. A Dendritic Cell-Based Vaccine Elicits T Cell Responses Associated with Control of HIV-1 Replication. Sci. Transl. Med. 2013, 5, 166ra2. [Google Scholar] [CrossRef]
- Teame, G.; Gebreyesus, A.; Tsegay, E.; Gebretsadik, M.; Adane, K. Hepatitis B and C Viral Coinfections and Their Association with HIV Viral Load Suppression among HIV-1 Infected Patients on ART at Mekelle Hospital, Northern Ethiopia. AIDS Res. Ther. 2022, 19, 57. [Google Scholar] [CrossRef] [PubMed]
- Seyoum, E.; Demissie, M.; Worku, A.; Mulu, A.; Abdissa, A.; Berhane, Y. HIV, Hepatitis B Virus, and Hepatitis C Virus Co-Infection among HIV Positives in Antiretroviral Treatment Program in Selected Hospitals in Addis Ababa: A Retrospective Cross-Sectional Study. PLoS ONE 2022, 17, e0267230. [Google Scholar] [CrossRef]
- Mulherkar, T.H.; Gómez, D.J.; Sandel, G.; Jain, P. Co-Infection and Cancer: Host-Pathogen Interaction between Dendritic Cells and HIV-1, HTLV-1, and Other Oncogenic Viruses. Viruses 2022, 14, 2037. [Google Scholar] [CrossRef]
- Khan, M.M.; Ali, M.J.; Hanif, H.; Maqsood, M.H.; Ahmad, I.; Alvarez, J.E.G.; Catana, M.A.; Lau, D.T.Y. The Dilemma of Cytomegalovirus and Hepatitis B Virus Interaction. Gastroenterol. Rep. 2022, 10, goac018. [Google Scholar] [CrossRef]
- Tang, C.Y.; Boftsi, M.; Staudt, L.; McElroy, J.A.; Li, T.; Duong, S.; Ohler, A.; Ritter, D.; Hammer, R.; Hang, J.; et al. SARS-CoV-2 and Influenza Co-Infection: A Cross-Sectional Study in Central Missouri during the 2021–2022 Influenza Season. Virology 2022, 576, 105–110. [Google Scholar] [CrossRef]
- Swets, M.C.; Russell, C.D.; Harrison, E.M.; Docherty, A.B.; Lone, N.; Girvan, M.; Hardwick, H.E.; Visser, L.G.; Openshaw, P.J.M.; Groeneveld, G.H.; et al. SARS-CoV-2 Co-Infection with Influenza Viruses, Respiratory Syncytial Virus, or Adenoviruses. Lancet 2022, 399, 1463–1464. [Google Scholar] [CrossRef]
- Mailly, L.; Xiao, F.; Lupberger, J.; Wilson, G.K.; Aubert, P.; Duong, F.H.T.; Calabrese, D.; Leboeuf, C.; Fofana, I.; Thumann, C.; et al. Clearance of Persistent Hepatitis C Virus Infection in Humanized Mice Using a Claudin-1-Targeting Monoclonal Antibody. Nat. Biotechnol. 2015, 33, 549–554. [Google Scholar] [CrossRef]
- Uchida, T.; Imamura, M.; Kan, H.; Hiraga, N.; Hayes, C.N.; Tsuge, M.; Abe-Chayama, H.; Aikata, H.; Makokha, G.N.; Miki, D.; et al. Usefulness of Humanized CDNA-UPA/SCID Mice for the Study of Hepatitis B Virus and Hepatitis C Virus Virology. J. Gen. Virol. 2017, 98, 1040–1047. [Google Scholar] [CrossRef]
- Wang, Z.; Wu, N.; Tesfaye, A.; Feinstone, S.; Kumar, A. HCV Infection-Associated Hepatocellular Carcinoma in Humanized Mice. Infect. Agent. Cancer 2015, 10, 1–9. [Google Scholar] [CrossRef]
- Washburn, M.L.; Bility, M.T.; Zhang, L.; Kovalev, G.I.; Buntzman, A.; Frelinger, J.A.; Barry, W.; Ploss, A.; Rice, C.M.; Su, L. A Humanized Mouse Model to Study Hepatitis C Virus Infection, Immune Response, and Liver Disease. Gastroenterology 2011, 140, 1334–1344. [Google Scholar] [CrossRef]
- Strick-Marchand, H.; Dusséaux, M.; Darche, S.; Huntington, N.D.; Legrand, N.; Masse-Ranson, G.; Corcuff, E.; Ahodantin, J.; Weijer, K.; Spits, H.; et al. A Novel Mouse Model for Stable Engraftment of a Human Immune System and Human Hepatocytes. PLoS ONE 2015, 10, e0119820. [Google Scholar] [CrossRef]
- Bility, M.T.; Zhang, L.; Washburn, M.L.; Curtis, T.A.; Kovalev, G.I.; Su, L. Generation of a Humanized Mouse Model with Both Human Immune System and Liver Cells to Model Hepatitis C Virus Infection and Liver Immunopathogenesis. Nat. Protoc. 2012, 7, 1608–1617. [Google Scholar] [CrossRef]

{kind=link}
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brunetti, J.E.; Kitsera, M.; Muñoz-Fontela, C.; Rodríguez, E. Use of Hu-PBL Mice to Study Pathogenesis of Human-Restricted Viruses. Viruses 2023, 15, 228. https://doi.org/10.3390/v15010228
Brunetti JE, Kitsera M, Muñoz-Fontela C, Rodríguez E. Use of Hu-PBL Mice to Study Pathogenesis of Human-Restricted Viruses. Viruses. 2023; 15(1):228. https://doi.org/10.3390/v15010228
Chicago/Turabian StyleBrunetti, Jesús Emanuel, Maksym Kitsera, César Muñoz-Fontela, and Estefanía Rodríguez. 2023. "Use of Hu-PBL Mice to Study Pathogenesis of Human-Restricted Viruses" Viruses 15, no. 1: 228. https://doi.org/10.3390/v15010228
APA StyleBrunetti, J. E., Kitsera, M., Muñoz-Fontela, C., & Rodríguez, E. (2023). Use of Hu-PBL Mice to Study Pathogenesis of Human-Restricted Viruses. Viruses, 15(1), 228. https://doi.org/10.3390/v15010228