
A Counterintuitive Neutrophil-Mediated Pattern in COVID-19 Patients Revealed through Transcriptomics Analysis
 , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
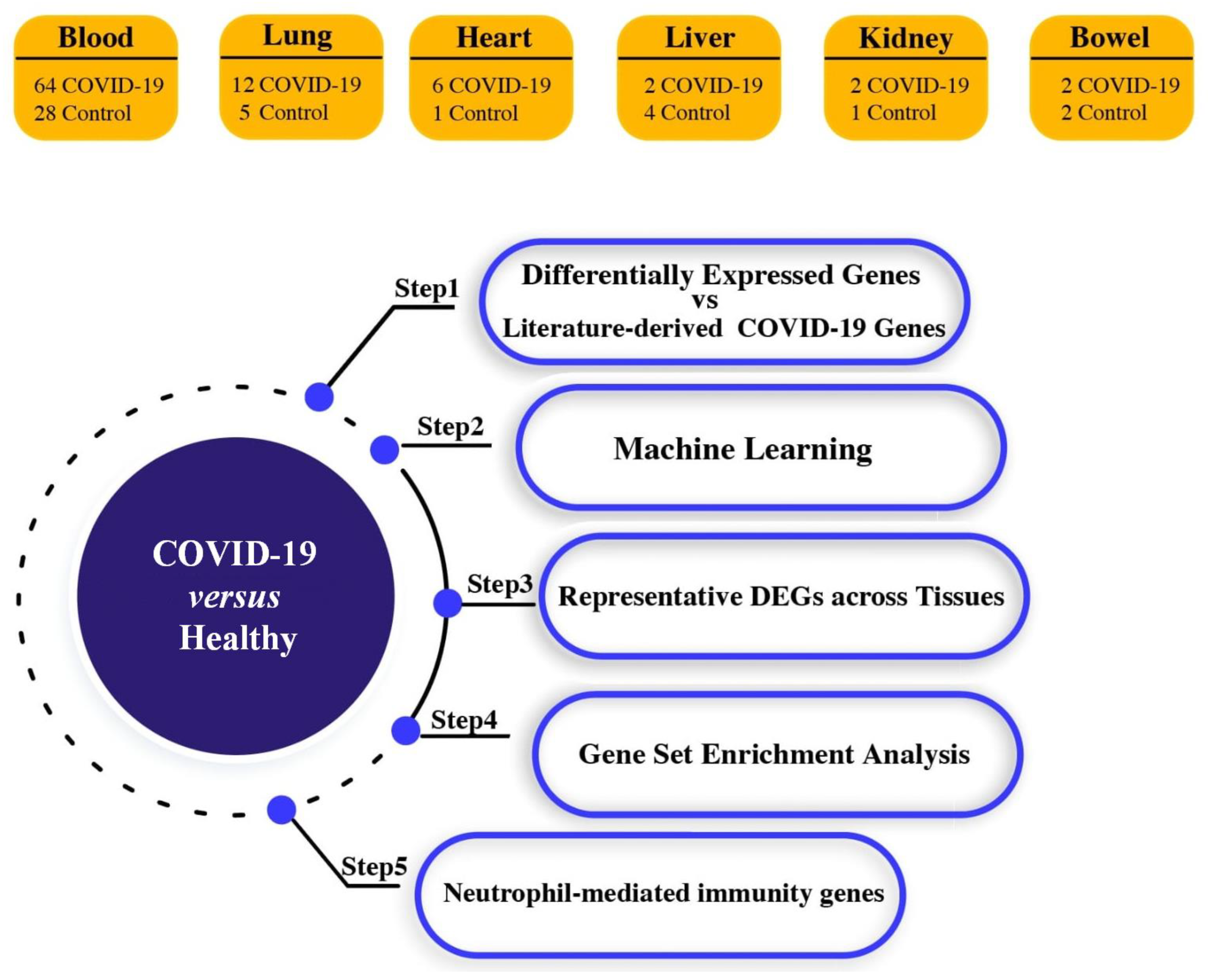
2.1. Acquisition of High-Throughput Transcriptome Data
2.2. RNA-Seq Data Processing
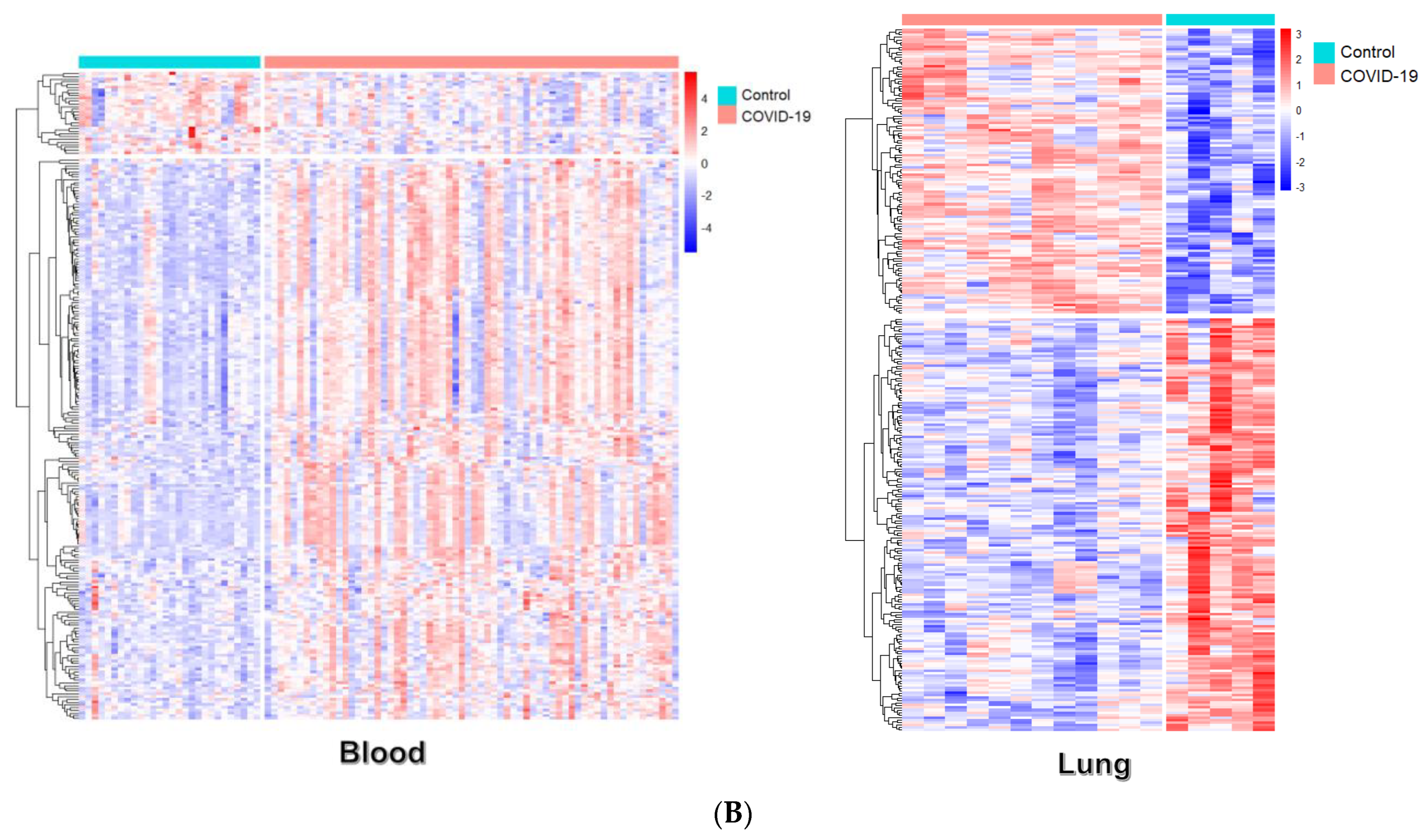
2.3. Differential Gene Expression Analysis
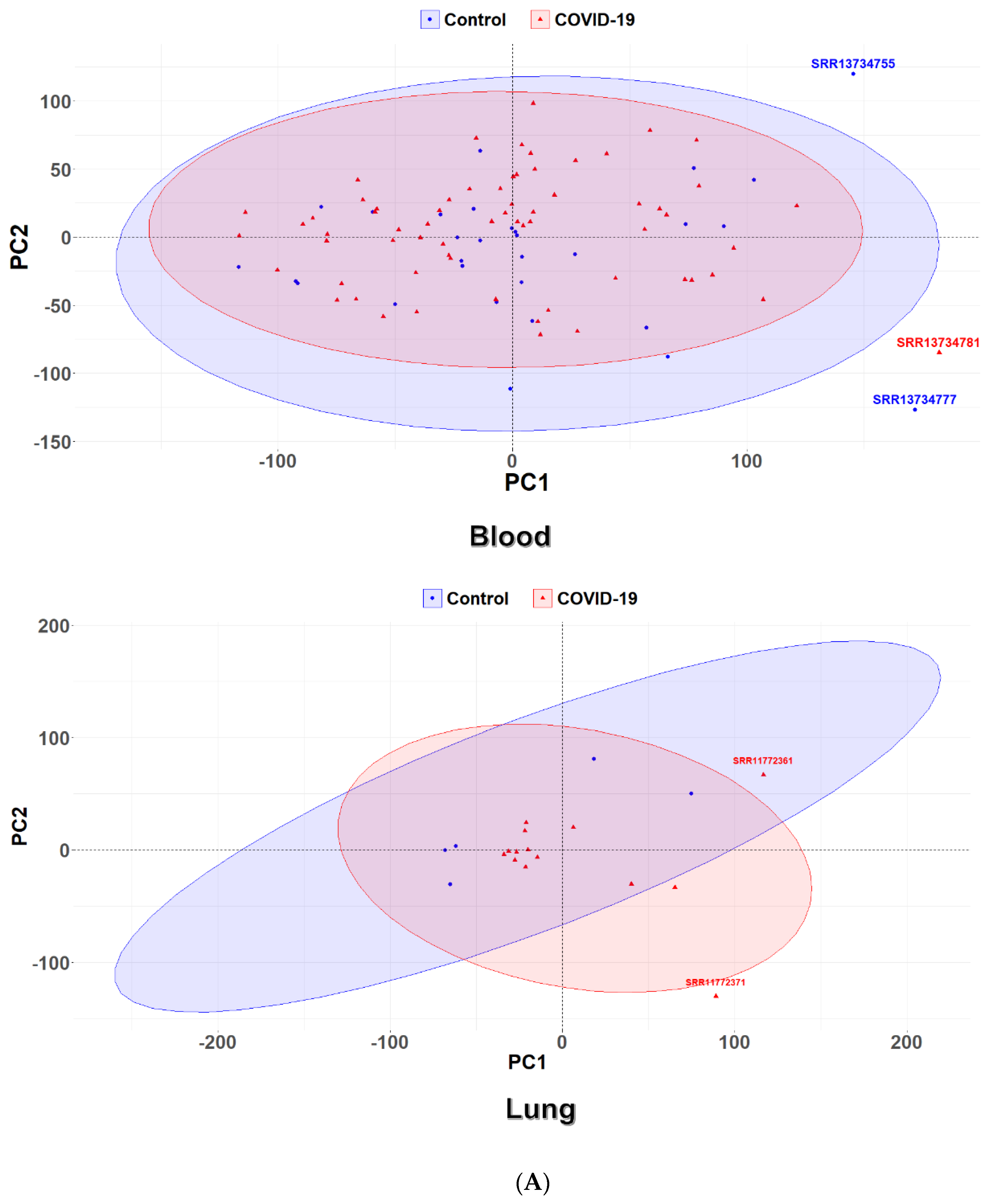
2.4. Principal Component Analysis
2.5. Ensemble Machine Learning
2.6. Functional Enrichment Analysis
2.7. Natural Language Processing
3. Results
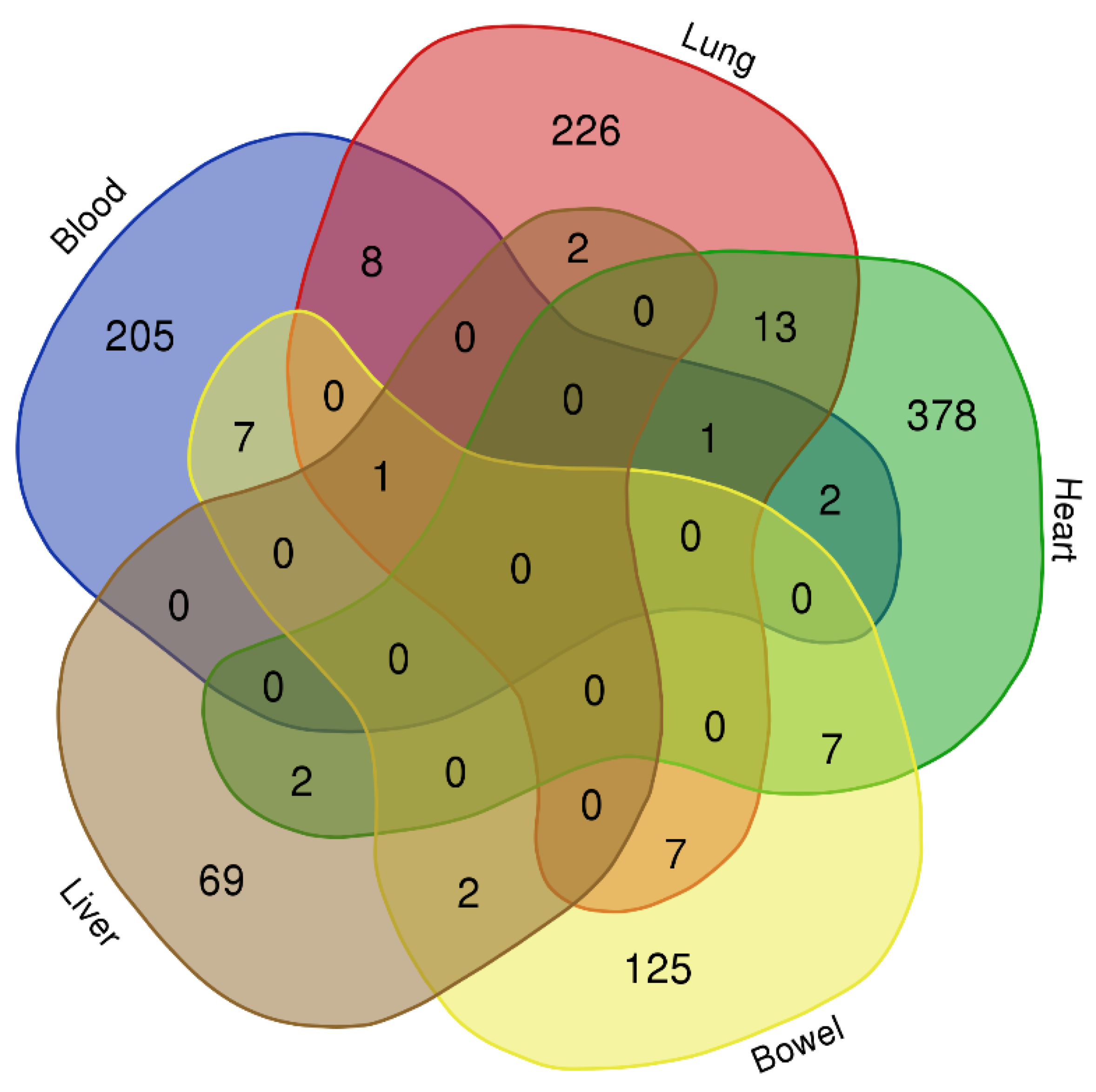
3.1. Identification of Differential Expression Patterns in SARS-CoV-2-İnfected vs. Healthy Tissues
3.2. Pathway Enrichment Analysis of Common Selected DEGs
4. Discussion
- The patients’ characteristics (ethnicity, locality, and demographics) and clinical histories (comorbidities and medications administered prior to sampling) were unclear in the source publications.
- The populations were small, and, hence, even if we knew the above characteristics, their stratification into smaller clusters of epidemiological interest would be questioned.
- The hypoxia-enhanced neutrophil degranulation mechanisms’ proteins were little-known.
- The variability in sample preparation could also have impacted the sequence findings.
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Lian, X.; Su, X.; Wu, W.; Marraro, G.A.; Zeng, Y. From SARS and MERS to COVID-19: A brief summary and comparison of severe acute respiratory infections caused by three highly pathogenic human coronaviruses. Respir. Res. 2020, 21, 224. [Google Scholar] [CrossRef] [PubMed]
- Hagemann, J.; Onorato, G.; Seifen, C.; Becker, S.; Huppertz, T.; Olze, H.; Kuna, P.; Mullol, J.; Salmi, S.T.; Fonseca, J.; et al. Presentation of airway and general symptoms in COVID-19 caused by dominant SARS-CoV-2 variants: A follow-up on ARIA consensus. Allergy 2022, 77, 3440–3444. [Google Scholar] [CrossRef]
- Lascarrou, J.B. COVID-19-related ARDS: One disease, two trajectories, and several unanswered questions. Lancet Respir. Med. 2021, 9, 1345–1347. [Google Scholar] [CrossRef] [PubMed]
- Camporota, L.; Cronin, J.N.; Busana, M.; Gattinoni, L.; Formenti, F. Pathophysiology of coronavirus-19 disease acute lung injury. Curr. Opin. Crit. Care 2022, 28, 9–16. [Google Scholar] [CrossRef]
- Fathi, N.; Rezaei, N. Lymphopenia in COVID-19: Therapeutic opportunities. Cell Biol. Int. 2020, 44, 1792–1797. [Google Scholar] [CrossRef]
- Reusch, N.; De Domenico, E.; Bonaguro, L.; Schulte-Schrepping, J.; Bassler, K.; Schultze, J.L.; Aschenbrenner, A.C. Neutrophils in COVID-19. Front. Immunol. 2021, 12, 652470. [Google Scholar] [CrossRef]
- Lopes-Pacheco, M.; Silva, P.L.; Cruz, F.F.; Battaglini, D.; Robba, C.; Pelosi, P.; Morales, M.M.; Caruso Neves, C.; Rocco, P.R.M. Pathogenesis of Multiple Organ Injury in COVID-19 and Potential Therapeutic Strategies. Front. Physiol. 2021, 12, 593223. [Google Scholar] [CrossRef]
- Mokhtari, T.; Hassani, F.; Ghaffari, N.; Ebrahimi, B.; Yarahmadi, A.; Hassanzadeh, G. COVID-19 and multiorgan failure: A narrative review on potential mechanisms. J. Mol. Histol. 2020, 51, 613–628. [Google Scholar] [CrossRef]
- Kocak Tufan, Z.; Kayaaslan, B.; Mer, M. COVID-19 and Sepsis. Turk. J. Med. Sci. 2021, 51, 3301–3311. [Google Scholar] [CrossRef]
- Buszko, M.; Nita-Lazar, A.; Park, J.H.; Schwartzberg, P.L.; Verthelyi, D.; Young, H.A.; Rosenberg, A.S. Lessons learned: New insights on the role of cytokines in COVID-19. Nat. Immunol. 2021, 22, 404–411. [Google Scholar] [CrossRef] [PubMed]
- Liuzzo, G.; Patrono, C. COVID-19: In the eye of the cytokine storm. Eur. Heart J. 2021, 42, 150–151. [Google Scholar] [CrossRef]
- Lucas, C.; Wong, P.; Klein, J.; Castro, T.B.R.; Silva, J.; Sundaram, M.; Ellingson, M.K.; Mao, T.; Oh, J.E.; Israelow, B.; et al. Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature 2020, 584, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.Y.C.; Hoiland, R.L.; Stukas, S.; Wellington, C.L.; Sekhon, M.S. Confronting the controversy: Interleukin-6 and the COVID-19 cytokine storm syndrome. Eur. Respir. J. 2020, 56, 2003006. [Google Scholar] [CrossRef] [PubMed]
- Santa Cruz, A.; Mendes-Frias, A.; Oliveira, A.I.; Dias, L.; Matos, A.R.; Carvalho, A.; Capela, C.; Pedrosa, J.; Castro, A.G.; Silvestre, R. Interleukin-6 Is a Biomarker for the Development of Fatal Severe Acute Respiratory Syndrome Coronavirus 2 Pneumonia. Front. Immunol. 2021, 12, 613422. [Google Scholar] [CrossRef]
- Shekhawat, J.; Gauba, K.; Gupta, S.; Purohit, P.; Mitra, P.; Garg, M.; Misra, S.; Sharma, P.; Banerjee, M. Interleukin-6 Perpetrator of the COVID-19 Cytokine Storm. Indian J. Clin.Biochem. IJCB 2021, 36, 440–450. [Google Scholar] [CrossRef]
- Geronikolou, S.A.; Takan, I.; Pavlopoulou, A.; Mantzourani, M.; Chrousos, G.P. Thrombocytopenia in COVID-19 and vaccine-induced thrombotic thrombocytopenia. Int. J. Mol. Med. 2022, 49, 1–19. [Google Scholar] [CrossRef]
- Wise, J. COVID-19: Symptomatic infection with omicron variant is milder and shorter than with delta, study reports. BMJ 2022, 377, o922. [Google Scholar] [CrossRef]
- Nealon, J.; Cowling, B.J. Omicron severity: Milder but not mild. Lancet 2022, 399, 412–413. [Google Scholar] [CrossRef]
- Suryawanshi, R.K.; Chen, I.P.; Ma, T.; Syed, A.M.; Brazer, N.; Saldhi, P.; Simoneau, C.R.; Ciling, A.; Khalid, M.M.; Sreekumar, B.; et al. Limited cross-variant immunity from SARS-CoV-2 Omicron without vaccination. Nature 2022, 607, 351–355. [Google Scholar] [CrossRef]
- Cron, R.Q.; Caricchio, R.; Chatham, W.W. Calming the cytokine storm in COVID-19. Nat. Med. 2021, 27, 1674–1675. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Xie, X.; Tu, Z.; Fu, J.; Xu, D.; Zhou, Y. The signal pathways and treatment of cytokine storm in COVID-19. Signal Transduct. Target. Ther. 2021, 6, 255. [Google Scholar] [CrossRef] [PubMed]
- Liao, M.; Liu, Y.; Yuan, J.; Wen, Y.; Xu, G.; Zhao, J.; Cheng, L.; Li, J.; Wang, X.; Wang, F.; et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat. Med. 2020, 26, 842–844. [Google Scholar] [CrossRef]
- Ong, E.Z.; Chan, Y.F.Z.; Leong, W.Y.; Lee, N.M.Y.; Kalimuddin, S.; Haja Mohideen, S.M.; Chan, K.S.; Tan, A.T.; Bertoletti, A.; Ooi, E.E.; et al. A Dynamic Immune Response Shapes COVID-19 Progression. Cell Host Microbe 2020, 27, 879–882.e872. [Google Scholar] [CrossRef]
- Desai, N.; Neyaz, A.; Szabolcs, A.; Shih, A.R.; Chen, J.H.; Thapar, V.; Nieman, L.T.; Solovyov, A.; Mehta, A.; Lieb, D.J.; et al. Temporal and spatial heterogeneity of host response to SARS-CoV-2 pulmonary infection. Nat. Commun. 2020, 11, 6319. [Google Scholar] [CrossRef]
- Galbraith, M.D.; Kinning, K.T.; Sullivan, K.D.; Baxter, R.; Araya, P.; Jordan, K.R.; Russell, S.; Smith, K.P.; Granrath, R.E.; Shaw, J.R.; et al. Seroconversion stages COVID-19 into distinct pathophysiological states. Elife 2021, 10, e65508. [Google Scholar] [CrossRef]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets--update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef]
- Clough, E.; Barrett, T. The Gene Expression Omnibus Database. Methods Mol. Biol. 2016, 1418, 93–110. [Google Scholar] [CrossRef]
- Alnasir, J.; Shanahan, H.P. Investigation into the annotation of protocol sequencing steps in the sequence read archive. GigaScience 2015, 4, 23. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Tweedie, S.; Braschi, B.; Gray, K.; Jones, T.E.M.; Seal, R.L.; Yates, B.; Bruford, E.A. Genenames.org: The HGNC and VGNC resources in 2021. Nucleic Acids Res. 2021, 49, D939–D946. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Society. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Breitling, R.; Armengaud, P.; Amtmann, A.; Herzyk, P. Rank products: A simple, yet powerful, new method to detect differentially regulated genes in replicated microarray experiments. FEBS Lett. 2004, 573, 83–92. [Google Scholar] [CrossRef]
- Breitling, R.; Herzyk, P. Rank-based methods as a non-parametric alternative of the T-statistic for the analysis of biological microarray data. J. Bioinform. Comput. Biol. 2005, 3, 1171–1189. [Google Scholar] [CrossRef]
- Heskes, T.; Eisinga, R.; Breitling, R. A fast algorithm for determining bounds and accurate approximate p-values of the rank product statistic for replicate experiments. BMC Bioinform. 2014, 15, 367. [Google Scholar] [CrossRef]
- Farrel, A.; Guo, J.T. An efficient algorithm for improving structure-based prediction of transcription factor binding sites. BMC Bioinform. 2017, 18, 342. [Google Scholar] [CrossRef]
- Mayr, A.; Binder, H.; Gefeller, O.; Schmid, M. The evolution of boosting algorithms. From machine learning to statistical modelling. Methods Inf. Med. 2014, 53, 419–427. [Google Scholar] [CrossRef]
- Alzubi, J.; Nayyar, A.; Kumar, A. Machine Learning from Theory to Algorithms: An Overview. J.Phys. Conf. Ser. 2018, 1142, 1–15. [Google Scholar] [CrossRef]
- Kirov, S.; Ji, R.; Wang, J.; Zhang, B. Functional annotation of differentially regulated gene set using WebGestalt: A gene set predictive of response to ipilimumab in tumor biopsies. Methods Mol. Biol. 2014, 1101, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Wang, J.; Jaehnig, E.J.; Shi, Z.; Zhang, B. WebGestalt 2019: Gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019, 47, W199–W205. [Google Scholar] [CrossRef]
- Neumann, M.; King, D.; Beltagy, I.; Ammar, W. ScispaCy: Fast and Robust Models for Biomedical Natural Language Processing. In Proceedings of the 18th BioNLP Workshop and Shared Task, Florence, Italy, 1 August 2019; pp. 319–327. [Google Scholar]
- Hickey, M.J.; Kubes, P. Intravascular immunity: The host-pathogen encounter in blood vessels. Nat. Rev. Immunol. 2009, 9, 364–375. [Google Scholar] [CrossRef]
- Rosales, C. Neutrophil: A Cell with Many Roles in Inflammation or Several Cell Types? Front. Physiol. 2018, 9, 113. [Google Scholar] [CrossRef]
- Li, Y.; Wang, W.; Yang, F.; Xu, Y.; Feng, C.; Zhao, Y. The regulatory roles of neutrophils in adaptive immunity. Cell Commun.Signal. CCS 2019, 17, 147. [Google Scholar] [CrossRef]
- Pillay, J.; Tak, T.; Kamp, V.M.; Koenderman, L. Immune suppression by neutrophils and granulocytic myeloid-derived suppressor cells: Similarities and differences. Cell. Mol. LifeSci. CMLS 2013, 70, 3813–3827. [Google Scholar] [CrossRef]
- Alipoor, S.D.; Mirsaeidi, M. SARS-CoV-2 cell entry beyond the ACE2 receptor. Mol. Biol. Rep. 2022, 49, 10715–10727. [Google Scholar] [CrossRef]
- del Valle-Mendoza, J.; Tarazona-Castro, Y.; Merino-Luna, A.; Carrillo-Ng, H.; Kym, S.; Aguilar-Luis, M.A.; del Valle, L.J.; Aquino-Ortega, R.; Martins-Luna, J.; Peña-Tuesta, I.; et al. Comparison of cytokines levels among COVID-19 patients living at sea level and high altitude. BMC Infect. Dis. 2022, 22, 96. [Google Scholar] [CrossRef]
- Stöwhas, A.C.; Latshang, T.D.; Lo Cascio, C.M.; Lautwein, S.; Stadelmann, K.; Tesler, N.; Ayers, L.; Berneis, K.; Gerber, P.A.; Huber, R.; et al. Effects of acute exposure to moderate altitude on vascular function, metabolism and systemic inflammation. PLoS ONE 2013, 8, e70081. [Google Scholar] [CrossRef]
- Stephens, K.E.; Chernyavskiy, P.; Bruns, D.R. Impact of altitude on COVID-19 infection and death in the United States: A modeling and observational study. PLoS ONE 2021, 16, e0245055. [Google Scholar] [CrossRef] [PubMed]
- Burtscher, M. Effects of living at higher altitudes on mortality: A narrative review. Aging Dis. 2014, 5, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Arias-Reyes, C.; Zubieta-DeUrioste, N.; Poma-Machicao, L.; Aliaga-Raduan, F.; Carvajal-Rodriguez, F.; Dutschmann, M.; Schneider-Gasser, E.M.; Zubieta-Calleja, G.; Soliz, J. Does the pathogenesis of SARS-CoV-2 virus decrease at high-altitude? Respir. Physiol. Neurobiol. 2020, 277, 103443. [Google Scholar] [CrossRef] [PubMed]
- Zubieta-Calleja, G.R.; Zubieta-DeUrioste, N.A. Extended longevity at high altitude: Benefits of exposure to chronic hypoxia. BLDE Univ. J. Health Sci. 2017, 2, 80. [Google Scholar] [CrossRef]
- Hunter, C.A.; Jones, S.A. IL-6 as a keystone cytokine in health and disease. Nat. Immunol. 2015, 16, 448–457. [Google Scholar] [CrossRef]
- Del Valle, D.M.; Kim-Schulze, S.; Huang, H.-H.; Beckmann, N.D.; Nirenberg, S.; Wang, B.; Lavin, Y.; Swartz, T.H.; Madduri, D.; Stock, A. An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat. Med. 2020, 26, 1636–1643. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, G.; Tschöp, M.; Fischer, R.; Bidlingmaier, C.; Riepl, R.; Tschöp, K.; Hautmann, H.; Endres, S.; Toepfer, M. High altitude increases circulating interleukin-6, interleukin-1 receptor antagonist and C-reactive protein. Cytokine 2000, 12, 246–252. [Google Scholar] [CrossRef]
- Cordon-Cardo, C.; Pujadas, E.; Wajnberg, A.; Sebra, R.; Patel, G.; Firpo-Betancourt, A.; Fowkes, M.; Sordillo, E.; Paniz-Mondolfi, A.; Gregory, J.; et al. COVID-19: Staging of a New Disease. Cancer Cell 2020, 38, 594–597. [Google Scholar] [CrossRef]
- Gandhi, M.; Yokoe, D.S.; Havlir, D.V. Asymptomatic Transmission, the Achilles’ Heel of Current Strategies to Control COVID-19. N. Engl. J. Med. 2020, 382, 2158–2160. [Google Scholar] [CrossRef]
- Huang, Y.; Zaas, A.K.; Rao, A.; Dobigeon, N.; Woolf, P.J.; Veldman, T.; Øien, N.C.; McClain, M.T.; Varkey, J.B.; Nicholson, B.; et al. Temporal dynamics of host molecular responses differentiate symptomatic and asymptomatic influenza a infection. PLoS Genet. 2011, 7, e1002234. [Google Scholar] [CrossRef]
- Cao, X. COVID-19: Immunopathology and its implications for therapy. Nat. Rev. Immunol. 2020, 20, 269–270. [Google Scholar] [CrossRef] [PubMed]
- Sarma, A.; Christenson, S.A.; Mick, E.; DeVoe, C.; Deiss, T.; Pisco, A.O.; Ghale, R.; Jauregui, A.; Byrne, A.; Moazed, F.; et al. COVID-19 ARDS is characterized by a dysregulated host response that differs from cytokine storm and is modified by dexamethasone. Res. Sq. 2021. [Google Scholar] [CrossRef]
- Edge, R.; Truscott, T.G. COVID-19 and the ethnicity link - is there a photochemical link? Photochem. Photobiol. Sci. 2021, 20, 183–188. [Google Scholar] [CrossRef]
- Remy, K.E.; Mazer, M.; Striker, D.A.; Ellebedy, A.H.; Walton, A.H.; Unsinger, J.; Blood, T.M.; Mudd, P.A.; Yi, D.J.; Mannion, D.A.; et al. Severe immunosuppression and not a cytokine storm characterizes COVID-19 infections. JCI Insight 2020, 5, e140329. [Google Scholar] [CrossRef] [PubMed]
- Mo, Y.; Eyre, D.W.; Lumley, S.F.; Walker, T.M.; Shaw, R.H.; O’Donnell, D.; Butcher, L.; Jeffery, K.; Donnelly, C.A.; Oxford COVID infection review team; et al. Transmission of community- and hospital-acquired SARS-CoV-2 in hospital settings in the UK: A cohort study. PLoS Med. 2021, 18, e1003816. [Google Scholar] [CrossRef]
- Ong, C.C.H.; Farhanah, S.; Linn, K.Z.; Tang, Y.W.; Poon, C.Y.; Lim, A.Y.; Tan, H.R.; Binte Hamed, N.H.; Huan, X.; Puah, S.H.; et al. Nosocomial infections among COVID-19 patients: An analysis of intensive care unit surveillance data. Antimicrob. Resist. Infect. Control 2021, 10, 119. [Google Scholar] [CrossRef]
- Povoa, P.; Martin-Loeches, I.; Nseir, S. Secondary pneumonias in critically ill patients with COVID-19: Risk factors and outcomes. Curr. Opin. Crit. Care 2021, 27, 468–473. [Google Scholar] [CrossRef]
- Vijay, S.; Bansal, N.; Rao, B.K.; Veeraraghavan, B.; Rodrigues, C.; Wattal, C.; Goyal, J.P.; Tadepalli, K.; Mathur, P.; Venkateswaran, R.; et al. Secondary Infections in Hospitalized COVID-19 Patients: Indian Experience. Infect. Drug Resist. 2021, 14, 1893–1903. [Google Scholar] [CrossRef]
- Francois, B.; Jeannet, R.; Daix, T.; Walton, A.H.; Shotwell, M.S.; Unsinger, J.; Monneret, G.; Rimmele, T.; Blood, T.; Morre, M.; et al. Interleukin-7 restores lymphocytes in septic shock: The IRIS-7 randomized clinical trial. JCI Insight 2018, 3, e98960. [Google Scholar] [CrossRef]
- Maas, S.L.; Soehnlein, O.; Viola, J.R. Organ-Specific Mechanisms of Transendothelial Neutrophil Migration in the Lung, Liver, Kidney, and Aorta. Front. Immunol. 2018, 9, 2739. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factor 1 (HIF-1) pathway. Sci. STKE 2007, 2007, cm8. [Google Scholar] [CrossRef] [PubMed]
- Tamura, D.Y.; Moore, E.E.; Partrick, D.A.; Johnson, J.L.; Offner, P.J.; Silliman, C.C. Acute hypoxemia in humans enhances the neutrophil inflammatory response. Shock 2002, 17, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Ong, C.W.M.; Fox, K.; Ettorre, A.; Elkington, P.T.; Friedland, J.S. Hypoxia increases neutrophil-driven matrix destruction after exposure to Mycobacterium tuberculosis. Sci. Rep. 2018, 8, 11475. [Google Scholar] [CrossRef] [PubMed]
- Lodge, K.M.; Cowburn, A.S.; Li, W.; Condliffe, A.M. The Impact of Hypoxia on Neutrophil Degranulation and Consequences for the Host. Int. J. Mol. Sci. 2020, 21, 1183. [Google Scholar] [CrossRef]
- Möllerherm, H.; Branitzki-Heinemann, K.; Brogden, G.; Elamin, A.A.; Oehlmann, W.; Fuhrmann, H.; Singh, M.; Naim, H.Y.; von Köckritz-Blickwede, M. Hypoxia Modulates the Response of Mast Cells to Staphylococcus aureus Infection. Front. Immunol. 2017, 8, 541. [Google Scholar] [CrossRef]
- Hirani, N.; Antonicelli, F.; Strieter, R.M.; Wiesener, M.S.; Ratcliffe, P.J.; Haslett, C.; Donnelly, S.C. The regulation of interleukin-8 by hypoxia in human macrophages—A potential role in the pathogenesis of the acute respiratory distress syndrome (ARDS). Mol. Med. 2001, 7, 685–697. [Google Scholar] [CrossRef]
- Thompson, A.A.; Dickinson, R.S.; Murphy, F.; Thomson, J.P.; Marriott, H.M.; Tavares, A.; Willson, J.; Williams, L.; Lewis, A.; Mirchandani, A.; et al. Hypoxia determines survival outcomes of bacterial infection through HIF-1alpha dependent re-programming of leukocyte metabolism. Sci. Immunol. 2017, 2, eaal2861. [Google Scholar] [CrossRef]
- Naruko, T.; Ueda, M.; Haze, K.; van der Wal, A.C.; van der Loos, C.M.; Itoh, A.; Komatsu, R.; Ikura, Y.; Ogami, M.; Shimada, Y.; et al. Neutrophil infiltration of culprit lesions in acute coronary syndromes. Circulation 2002, 106, 2894–2900. [Google Scholar] [CrossRef]
- Guan, W.J.; Ni, Z.Y.; Hu, Y.; Liang, W.H.; Ou, C.Q.; He, J.X.; Liu, L.; Shan, H.; Lei, C.L.; Hui, D.S.C.; et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef]
- Giamarellos-Bourboulis, E.J.; Netea, M.G.; Rovina, N.; Akinosoglou, K.; Antoniadou, A.; Antonakos, N.; Damoraki, G.; Gkavogianni, T.; Adami, M.E.; Katsaounou, P.; et al. Complex Immune Dysregulation in COVID-19 Patients with Severe Respiratory Failure. Cell Host Microbe 2020, 27, 992–1000.e1003. [Google Scholar] [CrossRef]
- Qin, C.; Zhou, L.; Hu, Z.; Zhang, S.; Yang, S.; Tao, Y.; Xie, C.; Ma, K.; Shang, K.; Wang, W.; et al. Dysregulation of Immune Response in Patients With Coronavirus 2019 (COVID-19) in Wuhan, China. Clin. Infect. Dis. 2020, 71, 762–768. [Google Scholar] [CrossRef] [PubMed]
- Meizlish, M.L.; Pine, A.B.; Bishai, J.D.; Goshua, G.; Nadelmann, E.R.; Simonov, M.; Chang, C.H.; Zhang, H.; Shallow, M.; Bahel, P.; et al. A neutrophil activation signature predicts critical illness and mortality in COVID-19. Blood Adv. 2021, 5, 1164–1177. [Google Scholar] [CrossRef] [PubMed]
- Chua, R.L.; Lukassen, S.; Trump, S.; Hennig, B.P.; Wendisch, D.; Pott, F.; Debnath, O.; Thürmann, L.; Kurth, F.; Völker, M.T.; et al. COVID-19 severity correlates with airway epithelium-immune cell interactions identified by single-cell analysis. Nat. Biotechnol. 2020, 38, 970–979. [Google Scholar] [CrossRef]
- Aschenbrenner, A.C.; Mouktaroudi, M.; Krämer, B.; Oestreich, M.; Antonakos, N.; Nuesch-Germano, M.; Gkizeli, K.; Bonaguro, L.; Reusch, N.; Baßler, K.; et al. Disease severity-specific neutrophil signatures in blood transcriptomes stratify COVID-19 patients. Genome Med. 2021, 13, 7. [Google Scholar] [CrossRef]
- Middleton, E.A.; He, X.Y.; Denorme, F.; Campbell, R.A.; Ng, D.; Salvatore, S.P.; Mostyka, M.; Baxter-Stoltzfus, A.; Borczuk, A.C.; Loda, M.; et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood 2020, 136, 1169–1179. [Google Scholar] [CrossRef]
- GeurtsvanKessel, C.H.; Geers, D.; Schmitz, K.S.; Mykytyn, A.Z.; Lamers, M.M.; Bogers, S.; Scherbeijn, S.; Gommers, L.; Sablerolles, R.S.G.; Nieuwkoop, N.N.; et al. Divergent SARS-CoV-2 Omicron–reactive T and B cell responses in COVID-19 vaccine recipients. Sci. Immunol. 2022, 7, eabo2202. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Blood ∩ Lung | Heart ∩ Bowel | Bowel ∩ Lung | Heart ∩ Lung | Blood ∩ Bowel |
| H2BC5, H2BC7, H4C8, IGKV2D-28, IGLV2-14, KIF11, PFKFB3, UPP1 | EIF3LP2, IL1RL1, MINCR, RHBDF2, S100A9, TPT1P5, ZBTB42 | CD300E, GK, HILPDA, HK2, IER3, PTX3, SNORA71C | FGG, H2AC16, H2AC18, H2AC19, H2AC21, H2BC17, H4C14, H4C2, HSPB6, MT1XP1, SCARNA6, SDCBP2-AS1, TLR2 | CD38, G0S2, IGHA1, IGLC2, IL18R1, TDRD9, TIMM10 |
| Blood ∩ Heart | Liver ∩ Lung | Liver ∩ Bowel | Blood ∩ Heart ∩ Lung | Liver ∩ Heart |
| HMGB2, IGKC | NDUFB1, RNU6-1016P | BCL2A1, MMP1 | MKI67 | PSMC1, ZNF524 |
| Gene Symbol | Gene Name | Function | ||
| BCL2A1 | BCL2 related protein A1 | An apoptosis-related gene and a direct target of the transcription factor NF-κB in inflammatory responses. | ||
| CD300E | CD300e molecule | It encodes a member of the CD300 family of cell surface receptors expressed on myeloid cells, and it is implicated in the modulation of immune responses. | ||
| CD38 | CD38 molecule | It is broadly expressed in immune system cells, facilitating effective immune responses. | ||
| EIF3LP2 | eukaryotic translation initiation factor 3 subunit L pseudogene 2 | Pseudogene with no known function. | ||
| FGG | fibrinogen gamma chain | It encodes a blood-borne glycoprotein suggested to contribute to immune responses. | ||
| G0S2 | G0/G1 switch 2 | It is associated with cell cycle progression and innate immune response. | ||
| GK | glycerol kinase | It encodes a protein implicated in the regulation of the uptake and metabolism of glycerol that has diverse cellular functions. | ||
| H2AC16 | H2A clustered histone 16 | The encoded protein constitutes a core component of nucleosomes, and it is expressed in B-cells. | ||
| H2AC18 | H2A clustered histone 18 | The encoded protein constitutes a core component of nucleosomes, and it is associated with the immune system. | ||
| H2AC19 | H2A clustered histone 19 | The encoded protein constitutes a core component of nucleosomes, and it is detected in all types of immune cells and is expressed in neutrophils. | ||
| H2AC21 | H2A clustered histone 21 | The encoded protein constitutes a core component of nucleosomes, and it is detected in many types of immune cells. | ||
| H2BC5 | H2B clustered histone 5 | The encoded protein constitutes a core component of nucleosomes, and it is detected in all types of immune cells. | ||
| H2BC7 | H2B clustered histone 7 | The encoded protein constitutes a core component of nucleosomes, and it is detected in some types of immune cells. | ||
| H2BC17 | H2B clustered histone 17 | The encoded protein constitutes a core component of nucleosomes, and it is expressed in eosinophils. | ||
| H4C2 | H4 clustered histone 2 | The encoded protein constitutes a core component of nucleosomes, and it is detected in many types of immune cells and is expressed in neutrophils. | ||
| H4C8 | H4 clustered histone 8 | The encoded protein constitutes a core component of nucleosomes, and it is expressed in eosinophils. | ||
| H4C14 | H4 clustered histone 14 | The encoded protein constitutes a core component of nucleosomes, and it is detected specifically in neutrophils. | ||
| HILPDA | hypoxia inducible lipid droplet associated | It is implicated in various cellular processes, and it is associated with immune cell infiltration. | ||
| HK2 | hexokinase 2 | The encoded protein catalyzes the phosphorylation of glucose to glucose-6-phosphate, and it is an innate immune receptor. | ||
| HMGB2 | High-mobility group box 2 | It encodes a chromatin-associated protein, and it is proposed to be implicated in the innate immune system response to immunogenic nucleic acids. | ||
| HSPB6 | heat shock protein family B (small) member 6 | It encodes a small heat shock protein which serves as a molecular chaperone. | ||
| IER3 | immediate early response 3 | It is involved in apoptosis and is associated with aberrant immune response. | ||
| IGHA1 | immunoglobulin heavy constant alpha 1 | It is linked to receptor-binding activities. | ||
| IGKC | immunoglobulin kappa constant | It participates in antigen binding; it is associated with COVID-19 disease severity. | ||
| IGKV2D-28 | immunoglobulin kappa variable 2D-28 | Effector phase of humoral immunity modulator. | ||
| IGLC2 | immunoglobulin lambda constant 2 | Effector phase of humoral immunity modulator. | ||
| IGLV2-14 | immunoglobulin lambda variable 2-14 | It participates in antigen recognition. | ||
| IL18R1 | interleukin 18 receptor 1 | It encodes a cytokine receptor necessary for IL18 signaling; it is associated with atherosclerosis. | ||
| IL1RL1 | interleukin 1 receptor-like 1 | The encoded protein serves as the receptor of IL33; it is associated with fibrosis and heart failure. | ||
| KIF11 | kinesin family member 11 | It encodes a motor protein implicated in several aspects of spindle dynamics, and it is considered a potential immunological pan-cancer biomarker. | ||
| MINCR | MYC-induced long noncoding RNA | Immune-related long noncoding RNA. | ||
| MKI67 | marker of proliferation Ki-67 | It encodes a protein essential for cell proliferation, and it is correlated with immune cell infiltration. | ||
| MMP1 | matrix metallopeptidase 1 | It encodes a protein involved in extracellular matrix degradation in pathophysiological processes, and it is related to cytokine signaling pathways. | ||
| MT1XP1 | metallothionein 1X pseudogene 1 | Pseudogene with no known function. | ||
| NDUFB1 | NADH:ubiquinone oxidoreductase subunit B1 | It encodes a protein implicated in the assembly of the mitochondrial respiratory chain complex I; it is involved in inflammatory responses, and it is mainly expressed in monocytes. | ||
| PFKFB3 | 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 | It encodes a protein involved in the synthesis and breakdown of fructose-2,6-bisphosphate. | ||
| PSMC1 | proteasome 26S subunit, ATPase 1 | It encodes one of the core subunits of the 19S proteasome complex, and it is involved in immune responses and is detected in all types of immune cells. | ||
| PTX3 | pentraxin 3 | It encodes a member of the pentraxin family of proteins, the expression of which is elicited by inflammatory cytokines. | ||
| RHBDF2 | rhomboid 5 homolog 2 | It facilitates protein transporter activity, and it is correlated with an immunosuppressive tumor microenvironment. | ||
| RNU6-1016P | RNA, U6 small nuclear 1016, pseudogene | Pseudogene with no known function. | ||
| S100A9 | S100 calcium-binding protein A9 | It encodes a member of the S100 protein family which is implicated in the regulation of diverse cellular processes; it plays a role in innate immunity and myeloid-derived suppressor-cell-mediated immune suppression. | ||
| SCARNA6 | small Cajal-body-specific RNA 6 | Small nucleolar RNA which is associated with inflammatory bowel disease. | ||
| SDCBP2-AS1 | SDCBP2 antisense RNA 1 | Long noncoding RNA. | ||
| SNORA71C | small nucleolar RNA, H/ACA box 71C | Small nucleolar RNA with no known function. | ||
| TDRD9 | tudor domain containing 9 | It has a potential role in RNA binding activity; it is involved in inflammatory responses and is mainly expressed in monocytes. | ||
| TIMM10 | translocase of inner mitochondrial membrane 10 | It encodes a component of a protein complex in the mitochondrial intermembrane space. | ||
| TLR2 | Toll-like receptor 2 | It encodes a member of the family of Toll-like receptors known to play a critical role in the innate immune recognition of pathogens. | ||
| TPT1P5 | TPT1 pseudogene 5 | Pseudogene with no known function. | ||
| UPP1 | uridine phosphorylase 1 | The encoded uridine phosphorylase is involved in pyrimidine ribonucleoside salvaging and degradation, and it is linked to immune and inflammatory responses. | ||
| ZBTB42 | zinc finger and BTB domain containing 42 | It encodes a member of the family of C2H2 zinc finger proteins. | ||
| ZNF524 | zinc finger protein 524 | The encoded protein is predicted to regulate RNA polymerase II transcription through sequence-specific DNA binding; it is detected in all types of immune cells and is expressed in eosinophils. | ||
| Gene Symbol | Gene Name | Expression |
|---|---|---|
| ALDOC | aldolase, fructose-bisphosphate C | Down |
| CD14 | CD14 molecule | Up |
| CEACAM1 | CEA cell adhesion molecule 1 | Up |
| CEACAM3 | CEA cell adhesion molecule 3 | Up |
| CYBA | cytochrome b-245 alpha chain | Up |
| DOCK2 | dedicator of cytokinesis 2 | Up |
| ENPP4 | ectonucleotide pyrophosphatase/phosphodiesterase 4 | Down |
| FABP5 | fatty-acid-binding protein 5 | Up |
| FCGR2A | Fc gamma receptor IIa | Up |
| GGH | gamma-glutamyl hydrolase | Up |
| HP | haptoglobin | Up |
| HVCN1 | hydrogen voltage-gated channel 1 | Up |
| IQGAP2 | IQ motif-containing GTPase activating protein 2 | Up |
| S100A9 | S100 calcium-binding protein A9 | Up |
| TLR2 | Toll-like receptor 2 | Down |
| TXNDC5 | thioredoxin domain containing 5 | Up |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Özbek, M.; Toy, H.I.; Takan, I.; Asfa, S.; Arshinchi Bonab, R.; Karakülah, G.; Kontou, P.I.; Geronikolou, S.A.; Pavlopoulou, A. A Counterintuitive Neutrophil-Mediated Pattern in COVID-19 Patients Revealed through Transcriptomics Analysis. Viruses 2023, 15, 104. https://doi.org/10.3390/v15010104
Özbek M, Toy HI, Takan I, Asfa S, Arshinchi Bonab R, Karakülah G, Kontou PI, Geronikolou SA, Pavlopoulou A. A Counterintuitive Neutrophil-Mediated Pattern in COVID-19 Patients Revealed through Transcriptomics Analysis. Viruses. 2023; 15(1):104. https://doi.org/10.3390/v15010104
Chicago/Turabian StyleÖzbek, Melih, Halil Ibrahim Toy, Işil Takan, Seyedehsadaf Asfa, Reza Arshinchi Bonab, Gökhan Karakülah, Panagiota I. Kontou, Styliani A. Geronikolou, and Athanasia Pavlopoulou. 2023. "A Counterintuitive Neutrophil-Mediated Pattern in COVID-19 Patients Revealed through Transcriptomics Analysis" Viruses 15, no. 1: 104. https://doi.org/10.3390/v15010104
APA StyleÖzbek, M., Toy, H. I., Takan, I., Asfa, S., Arshinchi Bonab, R., Karakülah, G., Kontou, P. I., Geronikolou, S. A., & Pavlopoulou, A. (2023). A Counterintuitive Neutrophil-Mediated Pattern in COVID-19 Patients Revealed through Transcriptomics Analysis. Viruses, 15(1), 104. https://doi.org/10.3390/v15010104